
Harnessing Natural Killer Cells in Cancer Immunotherapy: A Review of Mechanisms and Novel Therapies


Abstract
:Simple Summary
Abstract
1. Introduction
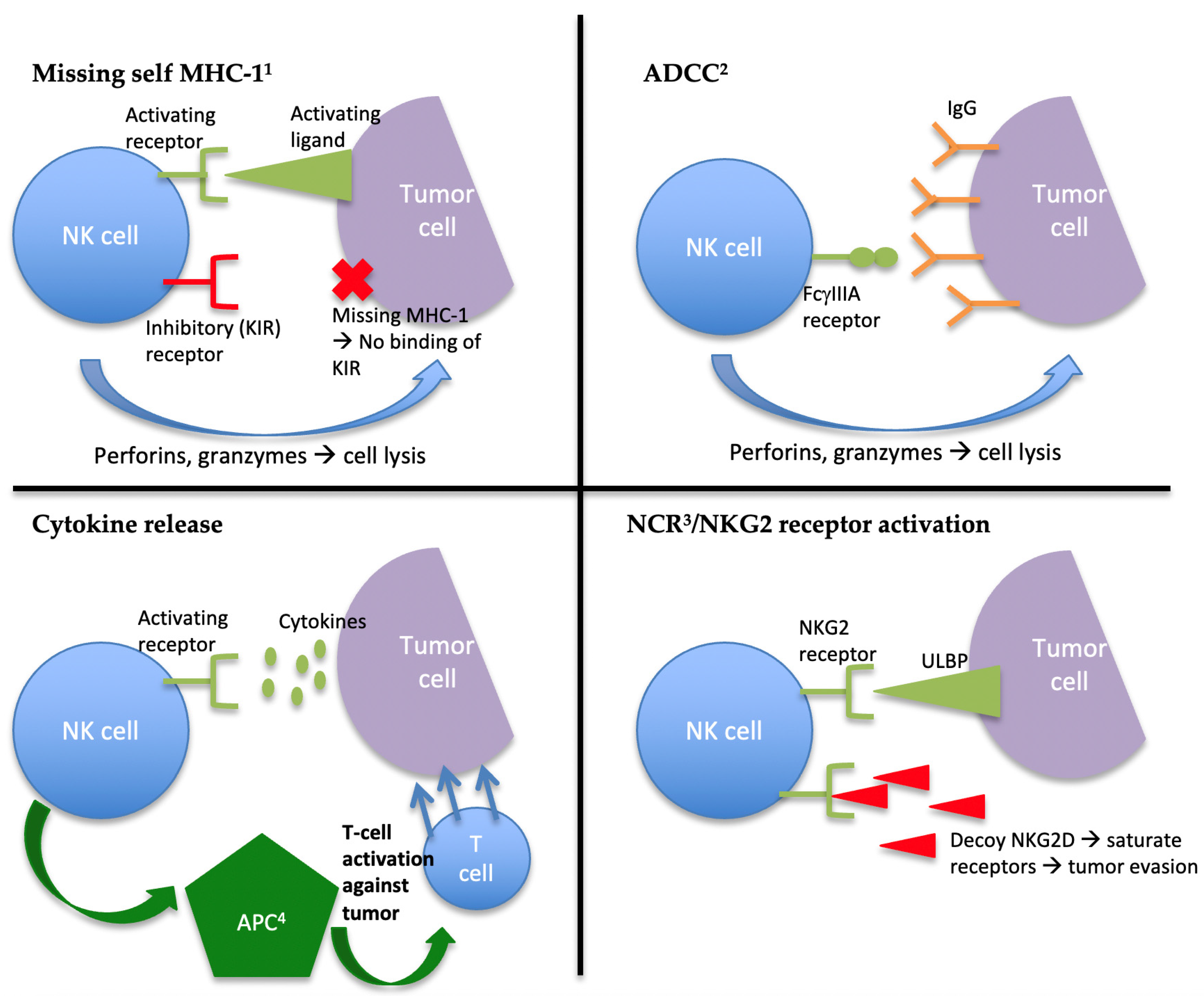
2. The Role of NK Cells in Innate Immunity and Tumor Suppression
3. Current Advances in NK Cell Targeted Therapy
3.1. Checkpoint Inhibitors
3.2. Targeting NK Cell Activation Through Cytokine Release
3.3. Promoting NK Cell ADCC
3.4. Adoptive NK Cell Immunotherapy
3.5. CAR-NK Therapy
3.6. Enhancing Drug Delivery
3.7. Overcoming Suppression by the Tumor Microenvironement
4. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.; Papneja, N.; Miller, W. A Review of Cancer Immunotherapy: From the Past, to the Present, to the Future. Curr. Oncol. 2020, 27, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Guo, F. Recent updates on cancer immunotherapy. Precis. Clin. Med. 2018, 1, 65–74. [Google Scholar] [CrossRef]
- Jinushi, M. The role of innate immune signals in antitumor immunity. OncoImmunology 2012, 1, 189–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Janeway, C.; Paul, T.; Walport, M.; Shlomchik, M. Immunobiology, 4th ed.; Garland Science: New York, NY, USA; London, UK, 2001. [Google Scholar]
- Yokoyama, W.M.; E Seaman, W. The Ly-49 and NKR-P1 Gene Families Encoding Lectin-Like Receptors on Natural Killer Cells: The NK Gene Complex. Annu. Rev. Immunol. 1993, 11, 613–635. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activatingreceptors andcoreceptorsinvolved inhumannaturalkillercell-mediatedcytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef]
- Ochoa, M.C.; Minute, L.; Rodriguez, I.; Garasa, S.; Perez-Ruiz, E.; Inogés, S.; Melero, I.; Berraondo, P. Antibody-dependent cell cytotoxicity: Immunotherapy strategies enhancing effector NK cells. Immunol. Cell Biol. 2017, 95, 347–355. [Google Scholar] [CrossRef]
- Moretta, A.; Bottino, C.; Pende, D.; Tripodi, G.; Tambussi, G.; Viale, O.; Orengo, A.; Barbaresi, M.; Merli, A.; Ciccone, E. Identification of four subsets of human CD3-CD16+ natural killer (NK) cells by the expression of clonally distributed functional surface molecules: Correlation between subset assignment of NK clones and ability to mediate specific alloantigen recognition. J. Exp. Med. 1990, 172, 1589–1598. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S. Natural killer cell cytotoxicity and its regulation by inhibitory receptors. Immunol. 2018, 154, 383–393. [Google Scholar] [CrossRef]
