
Telomere Architecture Correlates with Aggressiveness in Multiple Myeloma

 , ,
, , 
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Acquisition and Patient Population
2.2. Sample Preparation
2.3. Immunostaining and Telomere Hybridization
2.4. Identification of Malignant Plasma Cells
2.5. Image Acquisition and Nuclear Architecture Analysis
2.6. Statistical Analyses
3. Results
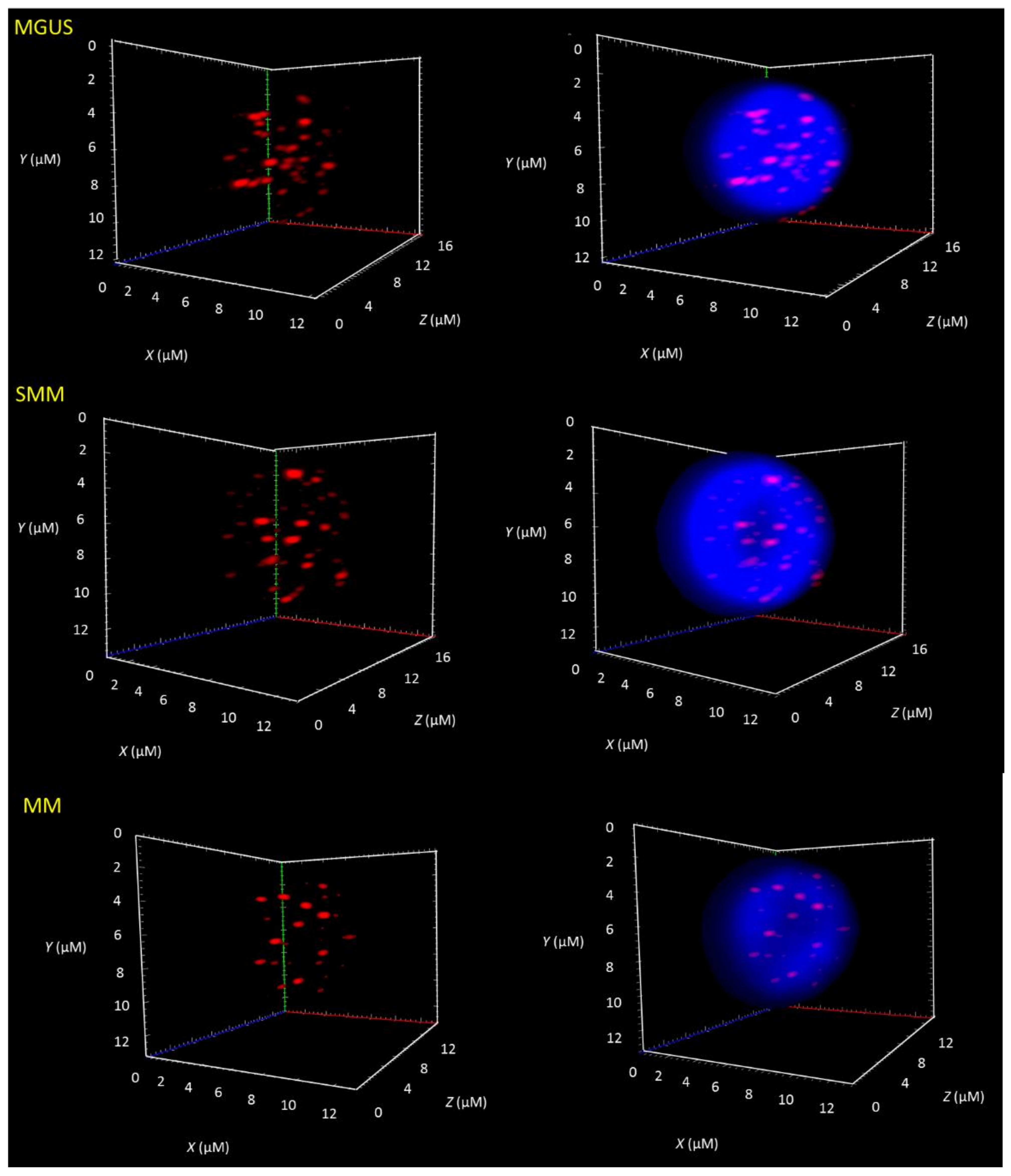
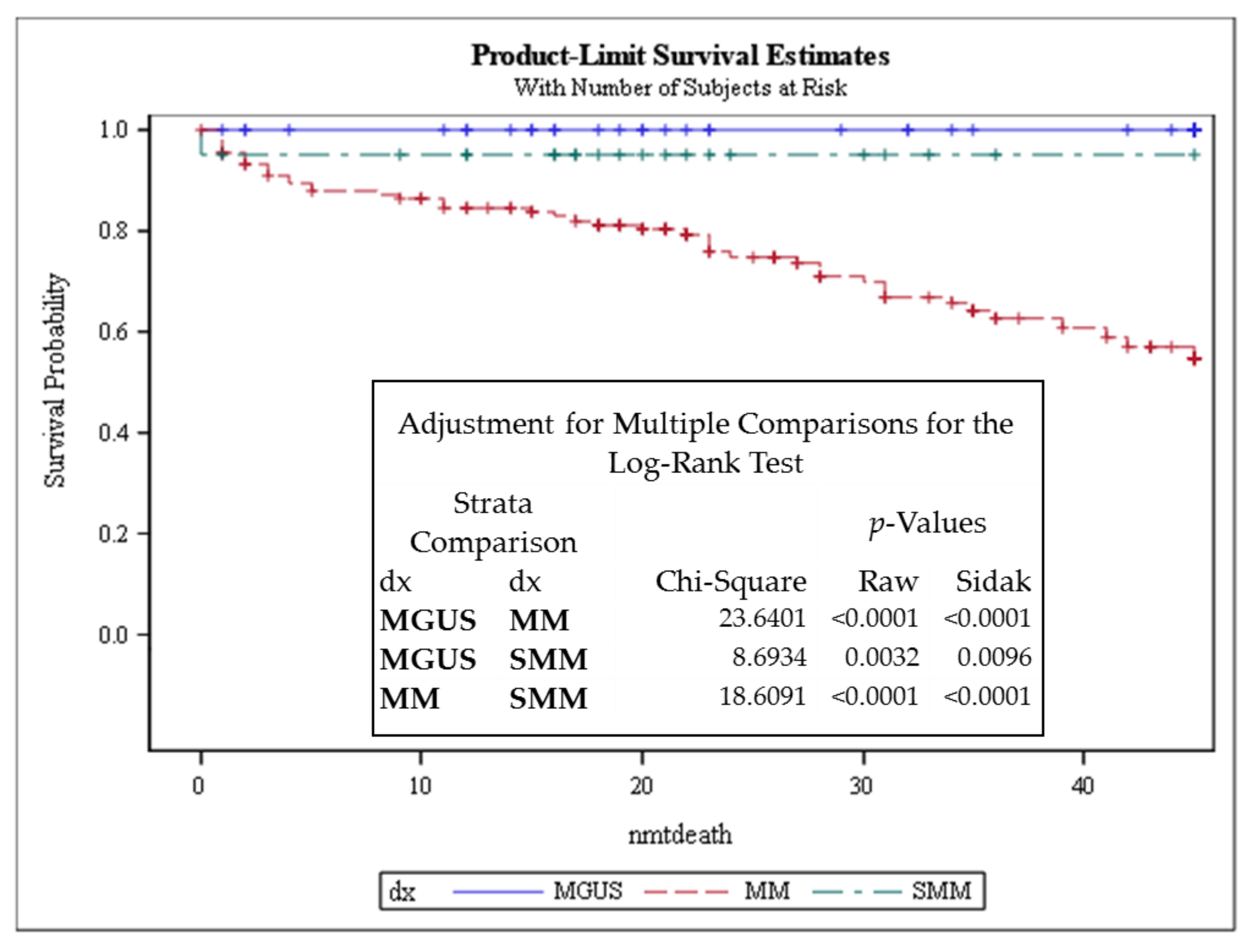
3.1. Telomere-Related Genomic Instability Differentiates Between MGUS, SMM, and MM and Identifies SMM and MM Patients with Progressive Disease
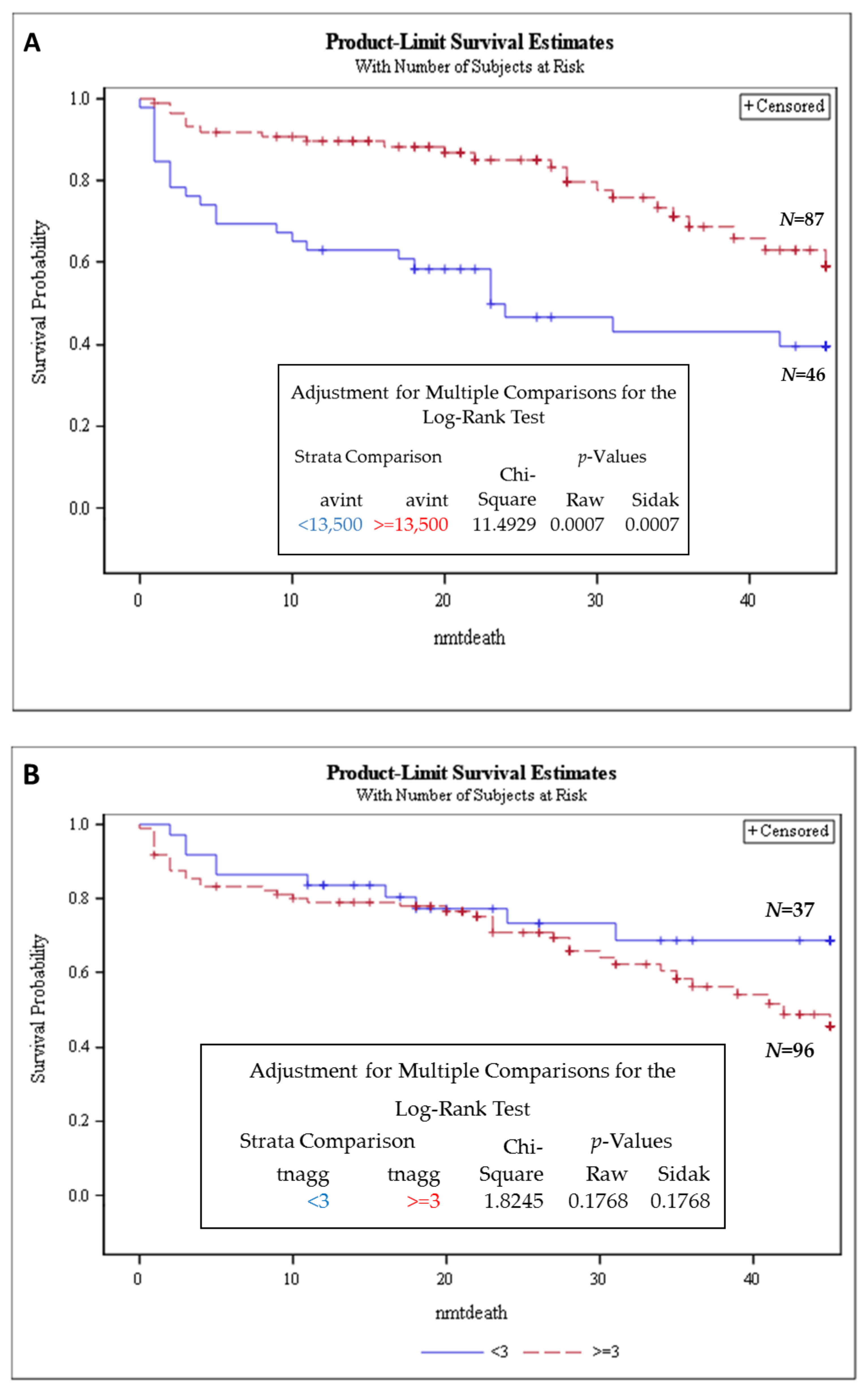
3.2. Stratifying SMM Patients and MM Patients Highlights High-Risk and Progression Groups
3.3. Telomere Intensity Was Significantly Associated with Shorter Overall Survival in MM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hallek, M.; Bergsagel, P.L.; Anderson, K.C. Multiple myeloma: Increasing evidence for a multistep transformation process. Blood 1998, 91, 3–21. [Google Scholar] [CrossRef] [Green Version]
- Dhodapkar, M.V. MGUS to myeloma: A mysterious gammopathy of underexplored significance. Blood 2016, 128, 2599–2606. [Google Scholar] [CrossRef] [PubMed]
- Albagoush, S.A.; Azevedo, A.M. Cancer, Multiple Myeloma; StatPearls Publishing: Tampa, FL, USA, 2018. [Google Scholar]
- Hill, E.; Dew, A.; Kazandjian, D. State of the science in smoldering myeloma: Should we be treating in the clinic? Semin. Oncol. 2019, 46, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispenzieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical Course and Prognosis of Smoldering (Asymptomatic) Multiple Myeloma. N. Engl. J. Med. 2007, 356, 2582–2590. [Google Scholar] [CrossRef]
