
Phenotypic Characteristics of the Tumour Microenvironment in Primary and Secondary Hepatocellular Carcinoma

 ,
, 
 , , ,
, , ,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Immunohistochemistry (IHC)
2.3. DNA and RNA Purification
2.4. Targeted Next Generation Sequencing (tNGS)
2.5. NanoString Immune Profiling
2.6. High-Resolution T-Cell Receptor Sequencing
2.7. Statistical Analysis
3. Results
3.1. Clinicopathologic Characteristics
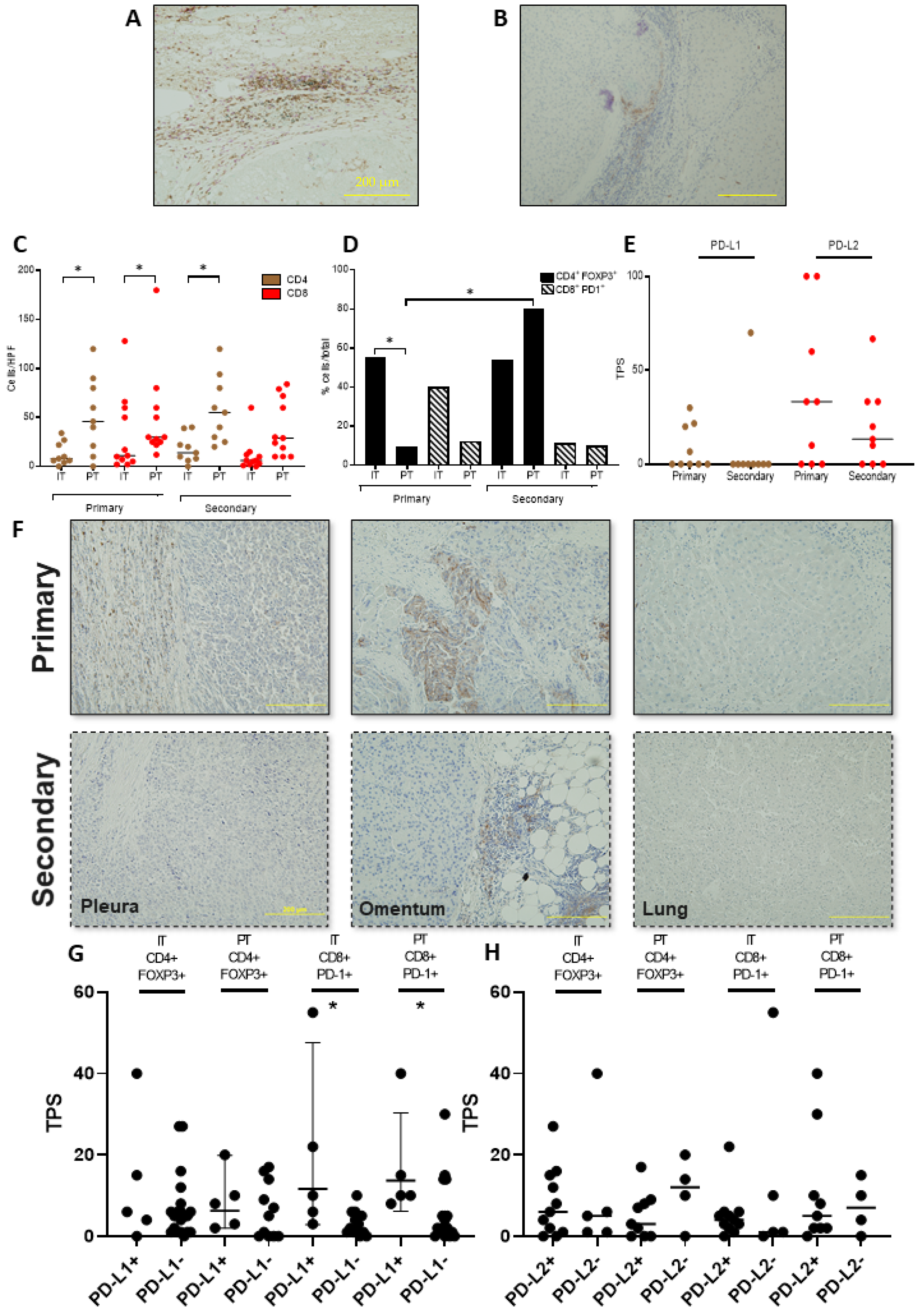
3.2. Functional Characterisation of the T-Cell Infiltrate across Primary and Secondary HCC
3.3. PD-L1/2 Expression Influences the Tumour Microenvironment in Primary and Secondary HCC
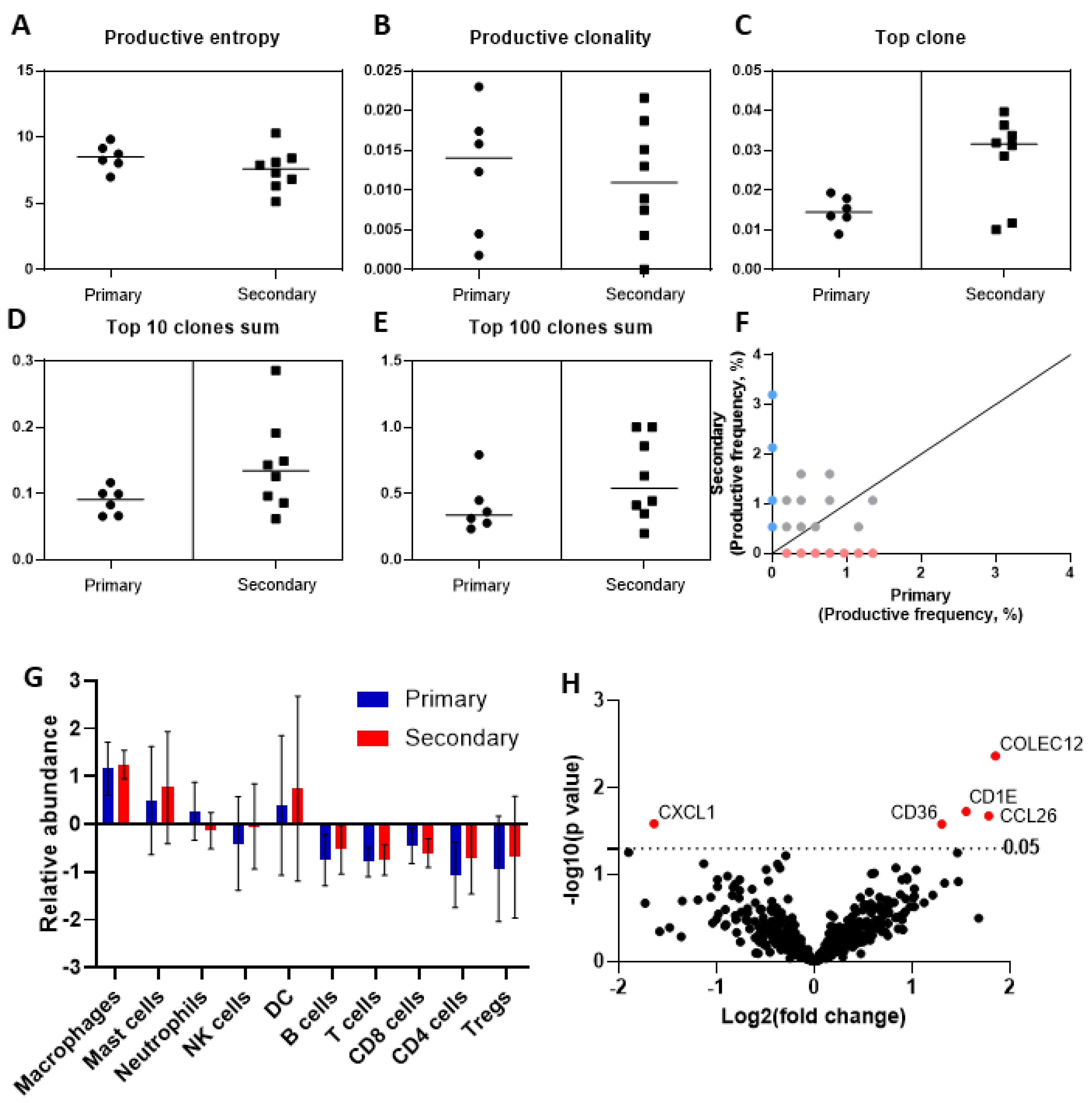
3.4. Deep Sequencing of the T-Cell Receptor T-Cell Infiltrate across Primary and Secondary HCC
3.5. Phenotypic Differences of the Immune Cell Infiltrate by Targeted Gene Expression Profiling
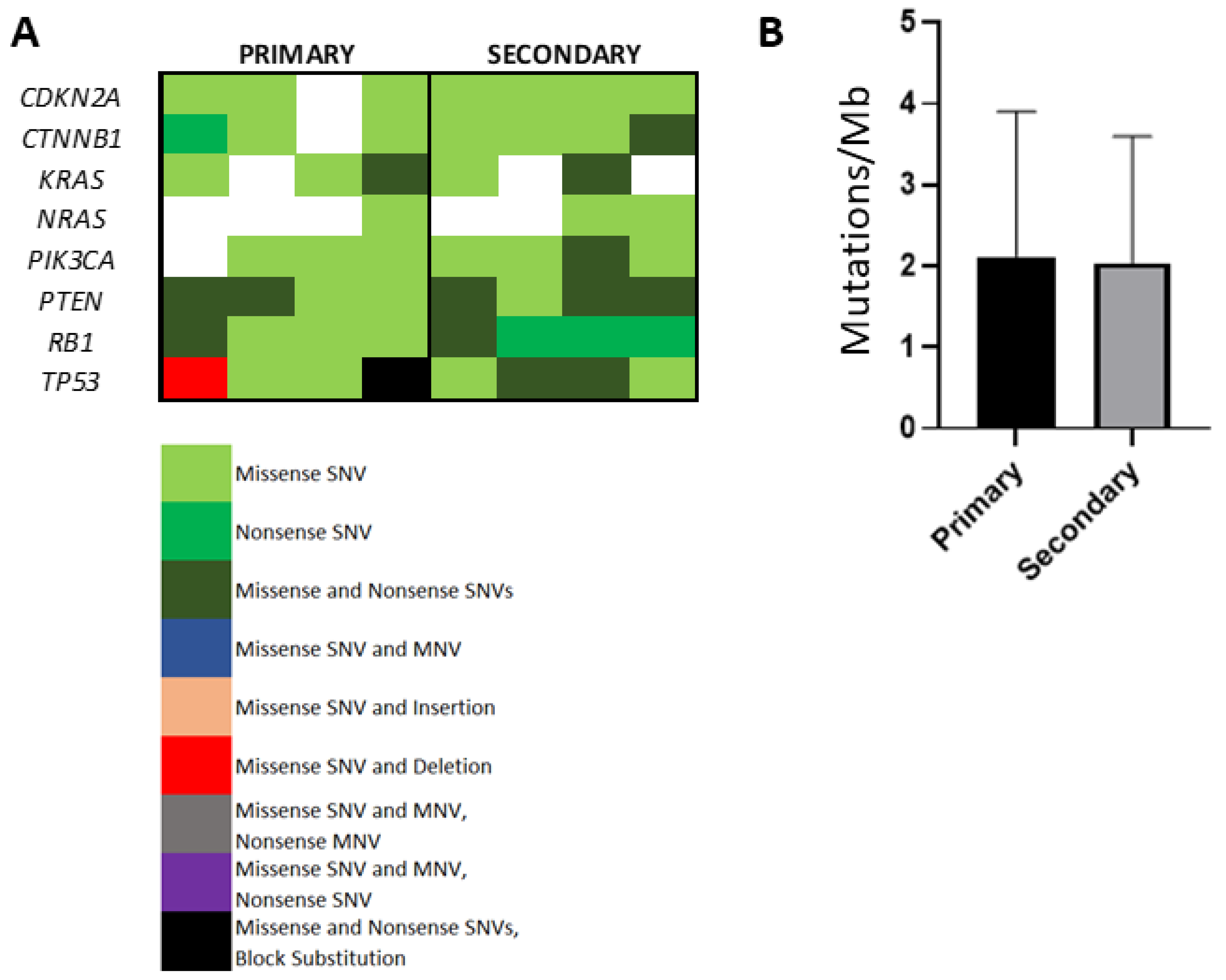
3.6. Evaluation of Somatic Mutational Burden in Primary Versus Secondary HCC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Ethics Approval
References