- Frazao, A.; Rethacker, L.; Messaoudene, M.; Avril, M.-F.; Toubert, A.; Dulphy, N.; Caignard, A. NKG2D/NKG2-Ligand Pathway Offers New Opportunities in Cancer Treatment. Front. Immunol. 2019, 10, 661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braud, V.M.; Allan, D.S.J.; O’Callaghan, C.A.; Söderström, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nat. Cell Biol. 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Biassoni, R.; Mingari, M.C.; Moretta, L. Receptors for HLA class-I molecules in human natural killer cells. Annu. Rev. Immunol. 1996, 14, 619–648. [Google Scholar] [CrossRef] [PubMed]
- Pesce, S.; Greppi, M.; Grossi, F.; Del Zotto, G.; Moretta, L.; Sivori, S.; Genova, C.; Marcenaro, E. PD/1-PD-Ls Checkpoint: Insight on the Potential Role of NK Cells. Front. Immunol. 2019, 10, 1242. [Google Scholar] [CrossRef]
- Minetto, P.; Guolo, F.; Pesce, S.; Greppi, M.; Obino, V.; Ferretti, E.; Sivori, S.; Genova, C.; Lemoli, R.M.; Marcenaro, E. Harnessing NK Cells for Cancer Treatment. Front. Immunol. 2019, 10, 2836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGilvray, R.W.; Eagle, R.A.; Watson, N.F.; Al-Attar, A.; Ball, G.; Jafferji, I.; Trowsdale, J.; Durrant, L.G. NKG2D Ligand Expression in Human Colorectal Cancer Reveals Associations with Prognosis and Evidence for Immunoediting. Clin. Cancer Res. 2009, 15, 6993–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melero, I.; Rouzaut, A.; Motz, G.T.; Coukos, G. T-Cell and NK-Cell Infiltration into Solid Tumors: A Key Limiting Factor for Efficacious Cancer Immunotherapy. Cancer Discov. 2014, 4, 522–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, M.; Cantoni, C.; Pietra, G.; Mingari, M.C.; Moretta, L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 2014, 44, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Sarhan, D.; Steven, A.; Seliger, B.; Kiessling, R.; Lundqvist, A. Inhibition of Tumor-Derived Prostaglandin-E2 Blocks the Induction of Myeloid-Derived Suppressor Cells and Recovers Natural Killer Cell Activity. Clin. Cancer Res. 2014, 20, 4096–4106. [Google Scholar] [CrossRef] [Green Version]
