
Oxaliplatin-Induced Senescence in Colorectal Cancer Cells Depends on p14ARF-Mediated Sustained p53 Activation

 , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Drug Treatment, siRNA-Mediated Knockdown, and Plasmid Transfection
2.2. Determination of Cell Death, Cell Cycle Progression, and Colony Formation Capacity
2.3. Determination of Senescence
2.4. Preparation of RNA and RT-qPCR
2.5. Preparation of Genomic DNA and Methylation-Specific PCR (MSP)
2.6. Preparation of Cell Extracts and Western-Blot Analysis
2.7. Statistical Analysis
3. Results
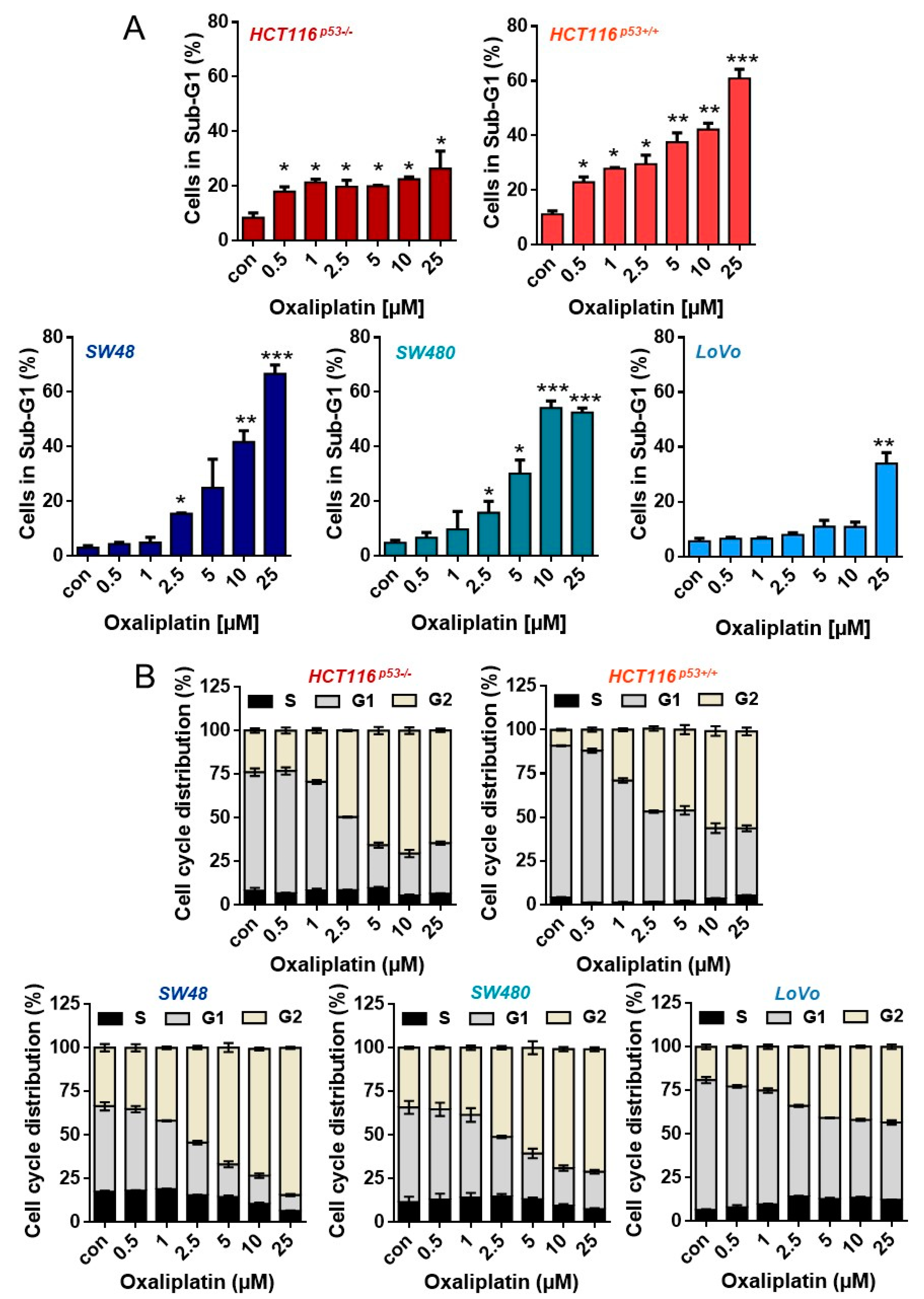
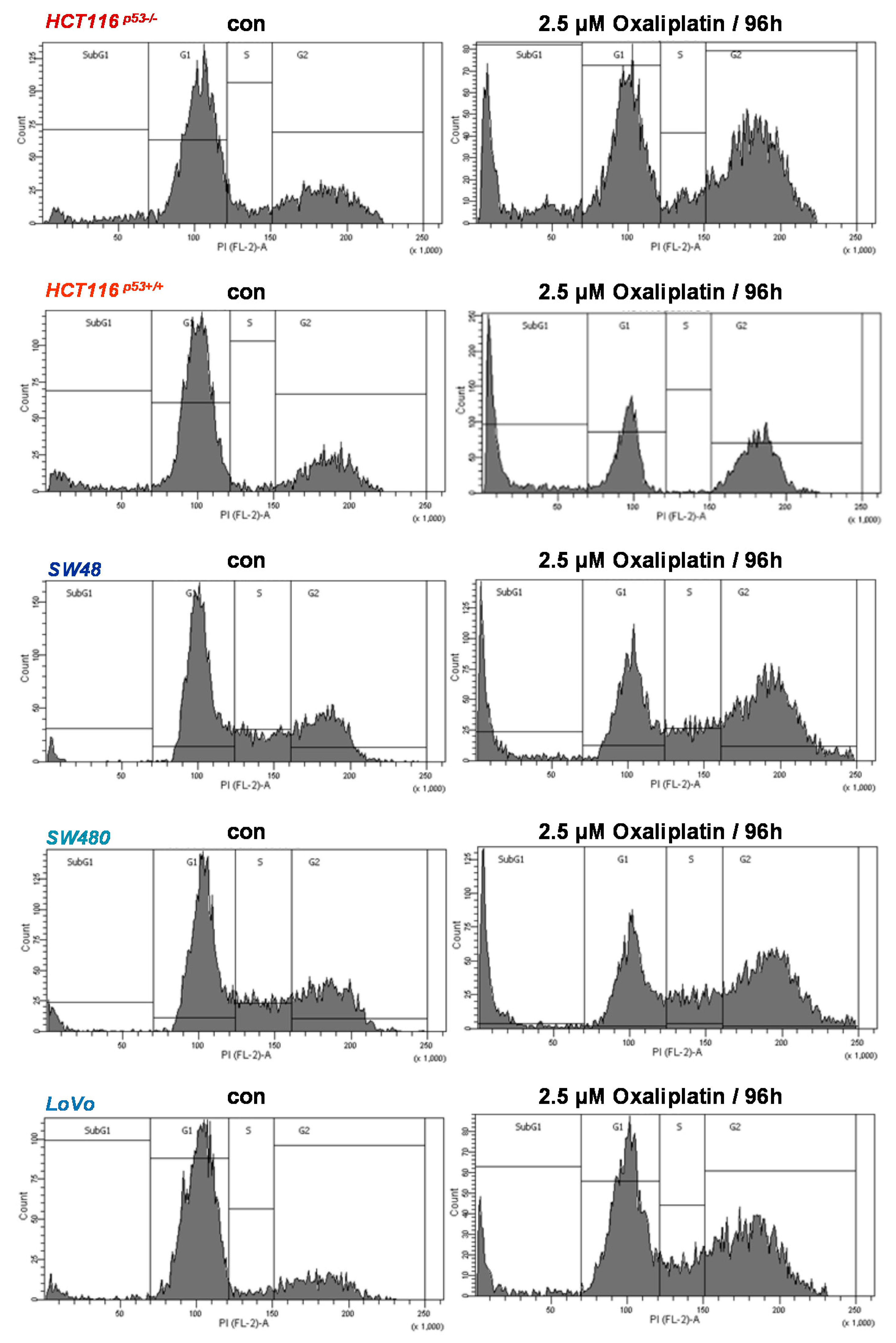
3.1. Oxaliplatin Induces Cell Death and Cell Cycle Arrest
3.2. Oxaliplatin Induces Proliferation Arrest and Senescence
3.3. Oxaliplatin Induces Cell Death, Cell Cycle Arrest, and Senescence at Late Time Points
3.4. Oxaliplatin Induces Upregulation of SASP and SCAP Factors
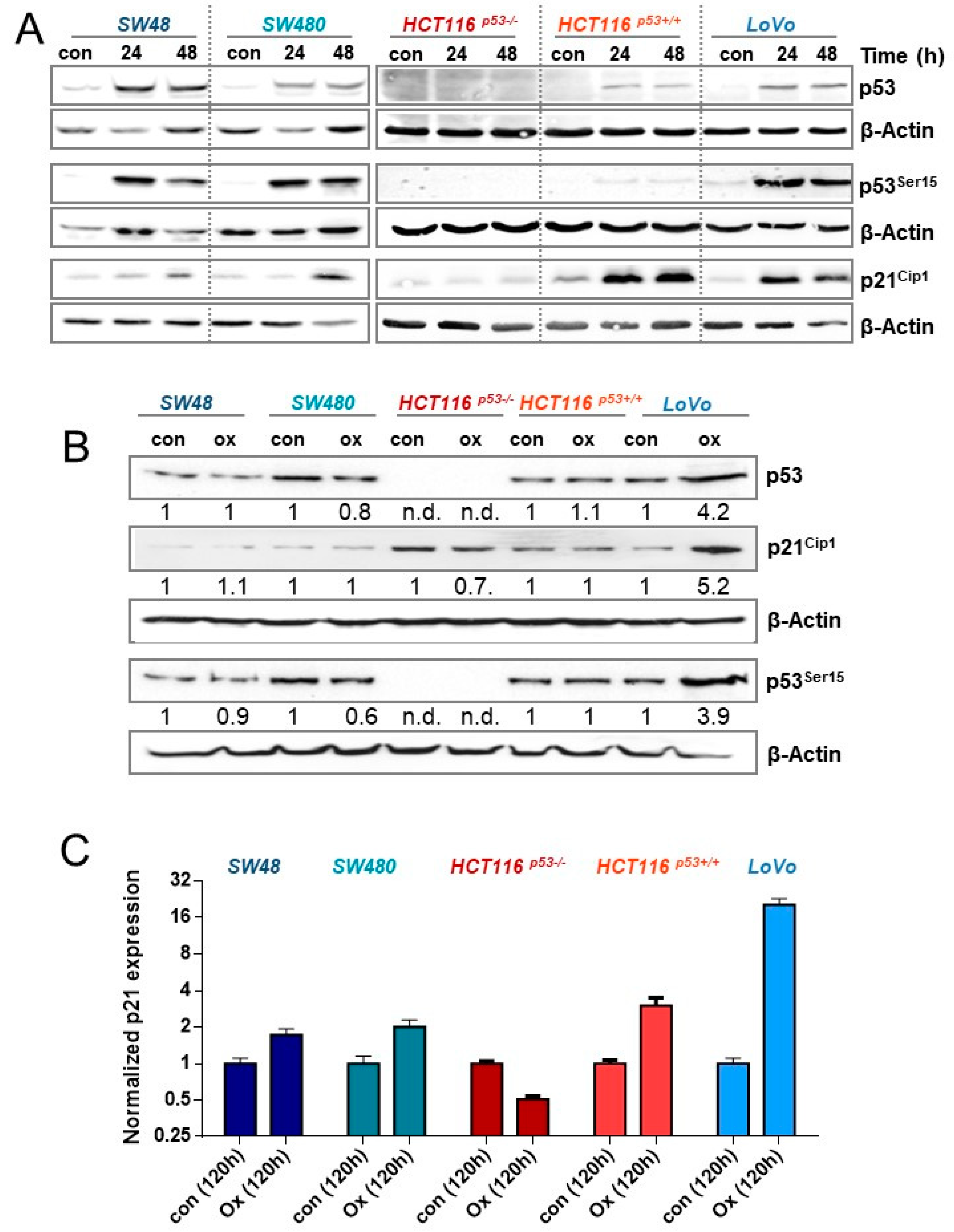
3.5. Activation of p53 and p21CIP1 upon Oxaliplatin Treatment
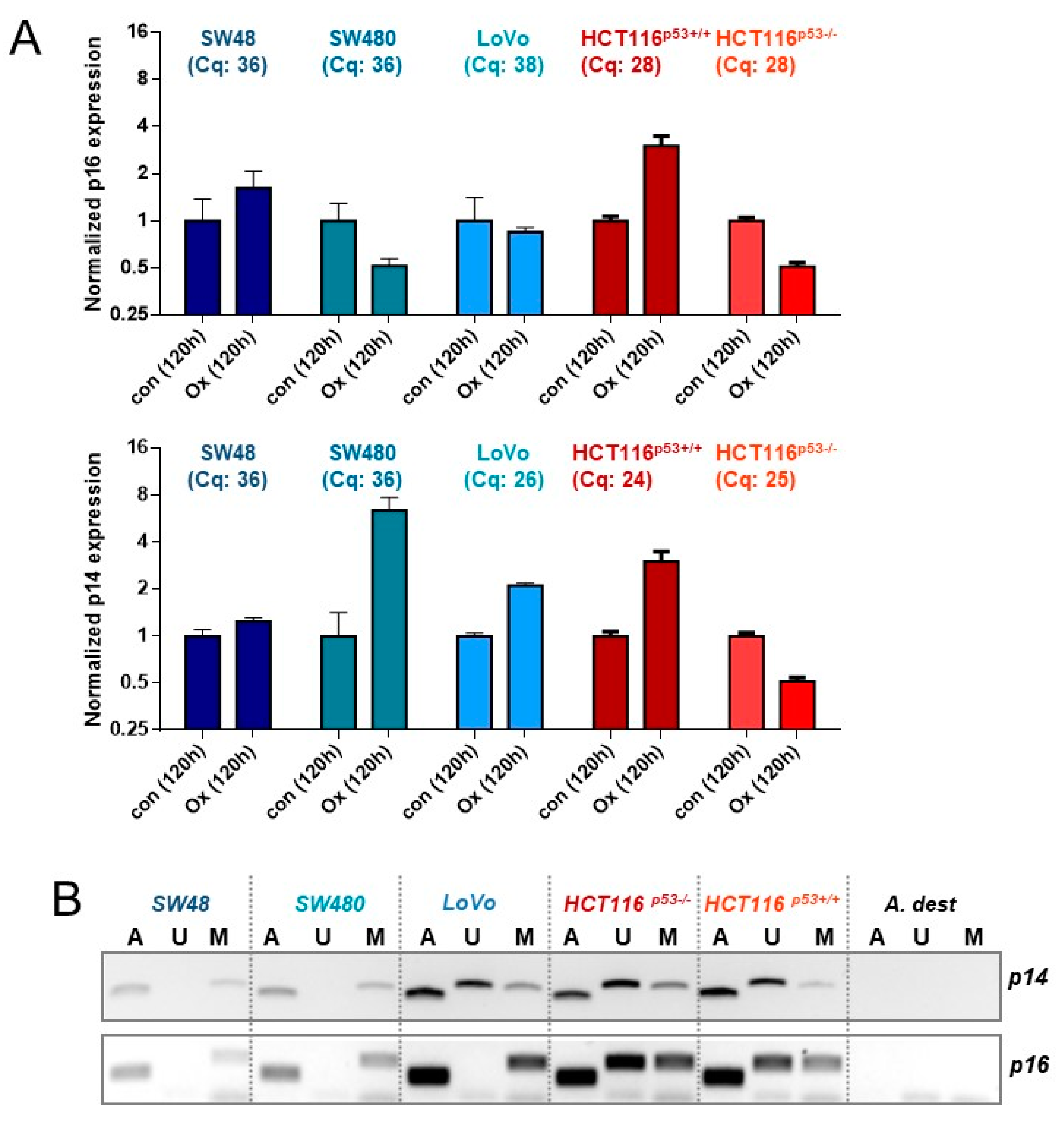
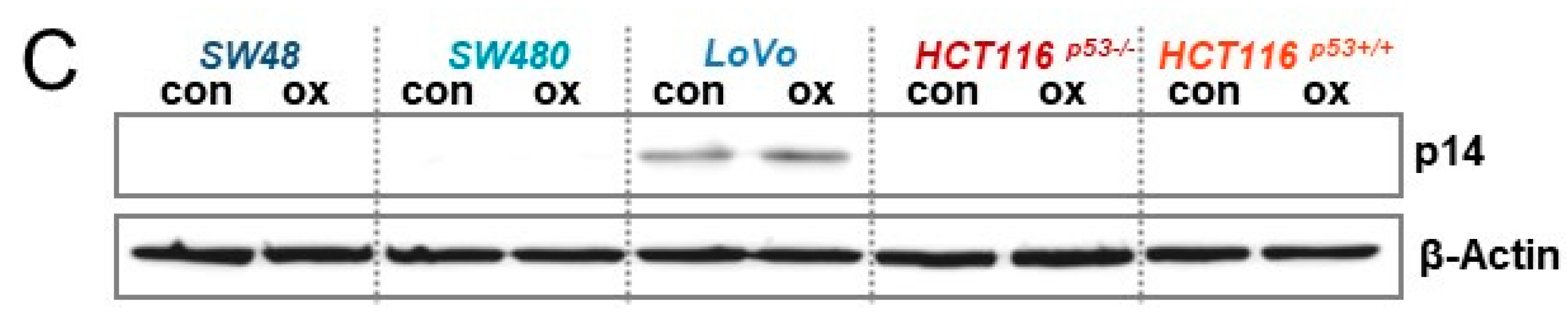
3.6. Expression of p14ARF and p16INK4A upon Oxaliplatin Treatment
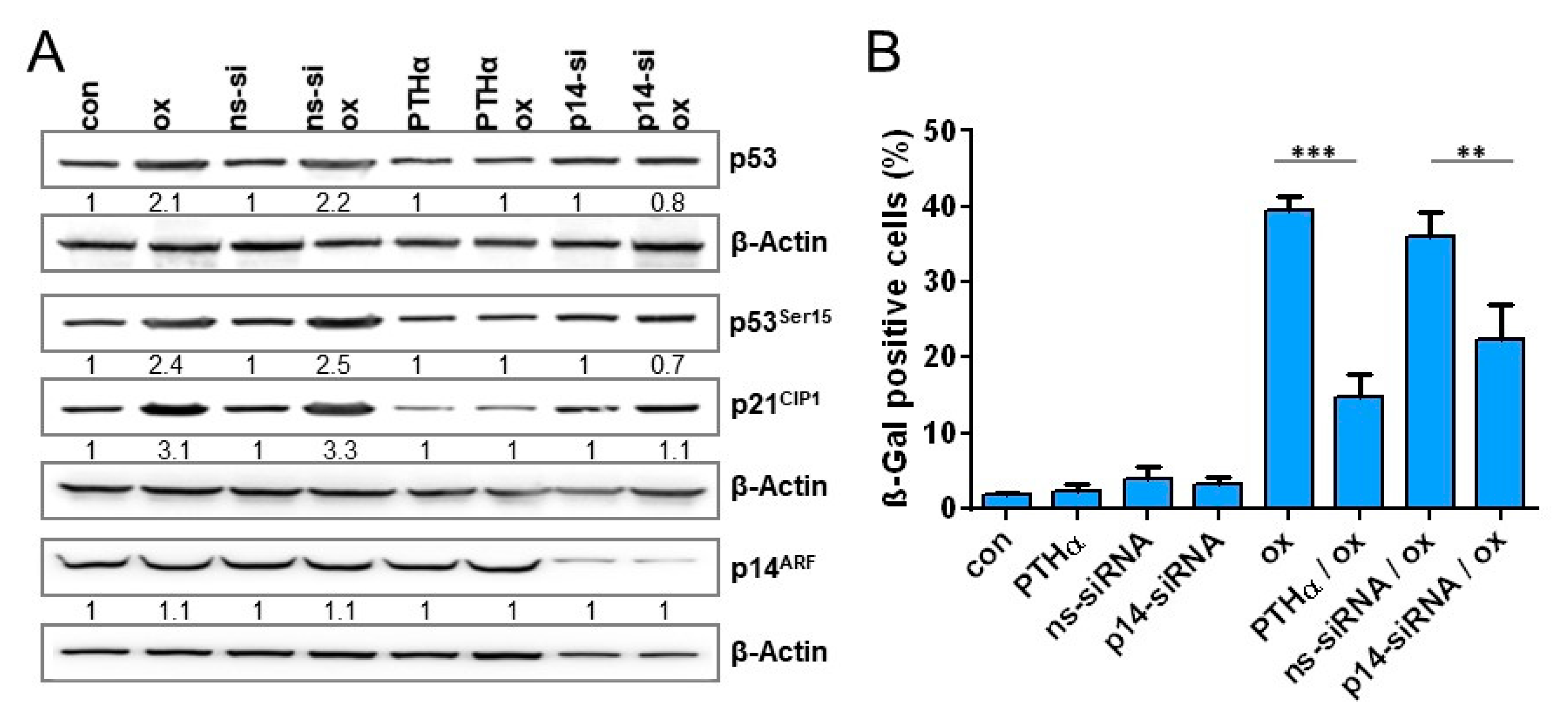
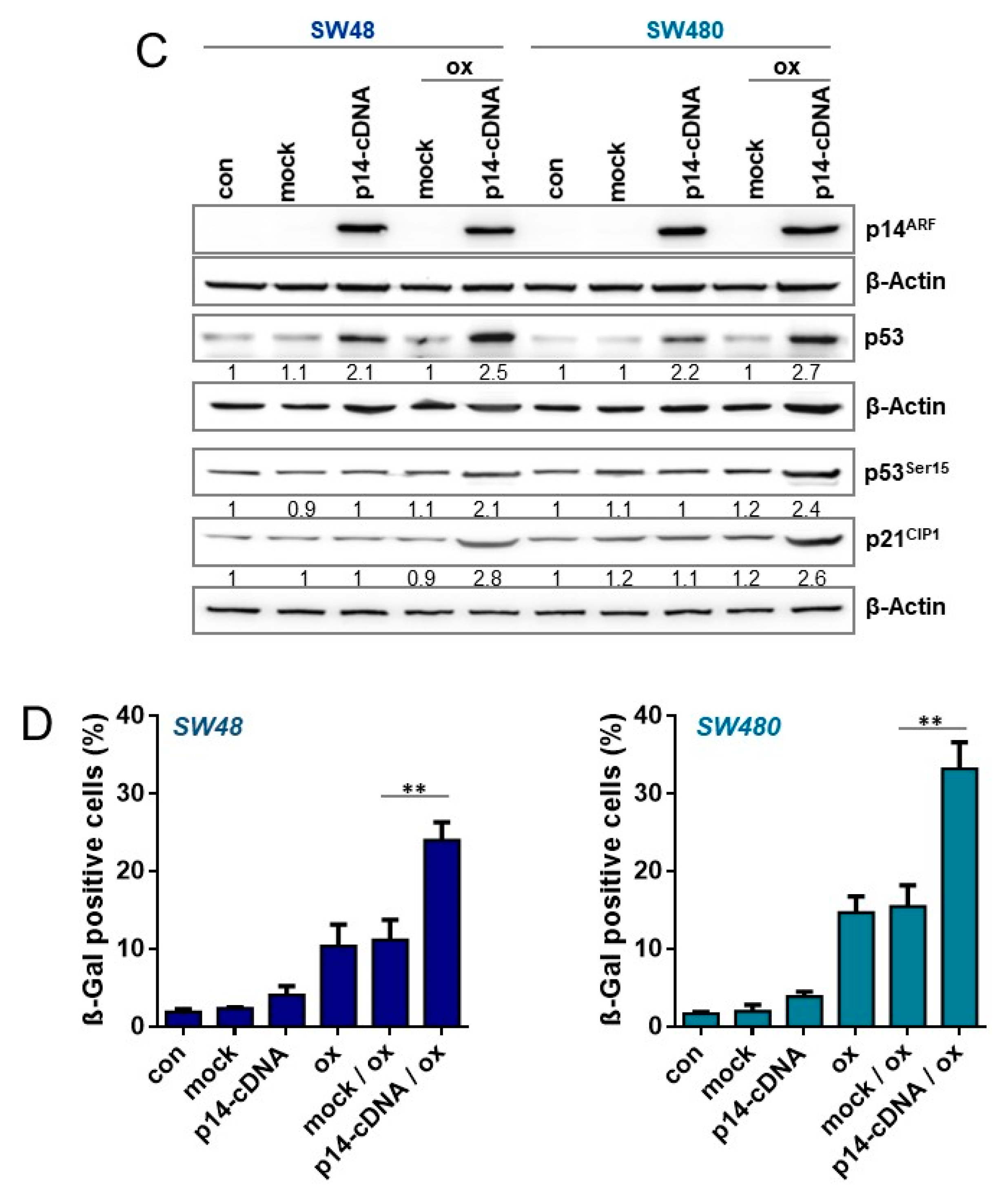
3.7. Impact of p53 and p14ARF on Oxaliplatin-Induced Cellular Senescence
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5‘-3‘) |
|---|---|
| ACTB-fw | TGGCATCCACGAAACTACC |
| ACTB-rev | GTGTTGGCGTACAGGTCTT |
| BAX-fw | CAGAAGGCACTAATCAAG |
| BAX-rev | ATCAGATGTGGTCTATAATG |
| Bcl2-fw | TTCAGAGACAGCCAGGAGAAA |
| Bcl2-rev | AGTACCTGAACCGGCACCT |
| BCLxL-fw | AAGCGTAGACAAGGAGAT |
| BCLxL-rev | TAGGTGGTCATTCAGGTAA |
| BIRC2/c-IAP1-fw | TTCCCAGGTCCCTCGTATCA |
| BIRC2/c-IAP1-rev | CCGGCGGGGAAAGTTGAATA |
| BIRC3/c-IAP2-fw | TCACTCCCAGACTCTTTCCA |
| BIRC3/c-IAP2-rev | CCCCGTGTTCTACAAGTGTC |
| BIRC5/Survivin-fw | ATGACTTGTGTGTGATGA |