- Cagnetta, A.; Lovera, D.; Grasso, R.; Colombo, N.; Canepa, L.; Ballerini, F.; Calvio, M.; Miglino, M.; Gobbi, M.; Lemoli, R.; et al. Mechanisms and Clinical Applications of Genome Instability in Multiple Myeloma. Biomed Res. Int. 2015, 2015, 1–8. [Google Scholar] [CrossRef]
- Capp, J.P.; Bataille, R. Multiple myeloma exemplifies a model of cancer based on tissue disruption as the initiator event. Front. Oncol. 2018, 8, 355. [Google Scholar] [CrossRef]
- Ziogas, D.C.; Dimopoulos, M.A.; Kastritis, E. Prognostic factors for multiple myeloma in the era of novel therapies. Expert Rev. Hematol. 2018, 11, 863–879. [Google Scholar] [CrossRef]
- Greipp, P.R.; Miguel, J.S.; Dune, B.G.M.; Crowley, J.J.; Barlogie, B.; Bladé, J.; Boccadoro, M.; Child, J.A.; Harousseau, J.L.; Kyle, R.A.; et al. International staging system for multiple myeloma. J. Clin. Oncol. 2005, 23, 3412–3420. [Google Scholar] [CrossRef]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Neri, P.; Bahlis, N.J. Genomic instability in multiple myeloma: Mechanisms and therapeutic implications. Expert Opin. Biol. Ther. 2013, 13, S69–S82. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Gadji, M.; Pozzo, A.R. From cellular morphology to molecular and epigenetic anomalies of myelodysplastic syndromes. Genes Chromosom. Cancer 2019, 58, 474–483. [Google Scholar] [CrossRef] [Green Version]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mender, I.; Shay, J. Telomere Dysfunction Induced Foci (TIF) Analysis. Bio Protoc. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, S.; Jones, R.E.; Heppel, N.H.; Grimstead, J.W.; Fegan, C.; Jackson, G.H.; Hills, R.; Allan, J.M.; Pratt, G.; Pepper, C.; et al. Telomere length is a critical determinant for survival in multiple myeloma. Br. J. Haematol. 2017, 178, 94–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai, S. Initiation of telomere-mediated chromosomal rearrangements in cancer. J. Cell. Biochem. 2010, 109, 1095–1102. [Google Scholar] [CrossRef]