- Cekic, C.; Day, Y.-J.; Sag, D.; Linden, J. Myeloid Expression of Adenosine A2A Receptor Suppresses T and NK Cell Responses in the Solid Tumor Microenvironment. Cancer Res. 2014, 74, 7250–7259. [Google Scholar] [CrossRef] [Green Version]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite l-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef]
- Moretta, L.; Bottino, C.; Pende, D.; Vitale, M.; Mingari, M.; Moretta, A. Different checkpoints in human NK-cell activation. Trends Immunol. 2004, 25, 670–676. [Google Scholar] [CrossRef]
- Romagné, F.; André, P.; Spee, P.; Zahn, S.; Anfossi, N.; Gauthier, L.; Capanni, M.; Ruggeri, L.; Benson, J.D.M.; Blaser, B.W.; et al. Preclinical characterization of 1-7F9, a novel human anti–KIR receptor therapeutic antibody that augments natural killer–mediated killing of tumor cells. Blood 2009, 114, 2667–2677. [Google Scholar] [CrossRef]
- Zaghi, E.; Calvi, M.; Marcenaro, E.; Mavilio, D.; Di Vito, C. Targeting NKG2A to elucidate natural killer cell ontogenesis and to develop novel immune-therapeutic strategies in cancer therapy. J. Leukoc. Biol. 2019, 105, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Vey, N.; Karlin, L.; Sadot-Lebouvier, S.; Broussais, F.; Berton-Rigaud, D.; Rey, J.; Charbonnier, A.; Marie, D.; André, P.; Paturel, C.; et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget 2018, 9, 17675–17688. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Infante, J.R.; Gibney, G.T.; Bertino, E.M.; Cooley, S.A.; Lekatis, K.; Wigginton, J.M.; Gutierrez, A.A.; Gupta, A.K.; Kim, S.Y.; et al. A phase I study of lirilumab (BMS-986015), an anti-KIR monoclonal antibody, administered in combination with ipilimumab, an anti-CTLA4 monoclonal antibody, in patients (Pts) with select advanced solid tumors. J. Clin. Oncol. 2013, 31, TPS3106. [Google Scholar] [CrossRef]
- Segal, N.; Infante, J.; Sanborn, R.; Gibney, G.; Lawrence, D.; Rizvi, N.; Leidner, R.; Gajewski, T.; Bertino, E.; Sharfman, W.; et al. Safety of the natural killer (NK) cell-targeted anti-KIR antibody, lirilumab (liri), in combination with nivolumab (nivo) or ipilimumab (ipi) in two phase 1 studies in advanced refractory solid tumors. Ann. Oncol. 2016, 27, vi372. [Google Scholar] [CrossRef]
- McKee, S. Lirilimumab/Opdivo Combo Shows “No Clear Evidence of Benefit”. 2017. Available online: http://www.pharmatimes.com/news/lirilumabopdivo_combo_shows_no_clear_evidence_of_benefit_1212529 (accessed on 31 March 2021).