| BIRC5/Survivin-rev | GTTTGTGCTATTCTGTGAA |
| CCL2-fw | CCTTCATTCCCCAAGGGCTC |
| CCL2-rev | CTTCTTTGGGACACTTGCTGC |
| CCL8-fw | TGTCCCAAGGAAGCTGTGAT |
| CCL8-rev | TGGAATCCCTGACCCATCTCT |
| CXCL1-fw | CTGGCTTAGAACAAAGGGGCT |
| CXCL1-rev | TAAAGGTAGCCCTTGTTTCCCC |
| IL-1α-fw | TCTTCTGGGAAACTCACGGC |
| IL-1α-rev | GCACACCCAGTAGTCTTGCT |
| IL-1β-fw | TGAGCTCGCCAGTGAAATGA |
| IL-1β-rev | AGATTCGTAGCTGGATGCCG |
| IL-6-fw | GCTGCAGGACATGACAACTC |
| IL-6-rev | AACAACAATCTGAGGTGCCC |
| IL-8-fw | CCAAACCTTTCCACCCCAAA |
| IL-8-rev | CTCTGCACCCAGTTTTCCTT |
| FASR-fw | AGAACTTGGAAGGCCTGCAT |
| FASR-rev | CTGGTTCATCCCCATTGACT |
| FASL-fw | GGGATGTTTCAGCTCTTCCA |
| FASL-rev | TAAATGGGCCACTTTCCTCA |
| MDM2-fw | ATCTTGATGCTGGTGTAA |
| MDM2-rev | AGGCTATAATCTTCTGAGTC |
| PUMA-fw | GACGACCTCAACGCACAGTA |
| PUMA-rev | CTGGGTAAGGGCAGGAGTC |
| NOXA-fw | ACACGGTGGACGTCCTGT |
| NOXA-rev | ACGAAGCACTTGGGGAAGAT |
| p14-fw | CCCTCGTGCTGATGCTACTG |
| p14-rev | CATCATGACCTGGTCTTCTAGGAA |
| p16-fw | GGAGCAGCATGGAGCCTTC |
| p16-rev | CATCATCATGACCTGGATCG |
| p21-fw | TACATCTTCTGCCTTAGT |
| p21-rev | TCTTAGGAACCTCTCATT |
| TNFα-fw | CAGCCTCTTCTCCTTCCTGAT |
| TNFα-rev | GCCAGGGGCTGATTAGAGA |
| XIAP-fw | CCGAAGAGAAACCACATTT |
| XIAP-rev | CTGAGCCAGATCAAAGTATG |
| Protein | Molecular Weight (kDa) | Antibody | RRID | Company |
|---|---|---|---|---|
| ß-Actin | 42 | sc-47778 | AB_2714189 | Santa Cruz Biotechnology |
| HSP90 | 90 | sc-13119 | AB_675659 | Santa Cruz Biotechnology |
| p14 | 14 | sc-53640 | AB_785015 | Santa Cruz Biotechnology |
| p21 | 21 | sc-817 | AB_628072 | Santa Cruz Biotechnology |
| p53 | 53 | sc-100 | AB_628087 | Santa Cruz Biotechnology |
| p53Ser15 | 53 | #9286 | AB_331741 | Cell Signaling Technology |
| HRP conjugated anti-mouse | KCB002 | AB_10703407 | Rockland | |
| HRP conjugated anti-rabbit | KCB003 | AB_10702763 | Rockland |




References