- Yu, P.L.I.; Wang, R.R.; Johnston, G.; Wang, Y.; Tammur, P.; Tamm, A.; Mai, S. Distinct Nuclear Organization of Telomeresand Centromeres in Monoclonal Gammopathyof Undetermined Significance and Multiple Myeloma. Cells 2019, 8, 723. [Google Scholar] [CrossRef] [Green Version]
- Klewes, L.; Vallente, R.; Dupas, E.; Brand, C.; Grün, D.; Guffei, A.; Sathitruangsak, C.; Awe, J.A.; Kuzyk, A.; Lichtensztejn, D.; et al. Three-dimensional Nuclear Telomere Organization in Multiple Myeloma. Transl. Oncol. 2013, 6, 749-IN36. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, L.H.; Schuster, D.; Herz, H. Generalized approach for accelerated maximum likelihood based image restoration applied to three-dimensional fluorescence microscopy. J. Microsc. 2002, 204, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermolen, B.J.; Garini, Y.; Mai, S.; Mougey, V.; Fest, T.; Chuang, T.C.-Y.; Chuang, A.Y.-C.; Wark, L.; Young, I.T. Characterizing the three-dimensional organization of telomeres. Cytom. Part A 2005, 67, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.H.; Pepper, C.; Baird, D.M. Telomere dysfunction and its role in haematological cancer. Br. J. Haematol. 2012, 156, 573–587. [Google Scholar] [CrossRef]
- Wark, L.; Quon, H.; Ong, A.; Drachenberg, D.; Rangel-Pozzo, A.; Mai, S. Long-Term Dynamics of Three Dimensional Telomere Profiles in Circulating Tumor Cells in High-Risk Prostate Cancer Patients Undergoing Androgen-Deprivation and Radiation Therapy. Cancers 2019, 11, 1165. [Google Scholar] [CrossRef] [Green Version]
- Drachenberg, D.; Awe, J.A.; Pozzo, A.R.; Saranchuk, J.; Mai, S. Advancing risk assessment of intermediate risk prostate cancer patients. Cancers 2019, 11, 855. [Google Scholar] [CrossRef] [Green Version]