- Armand, P.; Lesokhin, A.; Borrello, I.; Timmerman, J.; Gutierrez, M.; Zhu, L.; McKiver, M.P.; Ansell, S.M. A phase 1b study of dual PD-1 and CTLA-4 or KIR blockade in patients with relapsed/refractory lymphoid malignancies. Leuk. 2021, 35, 777–786. [Google Scholar] [CrossRef]
- Vey, N.; Dumas, P.-Y.; Recher, C.; Gastaud, L.; Lioure, B.; Bulabois, C.-E.; Pautas, C.; Marolleau, J.-P.; Leprêtre, S.; Raffoux, E.; et al. Randomized Phase 2 Trial of Lirilumab (anti-KIR monoclonal antibody, mAb) As Maintenance Treatment in Elderly Patients (pts) with Acute Myeloid Leukemia (AML): Results of the Effikir Trial. Blood 2017, 130, 889. [Google Scholar] [CrossRef]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef] [Green Version]
- McWilliams, E.M.; Mele, J.M.; Cheney, C.; Timmerman, E.A.; Fiazuddin, F.; Strattan, E.J.; Mo, X.; Byrd, J.C.; Muthusamy, N.; Awan, F.T. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. OncoImmunology 2016, 5, e1226720. [Google Scholar] [CrossRef] [Green Version]
- Seymour, L.; Tinker, A.; Hirte, H.; Wagtmann, N.; Dodion, P. Phase I and dose ranging, phase II studies with IPH2201, a humanized monoclonal antibody targeting HLA-E receptor CD94/NKG2A. Ann. Oncol. 2015, 26, ii3. [Google Scholar] [CrossRef]
- Tinker, A.V.; Hirte, H.W.; Provencher, D.; Butler, M.; Ritter, H.; Tu, D.; Azim, H.A.; Paralejas, P.; Grenier, N.; Hahn, S.-A.; et al. Dose-Ranging and Cohort-Expansion Study of Monalizumab (IPH2201) in Patients with Advanced Gynecologic Malignancies: A Trial of the Canadian Cancer Trials Group (CCTG): IND221. Clin. Cancer Res. 2019, 25, 6052–6060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Hu, Y.; Shi, C. Targeting Natural Killer Cells for Tumor Immunotherapy. Front. Immunol. 2020, 11, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Sun, H.; Huang, Q.; Huang, M.; Wen, H.; Lin, R.; Zheng, M.; Qu, K.; Li, K.; Wei, H.; Xiao, W.; et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatol. 2019, 70, 168–183. [Google Scholar] [CrossRef]
- Fang, F.; Xiao, W.; Tian, Z. NK cell-based immunotherapy for cancer. Semin. Immunol. 2017, 31, 37–54. [Google Scholar] [CrossRef]
- Ito, S.; Bollard, C.M.; Carlsten, M.; Melenhorst, J.J.; Biancotto, A.; Wang, E.; Chen, J.; Kotliarov, Y.; Cheung, F.; Xie, Z.; et al. Ultra-low Dose Interleukin-2 Promotes Immune-modulating Function of Regulatory T Cells and Natural Killer Cells in Healthy Volunteers. Mol. Ther. 2014, 22, 1388–1395. [Google Scholar] [CrossRef] [Green Version]
- Levin, A.M.; Bates, D.L.; Ring, A.M.; Krieg, C.; Lin, J.T.; Su, L.; Moraga, I.L.; Raeber, M.E.; Bowman, G.R.; Novick, P.; et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nat. Cell Biol. 2012, 484, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.S.; Morishima, C.; McNeel, D.G.; Patel, M.R.; Kohrt, H.E.; Thompson, J.A.; Sondel, P.M.; Wakelee, H.A.; Disis, M.L.; Kaiser, J.C.; et al. A First-in-Human Phase I Study of Subcutaneous Outpatient Recombinant Human IL15 (rhIL15) in Adults with Advanced Solid Tumors. Clin. Cancer Res. 2017, 24, 1525–1535. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, T.A.; Dubois, S.; Miljkovic, M.D.; Conlon, K.C. IL-15 in the Combination Immunotherapy of Cancer. Front. Immunol. 2020, 11, 868. [Google Scholar] [CrossRef]