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Muller, M. Cellular senescence: Molecular mechanisms, in vivo significance, and redox considerations. Antioxid. Redox Signal. 2009, 11, 59–98. [Google Scholar] [CrossRef] [PubMed]
- Fridman, A.L.; Tainsky, M.A. Critical pathways in cellular senescence and immortalization revealed by gene expression profiling. Oncogene 2008, 27, 5975–5987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cichowski, K.; Hahn, W.C. Unexpected pieces to the senescence puzzle. Cell 2008, 133, 958–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Zglinicki, T.; Saretzki, G.; Ladhoff, J.; d’Adda di Fagagna, F.; Jackson, S.P. Human cell senescence as a DNA damage response. Mech. Ageing Dev. 2005, 126, 111–117. [Google Scholar] [CrossRef] [PubMed]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Dimova, D.K.; Dyson, N.J. The E2F transcriptional network: Old acquaintances with new faces. Oncogene 2005, 24, 2810–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, E.; Smith, R.; Parry, D.; Tahara, H.; Stone, S.; Peters, G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol. Cell. Biol. 1996, 16, 859–867. [Google Scholar] [CrossRef] [Green Version]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulic, V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, J.; Schreiber-Agus, N.; Liegeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2′s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.C.; Wang, Q.; Grobman, L.; Chu, E.; Wu, D.Y. Accelerated cellular senescence in solid tumor therapy. Exp. Oncol. 2012, 34, 298–305. [Google Scholar] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, B.D.; Broude, E.V.; Dokmanovic, M.; Zhu, H.; Ruth, A.; Xuan, Y.; Kandel, E.S.; Lausch, E.; Christov, K.; Roninson, I.B. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999, 59, 3761–3767. [Google Scholar] [PubMed]