- Rangel-Pozzo, A.; Corrêa de Souza, D.; Schmid-Braz, A.T.; de Azambuja, A.P.; Ferraz-Aguiar, T.; Borgonovo, T.; Mai, S. 3D Telomere Structure Analysis to DetectGenomic Instability and Cytogenetic Evolutionin Myelodysplastic Syndromes. Cells 2019, 8, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai, S.; Garini, Y. The significance of telomeric aggregates in the interphase nuclei of tumor cells. J. Cell. Biochem. 2006, 97, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Freitas, M.O.; Gartner, J.; Rangel-Pozzo, A.; Mai, S. Genomic instability in circulating tumor cells. Cancers 2020, 12, 3001. [Google Scholar] [CrossRef]
- Rangel-Pozzo, A.; Sisdelli, L.; Cordioli, M.I.V.; Vaisman, F.; Caria, P.; Mai, S.; Cerutti, J.M. Genetic landscape of papillary thyroid carcinoma and nuclear architecture: An overview comparing pediatric and adult populations. Cancers 2020, 12, 3146. [Google Scholar] [CrossRef]
- Rangel-Pozzo, A.; Booth, S.; Yu, P.L.I.; Singh, M.; Selivanova, G.; Mai, S. p53 CRISPR Deletion Affects DNA Structure and Nuclear Architecture. J. Clin. Med. 2020, 9, 598. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.D.; Orme, L.M.; Shaughnessy, J.; Jacobson, J.; Barlogie, B.; Moore, M.A.S. Telomerase and telomere length in multiple myeloma: Correlations with disease heterogeneity, cytogenetic status, and overall survival. Blood 2003, 101, 4982–4989. [Google Scholar] [CrossRef] [PubMed]
- Cottliar, A.; Pedrazzini, E.; Corrado, C.; Engelberger, M.I.; Narbaitz, M.; Slavutsky, I. Telomere shortening in patients with plasma cell disorders. Eur. J. Haematol. 2003, 71, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Maura, F.; Minvielle, S.; Gloznik, D.; Szalat, R.; Fullam, A.; Martincorena, I.; Dawson, K.J.; Samur, M.K.; Zamora, J.; et al. Genomic patterns of progression in smoldering multiple myeloma. Nat. Commun. 2018, 9, 3363. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateos, M.V.; Kumar, S.; Dimopoulos, M.A.; González-Calle, V.; Kastritis, E.; Hajek, R.; De Larrea, C.F.; Morgan, G.J.; Merlini, G.; Goldschmidt, H.; et al. International Myeloma Working Group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer J. 2020, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Alagpulinsa, D.A.; Szalat, R.E.; Poznansky, M.C.; Shmookler Reis, R.J. Genomic Instability in Multiple Myeloma. Trends Cancer 2020, 6, 858–873. [Google Scholar] [CrossRef]
- Beksac, M.; Balli, S.; Akcora Yildiz, D. Drug Targeting of Genomic Instability in Multiple Myeloma. Front. Genet. 2020, 11, 228. [Google Scholar] [CrossRef] [PubMed]
- Schrank, Z.; Khan, N.; Osude, C.; Singh, S.; Miller, R.; Merrick, C.; Mabel, A.; Kuckovic, A.; Puri, N. Oligonucleotides Targeting Telomeres and Telomerase in Cancer. Molecules 2018, 23, 2267. [Google Scholar] [CrossRef] [Green Version]
- Shammas, M.A.; Koley, H.; Bertheau, R.C.; Neri, P.; Fulciniti, M.; Tassone, P.; Blotta, S.; Protopopov, A.; Mitsiades, C.; Batchu, R.B.; et al. Telomerase inhibitor GRN163L inhibits myeloma cell growth in vitro and in vivo. Leukemia 2008, 22, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Neri, P.; Ren, L.; Gratton, K.; Stebner, E.; Johnson, J.; Klimowicz, A.; Duggan, P.; Tassone, P.; Mansoor, A.; Stewart, D.A.; et al. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood 2011, 118, 6368–6379. [Google Scholar] [CrossRef] [Green Version]
- Botrugno, O.A.; Bianchessi, S.; Zambroni, D.; Frenquelli, M.; Belloni, D.; Bongiovanni, L.; Girlanda, S.; Di Terlizzi, S.; Ferrarini, M.; Ferrero, E.; et al. ATR addiction in multiple myeloma: Synthetic lethal approaches exploiting established therapies. Haematologica 2019, 105, 2440–2447. [Google Scholar] [CrossRef]