- Finn, R.S.; Zhu, A.X. Evolution of Systemic Therapy for Hepatocellular Carcinoma. Hepatology 2021. Available online: https://pubmed.ncbi.nlm.nih.gov/32380571/ (accessed on 3 November 2020). [CrossRef]
- Ang, C.; Miura, J.T.; Gamblin, T.C.; He, R.; Xiu, J.; Millis, S.Z.; Gatalica, Z.; Reddy, S.K.; Yee, N.S.; Abou-Alfa, G.K. Comprehensive multiplatform biomarker analysis of 350 hepatocellular carcinomas identifies potential novel therapeutic options. J. Surg. Oncol. 2016, 113, 55–61. Available online: https://pubmed.ncbi.nlm.nih.gov/26661118/ (accessed on 3 November 2020). [CrossRef]
- Swanton, C. Intratumor heterogeneity: Evolution through space and time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef] [Green Version]
- Stanta, G.; Bonin, S. Overview on clinical relevance of intra-tumor heterogeneity. Front. Med. 2018, 5. Available online: https://pubmed.ncbi.nlm.nih.gov/29682505/ (accessed on 1 October 2020). [CrossRef] [Green Version]
- Losic, B.; Craig, A.J.; Villacorta-Martin, C.; Martins-Filho, S.N.; Akers, N.; Chen, X.; Ahsen, M.E.; Von Felden, J.; Labgaa, I.; D’avola, D.; et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Kenmochi, K.; Sugihara, S.; Kojiro, M. Relationship