- Aasland, D.; Gotzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR-CHK1, p21, and NF-kappaB. Cancer Res. 2019, 79, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymond, E.; Faivre, S.; Chaney, S.; Woynarowski, J.; Cvitkovic, E. Cellular and molecular pharmacology of oxaliplatin. Mol. Cancer Ther. 2002, 1, 227–235. [Google Scholar] [PubMed]
- Mani, S.; Graham, M.A.; Bregman, D.B.; Ivy, P.; Chaney, S.G. Oxaliplatin: A review of evolving concepts. Cancer Investig. 2002, 20, 246–263. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, K.; Blasiak, J. Recognition and repair of DNA-cisplatin adducts. Acta Biochim. Pol. 2002, 49, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. Third row transition metals for the treatment of cancer. Philos. Trans. A Math. Phys. Eng. Sci. 2015, 373, 20140185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christmann, M.; Diesler, K.; Majhen, D.; Steigerwald, C.; Berte, N.; Freund, H.; Stojanovic, N.; Kaina, B.; Osmak, M.; Ambriovic-Ristov, A.; et al. Integrin alphaVbeta3 silencing sensitizes malignant glioma cells to temozolomide by suppression of homologous recombination repair. Oncotarget 2017, 8, 27754–27771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomicic, M.T.; Steigerwald, C.; Rasenberger, B.; Brozovic, A.; Christmann, M. Functional mismatch repair and inactive p53 drive sensitization of colorectal cancer cells to irinotecan via the IAP antagonist BV6. Arch. Toxicol. 2019, 93, 2265–2277. [Google Scholar] [CrossRef]
- Esteller, M.; Tortola, S.; Toyota, M.; Capella, G.; Peinado, M.A.; Baylin, S.B.; Herman, J.G. Hypermethylation-associated inactivation of p14(ARF) is independent of p16(INK4a) methylation and p53 mutational status. Cancer Res. 2000, 60, 129–133. [Google Scholar]