- Han, K.-P.; Zhu, X.; Liu, B.; Jeng, E.; Kong, L.; Yovandich, J.L.; Vyas, V.V.; Marcus, W.D.; Chavaillaz, P.-A.; Romero, C.A.; et al. IL-15:IL-15 receptor alpha superagonist complex: High-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine 2011, 56, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrangle, J.M.; Velcheti, V.; Patel, M.R.; Garrett-Mayer, E.; Hill, E.G.; Ravenel, J.G.; Miller, J.S.; Farhad, M.; Anderton, K.; Lindsey, K.; et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: A non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018, 19, 694–704. [Google Scholar] [CrossRef]
- Vasu, S.; Sharma, N.; Odonnell, L.; Bosse, K.; Lee, D.A. A phase I clinical trial testing the safety of IL-21-expanded, off-the-shelf, natural killer cells for relapsed/refractory acute myeloid leukemia and myelodysplastic syndrome. J. Clin. Oncol. 2020, 38, TPS7562. [Google Scholar] [CrossRef]
- Nguyen, K.G.; Vrabel, M.R.; Mantooth, S.M.; Hopkins, J.J.; Wagner, E.S.; Gabaldon, T.A.; Zaharoff, D.A. Localized Interleukin-12 for Cancer Immunotherapy. Front. Immunol. 2020, 11, 575597. [Google Scholar] [CrossRef] [PubMed]
- Varchetta, S.; Gibelli, N.; Oliviero, B.; Nardini, E.; Gennari, R.; Gatti, G.; Silva, L.S.; Villani, L.; Tagliabue, E.; Ménard, S.; et al. Elements Related to Heterogeneity of Antibody-Dependent Cell Cytotoxicity in Patients Under Trastuzumab Therapy for Primary Operable Breast Cancer Overexpressing Her2. Cancer Res. 2007, 67, 11991–11999. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Huang, K.; Liu, L.; Qu, Y.; Huang, Y.; Wu, Y.; Wei, J. Effects of complement and serum IgG on rituximab‑dependent natural killer cell‑mediated cytotoxicity against Raji cells. Oncol. Lett. 2018, 17, 339–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binyamin, L.; Alpaugh, P.R.K.; Campbell, P.K.S.; Borghaei, D.H.; Weiner, L.M. Rituximab-Mediated ADCC Is Augmented by Concomitant Interference with Inhibitory Self-Recognition by Human NK Cells. Blood 2005, 106, 2456. [Google Scholar] [CrossRef]
- Barth, M.J.; Mavis, C.; Czuczman, M.S.; Hernandez-Ilizaliturri, F.J. Ofatumumab Exhibits Enhanced In Vitro and In Vivo Activity Compared to Rituximab in Preclinical Models of Mantle Cell Lymphoma. Clin. Cancer Res. 2015, 21, 4391–4397. [Google Scholar] [CrossRef] [Green Version]
- Koerner, S.P.; André, M.C.; Leibold, J.S.; Kousis, P.C.; Kübler, A.; Pal, M.; Haen, S.P.; Bühring, H.-J.; Grosse-Hovest, L.; Jung, G.; et al. An Fc-optimized CD133 antibody for induction of NK cell reactivity against myeloid leukemia. Leuk. 2017, 31, 459–469. [Google Scholar] [CrossRef]
- Kim, Y.M.; Park, J.S.; Kim, S.K.; Jung, K.M.; Hwang, Y.S.; Han, M.; Lee, H.J.; Seo, H.W.; Suh, J.-Y.; Han, B.K.; et al. The transgenic chicken derived anti-CD20 monoclonal antibodies exhibits greater anti-cancer therapeutic potential with enhanced Fc effector functions. Biomater. 2018, 167, 58–68. [Google Scholar] [CrossRef]
- Jochems, C.; Hodge, J.W.; Fantini, M.; Fujii, R.; Ii, Y.M.M.; Greiner, J.W.; Padget, M.R.; Tritsch, S.R.; Tsang, K.Y.; Campbell, K.S.; et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget 2016, 7, 86359–86373. [Google Scholar] [CrossRef] [Green Version]