of histologic grade of hepatocellular carcinoma (HCC) to tumor size, and demonstration of tumor cells of multiple different grades in single small HCC. Liver Int. 2008, 7, 18–26. Available online: https://pubmed.ncbi.nlm.nih.gov/3033422/ (accessed on 2 October 2020). [CrossRef]
- Pinato, D.J.; Vallipuram, A.; Evans, J.S.; Wong, C.; Zhang, H.; Brown, M.; Dina, R.E.; Trivedi, P.; Akarca, A.U.; Marafioti, T.; et al. Programmed cell death ligands expression drives immune tolerogenesis across the diverse subtypes of neuroendocrine tumours. Neuroendocrinology 2020. Available online: https://pubmed.ncbi.nlm.nih.gov/32097935/ (accessed on 3 November 2020). [CrossRef] [PubMed]
- Tsongalis, G.J.; Peterson, J.D.; De Abreu, F.B.; Tunkey, C.D.; Gallagher, T.L.; Strausbaugh, L.D.; Wells, W.A.; Amos, C.I.; et al. Routine use of the Ion Torrent AmpliSeqTM Cancer Hotspot Panel for identification of clinically actionable somatic mutations. Clin. Chem. Lab. Med. 2014, 52, 707–714. Available online: https://pubmed.ncbi.nlm.nih.gov/24334431/ (accessed on 6 October 2020). [CrossRef] [PubMed]