- Herman, J.G.; Graff, J.R.; Myohanen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [Green Version]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Quaas, M.; Nickel, A.; Engeland, K. Indirect p53-dependent transcriptional repression of Survivin, CDC25C, and PLK1 genes requires the cyclin-dependent kinase inhibitor p21/CDKN1A and CDE/CHR promoter sites binding the DREAM complex. Oncotarget 2015, 6, 41402–41417. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Pan, Y.; Ling, G.; Wang, S.; Huang, M.; Jiang, X.; Ke, Y. Escape of U251 glioma cells from temozolomide-induced senescence was modulated by CDK1/survivin signaling. Am. J. Transl. Res. 2017, 9, 2163–2180. [Google Scholar]
- Burri, N.; Shaw, P.; Bouzourene, H.; Sordat, I.; Sordat, B.; Gillet, M.; Schorderet, D.; Bosman, F.T.; Chaubert, P. Methylation silencing and mutations of the p14ARF and p16INK4a genes in colon cancer. Lab. Investig. J. Tech. Methods Pathol. 2001, 81, 217–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.; Chen, P.; McMillan, A.; Lafuente, A.; Lafuente, M.J.; Ballesta, A.; Trias, M.; Wiencke, J.K. Correlations of partial and extensive methylation at the p14(ARF) locus with reduced mRNA expression in colorectal cancer cell lines and clinicopathological features in primary tumors. Carcinogenesis 2000, 21, 2057–2064. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Nordlinger, B.; Arnold, D.; Group, E.G.W. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25 (Suppl. S3), iii1–iii9. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Nordlinger, B.; Cervantes, A.; Group, E.G.W. Advanced colorectal cancer: ESMO Clinical Practice Guidelines for treatment. Ann. Oncol. 2010, 21 (Suppl. S5), v93–v97. [Google Scholar] [CrossRef]
- Venook, A. Critical evaluation of current treatments in metastatic colorectal cancer. Oncologist 2005, 10, 250–261. [Google Scholar] [CrossRef]