- Bajpai, R.; Matulis, S.M.; Wei, C.; Nooka, A.K.; Von Hollen, H.E.; Lonial, S.; Boise, L.H.; Shanmugam, M. Targeting glutamine metabolism in multiple myeloma enhances BIM binding to BCL-2 eliciting synthetic lethality to venetoclax. Oncogene 2016, 35, 3955–3964. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | MGUS | SMM | MM |
|---|---|---|---|
| Number of Patients | 54 | 24 | 136 |
| Basic Clinical Characteristics | |||
| Age | 66.6 ± 10.0 | 70.8 ± 10.0 | 67.5 ± 11.5 |
| BMPC (%) | 2.8 ± 1.3 | 18.1 ± 6.3 | 37.9 ± 28.2 |
| M-protein | 5.5 ± 4.5 | 17.1 ± 10.6 | 26.9 ± 21.4 |
| IgG (%) | 12.5 | 25.5 | 30.9 |
| IgA (%) | 3.6 | 7.7 | 9.9 |
| IgM (%) | 3.3 | 0.7 | 0.3 |
| Lytic lesions (%) | 0/54 (0%) | 0/21 (0%) | 86/135 (63.7%) |
| Cytogenetic information | |||
| Patients with t(11;14) (%) | 1/36 (2.7%) | N/A | 20/25 (80.0%) |
| Patients with t(4;14) (%) | 1/8 (12.5) | N/A | 8/19 (42.1%) |
| Patients with del(14q1.3)/13qter (%) | 0/8 (0%) | N/A | 3/19 (15.8%) |
| Telomere Parameters | MGUS vs. MM (All Cases) | MGUS vs. SMM (All Cases) | SMM (All Cases) vs. MM (All Cases) | SMM Stable (stb) vs. SMM with High Risk to Progression (prg) | MM Stable (stb) vs. MM with Progressive Disease (prg) |
|---|---|---|---|---|---|
| Telomere numbers | p = 0.0193 (↑ MGUS × MM ↓) | ns | ns | p ≤ 0.0001 (↓ stb × prg ↑) | p ≤ 0.0001 (↓ stb × prg ↑) |
| Total telomere intensity | ns | p = 0.0370 (↓ MGUS × SMM ↑) | p = 0.0019 (↑ SMM × MM ↓) | p ≤ 0.0001 (↓ stb × prg ↑) | ns |
| Average telomere intensity | ns | p = 0.0009 (↓ MGUS × SMM ↑) | p = 0.0097 (↑ SMM × MM ↓) | p = 0.0493 (↓ stb × prg ↑) | p ≤ 0.0001 (↑ stb × prg ↓) |
| Telomere aggregates | ns | ns | ns | p = 0.0014 (↓ stb × prg ↑) | p = 0.0001 (↓ stb × prg ↑) |
| a/c ratio | p = 0.0112 (↓ MGUS × MM ↑) | ns | ns | p ≤ 0.0001 (↓ stb × prg ↑) | p ≤ 0.0001 (↓ stb × prg ↑) |
| Nuclear volume | ns | p = 0.05 (↓ MGUS × SMM ↑) | ns | p = 0.0033 (↓ stb × prg ↑) | p ≤ 0.0001 (↓ stb × prg ↑) |