- Reuben, A.; Gittelman, R.; Gao, J.; Zhang, J.; Yusko, E.C.; Wu, C.J.; Emerson, R.; Zhang, J.; Tipton, C.; Li, J.; et al. TCR repertoire intratumor heterogeneity in localized lung adenocarcinomas: An association with predicted neoantigen heterogeneity and postsurgical recurrence. Cancer Discov. 2017, 7, 1088–1097. Available online: https://pubmed.ncbi.nlm.nih.gov/28733428/ (accessed on 3 November 2020). [CrossRef] [Green Version]
- Monod, M.Y.; Giudicelli, V.; Chaume, D.; Lefranc, M.P. IMGT/JunctionAnalysis: The first tool for the analysis of the immunoglobulin and T cell receptor complex V-J and V-D-J JUNCTIONs. Bioinformatics 2004. Available online: https://pubmed.ncbi.nlm.nih.gov/15262823/ (accessed on 21 December 2020). [CrossRef] [Green Version]
- Kirsch, I.; Vignali, M.; Robins, H. T-cell receptor profiling in cancer. Mol. Oncol. 2015, 9, 2063–2070. Available online: https://pubmed.ncbi.nlm.nih.gov/26404496/ (accessed on 3 November 2020). [CrossRef]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 307–313. Available online: https://pubmed.ncbi.nlm.nih.gov/22378190/ (accessed on 24 November 2020). [CrossRef]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.M.; Lee, D. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1334. [Google Scholar] [CrossRef] [Green Version]