- Kellner, C.; Bruenke, J.; Horner, H.; Schubert, J.; Schwenkert, M.; Mentz, K.; Barbin, K.; Stein, C.; Peipp, M.; Stockmeyer, B. Heterodimeric bispecific antibody-derivatives against CD19 and CD16 induce effective antibody-dependent cellular cytotoxicity against B-lymphoid tumor cells. Cancer Lett. 2011, 303, 128–139. [Google Scholar] [CrossRef]
- Yang, C.; Li, Y.; Yang, Y.; Chen, Z. Overview of Strategies to Improve Therapy against Tumors Using Natural Killer Cell. J. Immunol. Res. 2020, 2020, 8459496. [Google Scholar] [CrossRef] [PubMed]
- Felices, M.; Kodal, B.; Hinderlie, P.; Kaminski, M.F.; Cooley, S.; Weisdorf, D.J.; Vallera, D.A.; Miller, J.S.; Bachanova, V. Novel CD19-targeted TriKE restores NK cell function and proliferative capacity in CLL. Blood Adv. 2019, 3, 897–907. [Google Scholar] [CrossRef]
- Au, K.M.; Park, S.I.; Wang, A.Z. Trispecific natural killer cell nanoengagers for targeted chemoimmunotherapy. Sci. Adv. 2020, 6, eaba8564. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Chang, A.E.; Avis, F.P.; Leitman, S.; Linehan, W.M.; Robertson, C.N.; Lee, R.E.; Rubin, J.T.; et al. A Progress Report on the Treatment of 157 Patients with Advanced Cancer Using Lymphokine-Activated Killer Cells and Interleukin-2 or High-Dose Interleukin-2 Alone. N. Engl. J. Med. 1987, 316, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Bachanova, V.; Miller, J.S. NK Cells in Therapy of Cancer. Crit. Rev. Oncog. 2014, 19, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Velardi, A.; Ruggeri, L.; Mancusi, A. Killer-cell immunoglobulin-like receptors reactivity and outcome of stem cell transplant. Curr. Opin. Hematol. 2012, 19, 319–323. [Google Scholar] [CrossRef]
- Ciurea, S.O.; Schafer, J.R.; Bassett, R.; Denman, C.J.; Cao, K.; Willis, D.; Rondon, G.; Chen, J.; Soebbing, D.; Kaur, I.; et al. Phase 1 clinical trial using mbIL21 ex vivo–expanded donor-derived NK cells after haploidentical transplantation. Blood 2017, 130, 1857–1868. [Google Scholar] [CrossRef]
- Ciurea, S.O.; Bassett, R.; Soebbing, D.; Rondon, G.; Cao, K.; Olson, A.L.; Bashir, Q.; Shpall, E.J.; Devine, S.; Pasquini, M.C.; et al. Improved Outcomes for Patients Receiving High-Doses of IL-21 Ex Vivo Expanded NK Cells after Haploidentical Transplantation (haploSCT): Long-Term Follow-up of a Phase 1/2 Clinical Trial with Comparison to CIBMTR Controls. Blood 2019, 134, 700. [Google Scholar] [CrossRef]
- Evert, J.S.H.-V.; Bekkers, R.; Ottevanger, N.; Jansen, J.H.; Massuger, L.; Dolstra, H. Harnessing natural killer cells for the treatment of ovarian cancer. Gynecol. Oncol. 2020, 157, 810–816. [Google Scholar] [CrossRef]
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. NK cell-based immunotherapy for malignant diseases. Cell. Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef]
- Dahlberg, C.I.M.; Sarhan, D.; Chrobok, M.; Duru, A.D.; Alici, E. Natural Killer Cell-Based Therapies Targeting Cancer: Possible Strategies to Gain and Sustain Anti-Tumor Activity. Front. Immunol. 2015, 6, 605. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Church, C.; Paulson, K.; Pierce, R.; Nghiem, P.; Lee, J.; Adcock, B.; Soon-Shiong, P.; Chandra, S. Final Results from a Phase 2 Study Using Off-the-Shelf Activated Natural Killer (aNK) Cells in Combination with N-803, an IL-15 Superagonist, in Patients with Metastatic Merkel Cell Carcinoma (MCC); Society for Immunotherapy of Cancer: National Harbor, MD, USA, 2019. [Google Scholar]