| Telomeres per nuclear volumes | ns | ns | ns | ns | ns |
| Analysis of Maximum Likelihood Estimates | ||||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | DF | Parameter Estimate | Standard Error | Chi-Square | Pr > ChiSq | Hazard Ratio | 95% Hazard Ratio Confidence Limits | |
| AVINT | 1 | −0.45436 | 0.13470 | 11.3781 | 0.0007 | 0.635 | 0.488 | 0.827 |
| TOTIN | 1 | 0.04296 | 0.02112 | 4.1358 | 0.0420 | 1.044 | 1.002 | 1.088 |
| MMDX | 1 | 2.07806 | 0.60041 | 11.9789 | 0.0005 | 7.989 | 2.463 | 25.916 |
| SMMDX | 1 | 2.01007 | 0.75049 | 7.1736 | 0.0074 | 7.464 | 1.715 | 32.492 |
| AGE | 1 | 0.04876 | 0.01310 | 13.8605 | 0.0002 | 1.050 | 1.023 | 1.077 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rangel-Pozzo, A.; Yu, P.L.I.; LaL, S.; Asbaghi, Y.; Sisdelli, L.; Tammur, P.; Tamm, A.; Punab, M.; Klewes, L.; Louis, S.; et al. Telomere Architecture Correlates with Aggressiveness in Multiple Myeloma. Cancers 2021, 13, 1969. https://doi.org/10.3390/cancers13081969
Rangel-Pozzo A, Yu PLI, LaL S, Asbaghi Y, Sisdelli L, Tammur P, Tamm A, Punab M, Klewes L, Louis S, et al. Telomere Architecture Correlates with Aggressiveness in Multiple Myeloma. Cancers. 2021; 13(8):1969. https://doi.org/10.3390/cancers13081969
Chicago/Turabian StyleRangel-Pozzo, Aline, Pak Lok Ivan Yu, Sadhana LaL, Yasmin Asbaghi, Luiza Sisdelli, Pille Tammur, Anu Tamm, Mari Punab, Ludger Klewes, Sherif Louis, and et al. 2021. "Telomere Architecture Correlates with Aggressiveness in Multiple Myeloma" Cancers 13, no. 8: 1969. https://doi.org/10.3390/cancers13081969
APA StyleRangel-Pozzo, A., Yu, P. L. I., LaL, S., Asbaghi, Y., Sisdelli, L., Tammur, P., Tamm, A., Punab, M., Klewes, L., Louis, S., Knecht, H., Olujohungbe, A., & Mai, S. (2021). Telomere Architecture Correlates with Aggressiveness in Multiple Myeloma. Cancers, 13(8), 1969. https://doi.org/10.3390/cancers13081969