- Hoff, P.M.; Saad, E.D.; Costa, F.; Coutinho, A.K.; Caponero, R.; Prolla, G.; Gansl, R.C. Literature review and practical aspects on the management of oxaliplatin-associated toxicity. Clin. Colorectal Cancer 2012, 11, 93–100. [Google Scholar] [CrossRef]
- De Gramont, A.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Boni, C.; Cortes-Funes, H.; Cervantes, A.; Freyer, G.; et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Wu, C.; Goldberg, R.M. Recent therapeutic advances in the treatment of colorectal cancer. Annu. Rev. Med. 2015, 66, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.; Chiu, C.C.; Li, C.H.; Chang, Y.T.; Hwang, H.T. Cisplatin-induced senescence and growth inhibition in human non-small cell lung cancer cells with ectopic transfer of p16INK4a. Oncol. Res. 2007, 16, 479–488. [Google Scholar] [CrossRef]
- Wang, X.; Wong, S.C.; Pan, J.; Tsao, S.W.; Fung, K.H.; Kwong, D.L.; Sham, J.S.; Nicholls, J.M. Evidence of cisplatin-induced senescent-like growth arrest in nasopharyngeal carcinoma cells. Cancer Res. 1998, 58, 5019–5022. [Google Scholar] [PubMed]
- Qu, K.; Lin, T.; Wei, J.; Meng, F.; Wang, Z.; Huang, Z.; Wan, Y.; Song, S.; Liu, S.; Chang, H.; et al. Cisplatin induces cell cycle arrest and senescence via upregulating P53 and P21 expression in HepG2 cells. Nan Fang Yi Ke Da Xue Xue Bao 2013, 33, 1253–1259. [Google Scholar]
- Sun, X.; Shi, B.; Zheng, H.; Min, L.; Yang, J.; Li, X.; Liao, X.; Huang, W.; Zhang, M.; Xu, S.; et al. Senescence-associated secretory factors induced by cisplatin in melanoma cells promote non-senescent melanoma cell growth through activation of the ERK1/2-RSK1 pathway. Cell Death Dis. 2018, 9, 260. [Google Scholar] [CrossRef]