- Luna, J.I.; Grossenbacher, S.K.; Sturgill, I.R.; Ames, E.; Judge, S.J.; Bouzid, L.A.; Darrow, M.A.; Murphy, W.J.; Canter, R.J. Bortezomib Augments Natural Killer Cell Targeting of Stem-Like Tumor Cells. Cancers 2019, 11, 85. [Google Scholar] [CrossRef] [Green Version]
- Shaim, H.; Sanabria, M.H.; Basar, R.; Wang, F.; Daher, M.; Zamler, D.; Gumin, J.; Gabrusiewicz, K.; Miao, Q.; Dou, J.; et al. Inhibition of the αv integrin-TGF-β axis improves natural killer cell function against glioblastoma stem cells. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Burga, R.A.; Yvon, E.; Chorvinsky, E.; Fernandes, R.; Cruz, C.R.Y.; Bollard, C.M. Engineering the TGFβ Receptor to Enhance the Therapeutic Potential of Natural Killer Cells as an Immunotherapy for Neuroblastoma. Clin. Cancer Res. 2019, 25, 4400–4412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.-I.; Chin, B.M.S.; Burden, B.A.T.; Sexton, S.; Wasko, K.; Nasser, J.M.; Antony, L.P.; Wong, K.K.; Borges, B.C.M.; Morgan, R.A.; et al. Generation of Natural Killer Cells with Enhanced Function from a CRISPR/Cas12a-Edited Induced Pluripotent Stem Cell Line. Blood 2020, 136, 8. [Google Scholar] [CrossRef]
- Zhu, H.; Blum, R.H.; Bernareggi, D.; Ask, E.H.; Wu, Z.; Hoel, H.J.; Meng, Z.; Wu, C.; Guan, K.-L.; Malmberg, K.-J.; et al. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-tumor Activity. Cell Stem Cell 2020, 27, 224–237.e6. [Google Scholar] [CrossRef] [PubMed]
- Basar, R.; Daher, M.; Rezvani, K. Next-generation cell therapies: The emerging role of CAR-NK cells. Blood Adv. 2020, 4, 5868–5876. [Google Scholar] [CrossRef] [PubMed]
- Hodgins, J.J.; Khan, S.T.; Park, M.M.; Auer, R.C.; Ardolino, M. Killers 2.0: NK cell therapies at the forefront of cancer control. J. Clin. Investig. 2019, 129, 3499–3510. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.-Y.; Fu, T.; Jiang, Y.-Z.; Shao, Z.-M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 1–26. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leuk. 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chu, J.; Chan, W.K.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.-H.; Grandi, P.; et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef] [PubMed]
- Kruschinski, A.; Moosmann, A.; Poschke, I.; Norell, H.; Chmielewski, M.; Seliger, B.; Kiessling, R.; Blankenstein, T.; Abken, H.; Charo, J. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc. Natl. Acad. Sci. USA 2008, 105, 17481–17486. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Liu, Q.; Zhong, M.; Wang, Z.; Chen, Z.; Zhang, Y.; Xing, H.; Tian, Z.; Tang, K.; Liao, X.; et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J. Hematol. Oncol. 2019, 12, 1–13. [Google Scholar] [CrossRef]
- Oelsner, S.; Waldmann, A.; Billmeier, A.; Röder, J.; Lindner, A.; Ullrich, E.; Marschalek, R.; Dotti, G.; Jung, G.; Große-Hovest, L.; et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int. J. Cancer 2019, 145, 1935–1945. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Kalimuthu, S.; Gangadaran, P.; Oh, J.M.; Lee, H.W.; Baek, S.H.; Jeong, S.Y.; Lee, S.-W.; Lee, J.; Ahn, B.-C. Exosomes derived from natural killer cells exert therapeutic effect in melanoma. Theranostics 2017, 7, 2732–2745. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Oh, J.M.; Gangadaran, P.; Kalimuthu, S.; Baek, S.H.; Jeong, S.Y.; Lee, S.-W.; Lee, J.; Ahn, B.-C. Targeting and Therapy of Glioblastoma in a Mouse Model Using Exosomes Derived From Natural Killer Cells. Front. Immunol. 2018, 9, 824. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lai, H.; Chen, T. Boosting Natural Killer Cell-Based Cancer Immunotherapy with Selenocystine/Transforming Growth Factor-Beta Inhibitor-Encapsulated Nanoemulsion. ACS Nano 2020, 14, 11067–11082. [Google Scholar] [CrossRef] [PubMed]
- Adjei, I.M.; Jordan, J.; Tu, N.; Le Trinh, T.; Kandell, W.; Wei, S.; Sharma, B. Functional recovery of natural killer cell activity by nanoparticle-mediated delivery of transforming growth factor beta 2 small interfering RNA. J. Interdiscip. Nanomed. 2019, 4, 98–112. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I Study of GC1008 (Fresolimumab): A Human Anti-Transforming Growth Factor-Beta (TGFβ) Monoclonal Antibody in Patients with Advanced Malignant Melanoma or Renal Cell Carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]


{kind=link}
| Type of Therapy | Studied Agents | Mechanism/Target Receptor |
|---|---|---|
| Checkpoint inhibitors | Lirilumab Ipilimumab Nivolumab Pembrolizumab Monalizumab | mAb 1 targeting KIR mAb 1 targeting CTLA4 mAb 1 targeting PD-1 mAb 1 targeting PD-1 mAb 1 targeting NKG2A |
| Cytokine activation | IL-2 IL-2/15Rβ IL-15 ALT-803 | IL-2 IL-2 (NK cells preferentially) IL-15 IL-15 (superagonist) |
| Enhancing ADCC2 | Trastuzumab Pertuzumab Cetuximab Rituximab Ofatumumab | mAb associated with enhanced function of CD16 (FcγIIIA) |
| NK-92 cell (haNK) | Engineered NK cell with IL-2 and the high-affinity CD16 FcγIIIA (158V) allele | |
| BiKEs 3 and TriKEs 4 | Fuse Fv domain of tumor cell Ag 5 with Fv domain binding to NK cell CD16 | |
| Adoptive transfer | Autologous NK cell infusion Allogeneic NK cells (HLA-haploidentical) CAR-NK 6 | NK cell repletion Targets specific tumor-associated Ag (CD19, CD7, CD5, CD123, EGFR, HER2, MUCI, ROBO1, mesothelin, etc.) |
| Nanoparticle technology | Nano-vesicles | Vesicles composed of lipid bilayer membrane with ability to carry NK-specific cytotoxic proteins and with potential for enhanced drug delivery |
| Optimizing the TME7 | Fresolimumab Galunisertib | mAb neutralizing TGF-β TGF-βR1 inhibitor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
St-Pierre, F.; Bhatia, S.; Chandra, S. Harnessing Natural Killer Cells in Cancer Immunotherapy: A Review of Mechanisms and Novel Therapies. Cancers 2021, 13, 1988. https://doi.org/10.3390/cancers13081988
St-Pierre F, Bhatia S, Chandra S. Harnessing Natural Killer Cells in Cancer Immunotherapy: A Review of Mechanisms and Novel Therapies. Cancers. 2021; 13(8):1988. https://doi.org/10.3390/cancers13081988
Chicago/Turabian StyleSt-Pierre, Frederique, Shailender Bhatia, and Sunandana Chandra. 2021. "Harnessing Natural Killer Cells in Cancer Immunotherapy: A Review of Mechanisms and Novel Therapies" Cancers 13, no. 8: 1988. https://doi.org/10.3390/cancers13081988
APA StyleSt-Pierre, F., Bhatia, S., & Chandra, S. (2021). Harnessing Natural Killer Cells in Cancer Immunotherapy: A Review of Mechanisms and Novel Therapies. Cancers, 13(8), 1988. https://doi.org/10.3390/cancers13081988