- Buchhalter, I.; Rempel, E.; Endris, V.; Allgäuer, M.; Neumann, O.; Volckmar, A.-L.; Kirchner, M.; Leichsenring, J.; Lier, A.; Von Winterfeld, M.; et al. Size matters: Dissecting key parameters for panel-based tumor mutational burden analysis. Int. J. Cancer 2019, 144, 848–858. Available online: https://pubmed.ncbi.nlm.nih.gov/30238975/ (accessed on 6 October 2020). [CrossRef] [Green Version]
- Tian, M.X.; Liu, W.R.; Wang, H.; Zhou, Y.F.; Jin, L.; Jiang, X.F.; Tao, C.Y.; Tang, Z.; Zhou, P.Y.; Fang, Y.; et al. Tissue-infiltrating lymphocytes signature predicts survival in patients with early/intermediate stage hepatocellular carcinoma. BMC Med. 2019, 17, 106. [Google Scholar] [CrossRef] [PubMed]
- Pai, S.I.; Cesano, A.; Marincola, F.M. The Paradox of Cancer Immune Exclusion: Immune Oncology Next Frontier. In Cancer Treatment and Research; Springer: Berlin/Heidelberg, Germany, 2020; pp. 173–195. Available online: https://pubmed.ncbi.nlm.nih.gov/32215870/ (accessed on 11 December 2020).