- Seignez, C.; Martin, A.; Rollet, C.E.; Racoeur, C.; Scagliarini, A.; Jeannin, J.F.; Bettaieb, A.; Paul, C. Senescence of tumor cells induced by oxaliplatin increases the efficiency of a lipid A immunotherapy via the recruitment of neutrophils. Oncotarget 2014, 5, 11442–11451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Was, H.; Czarnecka, J.; Kominek, A.; Barszcz, K.; Bernas, T.; Piwocka, K.; Kaminska, B. Some chemotherapeutics-treated colon cancer cells display a specific phenotype being a combination of stem-like and senescent cell features. Cancer Biol. Ther. 2018, 19, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Bojko, A.; Czarnecka-Herok, J.; Charzynska, A.; Dabrowski, M.; Sikora, E. Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells 2019, 8, 1501. [Google Scholar] [CrossRef] [Green Version]
- Toscano, F.; Parmentier, B.; Fajoui, Z.E.; Estornes, Y.; Chayvialle, J.A.; Saurin, J.C.; Abello, J. p53 dependent and independent sensitivity to oxaliplatin of colon cancer cells. Biochem. Pharmacol. 2007, 74, 392–406. [Google Scholar] [CrossRef]
- Dimri, G.P.; Itahana, K.; Acosta, M.; Campisi, J. Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Mol. Cell. Biol. 2000, 20, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Hemmer, R.M.; Sedivy, J.M. Role of p14(ARF) in replicative and induced senescence of human fibroblasts. Mol. Cell. Biol. 2001, 21, 6748–6757. [Google Scholar] [CrossRef] [Green Version]
- Groth, A.; Weber, J.D.; Willumsen, B.M.; Sherr, C.J.; Roussel, M.F. Oncogenic Ras induces p19ARF and growth arrest in mouse embryo fibroblasts lacking p21Cip1 and p27Kip1 without activating cyclin D-dependent kinases. J. Biol. Chem. 2000, 275, 27473–27480. [Google Scholar] [CrossRef]
- Palmero, I.; Pantoja, C.; Serrano, M. p19ARF links the tumour suppressor p53 to Ras. Nature 1998, 395, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Ries, S.; Biederer, C.; Woods, D.; Shifman, O.; Shirasawa, S.; Sasazuki, T.; McMahon, M.; Oren, M.; McCormick, F. Opposing effects of Ras on p53: Transcriptional activation of mdm2 and induction of p19ARF. Cell 2000, 103, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Ko, A.; Han, S.Y.; Choi, C.H.; Cho, H.; Lee, M.S.; Kim, S.Y.; Song, J.S.; Hong, K.M.; Lee, H.W.; Hewitt, S.M.; et al. Oncogene-induced senescence mediated by c-Myc requires USP10 dependent deubiquitination and stabilization of p14ARF. Cell Death Differ. 