- Pinato, D.J.; Shiner, R.J.; White, S.D.T.; Black, J.R.M.; Trivedi, P.; Stebbing, J.; Sharma, R.; Mauri, F.A.; et al. Intra-tumoral heterogeneity in the expression of programmed-death (PD) ligands in isogeneic primary and metastatic lung cancer: Implications for immunotherapy. Oncoimmunology 2016, 5. Available online: https://pubmed.ncbi.nlm.nih.gov/27757309/ (accessed on 21 December 2020). [CrossRef] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. Available online: https://pubmed.ncbi.nlm.nih.gov/25428505/ (accessed on 11 December 2020). [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-derived suppressor cells. Cancer Immunol. Res. 2017, 5, 3–8. Available online: https://pubmed.ncbi.nlm.nih.gov/28052991/ (accessed on 11 December 2020). [CrossRef] [PubMed] [Green Version]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. Available online: https://pubmed.ncbi.nlm.nih.gov/20856220/ (accessed on 11 December 2020). [CrossRef]
- Nath, A.; Li, I.; Roberts, L.R.; Chan, C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci. Rep. 2015, 5. Available online: https://pubmed.ncbi.nlm.nih.gov/26424075/ (accessed on 15 October 2020). [CrossRef] [Green Version]
- Tang, X.; Shu, Z.; Zhang, W.; Cheng, L.; Yu, J.; Zhang, M.; Zheng, S. Clinical significance of the immune cell landscape in hepatocellular carcinoma patients with different degrees of fibrosis. Ann. Transl. Med. 2019, 7, 528. Available online: https://pubmed.ncbi.nlm.nih.gov/31807510/ (accessed on 11 December 2020). [CrossRef]
- Koudelkova, P.; Costina, V.; Weber, G.; Dooley, S.; Findeisen, P.; Winter, P.; Agarwal, R.; Schlangen, K.; Mikulits, W. Transforming growth factor-β drives the transendothelial migration of hepatocellular carcinoma cells. Int. J. Mol. Sci. 2017, 18, 2119. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Baseline Characteristics | N = 11 [n (%)] |
|---|---|
| Age [median (range)] | 69 (44–82) |
| Year of diagnosis [median (range)] | 2006 (1996–2013) |
| Barcelona Clinic Liver Cancer stage at initial diagnosis | |
| A | 6 (54) |
| C | 5 (46) |
| Aetiology of chronic liver disease | |
| Hepatitis C virus | 5 (45.5) |
| Hepatitis B virus | 1 (9.1) |
| Alcohol Excess | 4 (36.4) |
| Cryptogenic | 2 (18.2) |
| Child-Pugh class at first diagnosis | |
| A | 11 (100) |
| Secondary site (n = 13) | |
| Abdominal wall | 1 (7.7) |
| Cutaneous and subcutaneous | 2 (15.4) |
| Liver relapse | 2 (15.4) |
| Lung | 4 (30.8) |
| Omentum | 2 (15.4) |
| Pleura | 2 (15.4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fessas, P.; Spina, P.; Boldorini, R.L.; Pirisi, M.; Minisini, R.; Mauri, F.A.; Simpson, F.; Olivieri, P.; Gennari, A.; Wong, C.N.; et al. Phenotypic Characteristics of the Tumour Microenvironment in Primary and Secondary Hepatocellular Carcinoma. Cancers 2021, 13, 2137. https://doi.org/10.3390/cancers13092137
Fessas P, Spina P, Boldorini RL, Pirisi M, Minisini R, Mauri FA, Simpson F, Olivieri P, Gennari A, Wong CN, et al. Phenotypic Characteristics of the Tumour Microenvironment in Primary and Secondary Hepatocellular Carcinoma. Cancers. 2021; 13(9):2137. https://doi.org/10.3390/cancers13092137
Chicago/Turabian StyleFessas, Petros, Paolo Spina, Renzo L. Boldorini, Mario Pirisi, Rosalba Minisini, Francesco A. Mauri, Fraser Simpson, Paola Olivieri, Alessandra Gennari, Ching Ngar Wong, and et al. 2021. "Phenotypic Characteristics of the Tumour Microenvironment in Primary and Secondary Hepatocellular Carcinoma" Cancers 13, no. 9: 2137. https://doi.org/10.3390/cancers13092137
APA StyleFessas, P., Spina, P., Boldorini, R. L., Pirisi, M., Minisini, R., Mauri, F. A., Simpson, F., Olivieri, P., Gennari, A., Wong, C. N., Siddique, A., Goldin, R. D., Akarca, A. U., Marafioti, T., & Pinato, D. J. (2021). Phenotypic Characteristics of the Tumour Microenvironment in Primary and Secondary Hepatocellular Carcinoma. Cancers, 13(9), 2137. https://doi.org/10.3390/cancers13092137