2018, 25, 1050–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Guevara, C.; Fujii, G.; Parry, D. p14ARF is a component of the p53 response following ionizing irradiation of normal human fibroblasts. Oncogene 2004, 23, 6040–6046. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Zhang, H.; Lai, J.; Diao, D.; Li, W.; Dang, C.; Song, Y. Relationships between p14ARF Gene Methylation and Clinicopathological Features of Colorectal Cancer: A Meta-Analysis. PLoS ONE 2016, 11, e0152050. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomicic, M.T.; Krämer, F.; Nguyen, A.; Schwarzenbach, C.; Christmann, M. Oxaliplatin-Induced Senescence in Colorectal Cancer Cells Depends on p14ARF-Mediated Sustained p53 Activation. Cancers 2021, 13, 2019. https://doi.org/10.3390/cancers13092019
Tomicic MT, Krämer F, Nguyen A, Schwarzenbach C, Christmann M. Oxaliplatin-Induced Senescence in Colorectal Cancer Cells Depends on p14ARF-Mediated Sustained p53 Activation. Cancers. 2021; 13(9):2019. https://doi.org/10.3390/cancers13092019
Chicago/Turabian StyleTomicic, Maja T., Franziska Krämer, Alexandra Nguyen, Christian Schwarzenbach, and Markus Christmann. 2021. "Oxaliplatin-Induced Senescence in Colorectal Cancer Cells Depends on p14ARF-Mediated Sustained p53 Activation" Cancers 13, no. 9: 2019. https://doi.org/10.3390/cancers13092019
APA StyleTomicic, M. T., Krämer, F., Nguyen, A., Schwarzenbach, C., & Christmann, M. (2021). Oxaliplatin-Induced Senescence in Colorectal Cancer Cells Depends on p14ARF-Mediated Sustained p53 Activation. Cancers, 13(9), 2019. https://doi.org/10.3390/cancers13092019