
Folate Transport and One-Carbon Metabolism in Targeted Therapies of Epithelial Ovarian Cancer

Abstract
:Simple Summary
Abstract
1. Introduction
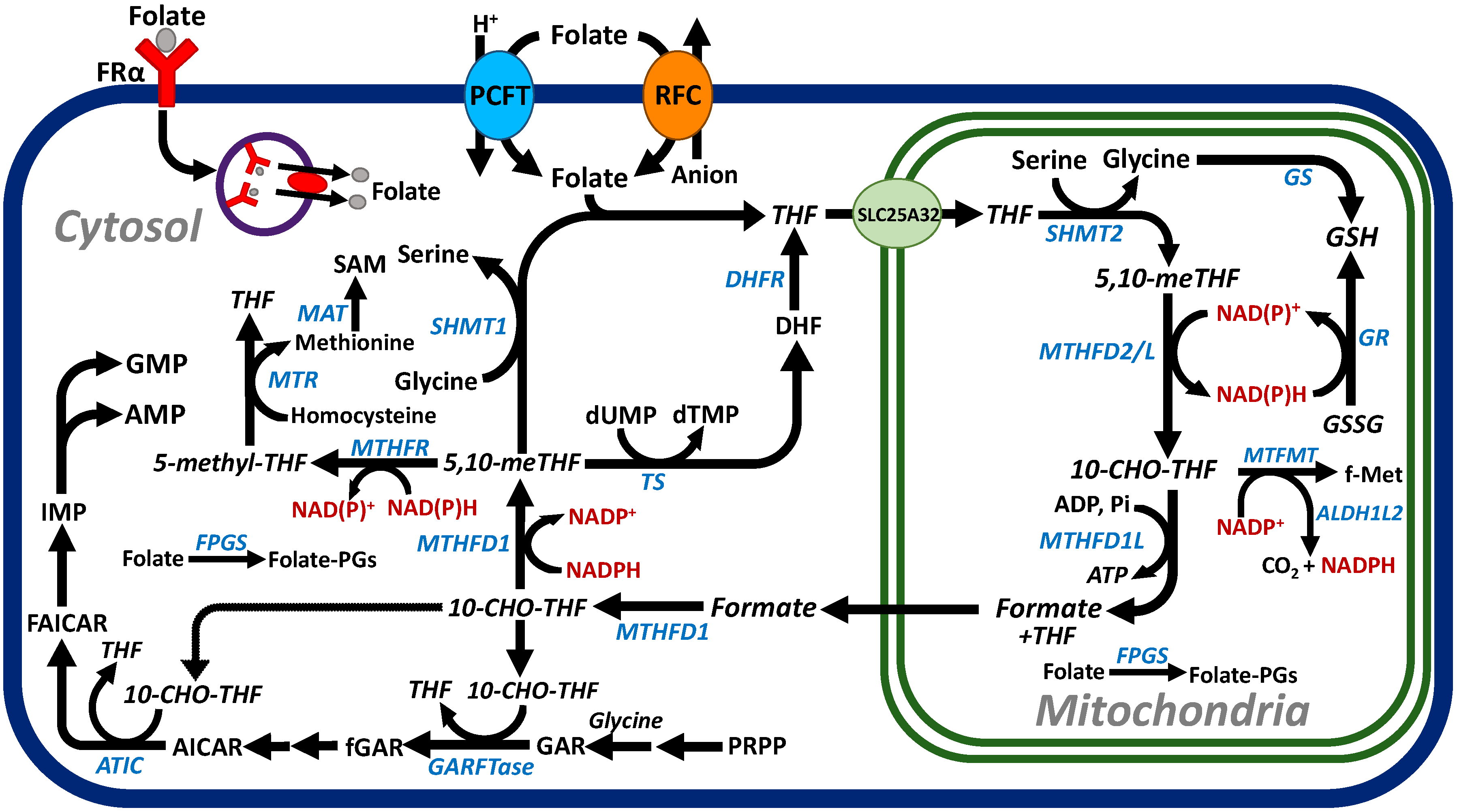
2. Folate Homeostasis, Folate Transport, and One-Carbon Metabolism
2.1. Folate Homeostasis and Transport
2.2. C1 Metabolism
3. Therapeutic Challenges of Treating Ovarian Cancer
4. The Role of FRα in the Treatment of EOC

{kind=link}
{kind=link}
| Farlatuzumab | |||
|---|---|---|---|
| Title | Patients | Results | Reference |
| Randomized, double-blind, placebo-controlled, phase III study to assess the efficacy and safety of weekly MORAb-003 in combination with carboplatin and taxane in subjects with platinum-sensitive ovarian cancer in first relapse (MORAb-003-004; NCT00849667) | Platinum-sensitive EOC (1100 patients) in first relapse | No significant differences in PFS among the treatment arms were observed. The primary end point of PFS was not met. | [116,118] |
| Phase III randomized clinical trial of weekly paclitaxel with or without farletuzumab (MORAb-003-003; NCT00738699) | Platinum-resistant ovarian cancer (417 patients) | Study was terminated due to failure to meet pre-specified criteria. | [117] |
| Vintafolide (EC145) | |||
| Study for women with platinum resistant ovarian cancer evaluating EC145 in combination with Doxil® (PROCEED) (EC-FV-06; NCT01170650) | FRα-positive platinum-resistant ovarian cancer (640 patients) | Trial was terminated owing to failure to meet pre-specified PFS criteria. | [125] |
| Mirvetuximab soravtansine | |||
| Phase III RCT (FORWARD I) evaluating chemotherapy (paclitaxel, pegylated liposomal doxorubicin, or topotecan) vs. mirvetuximab soravtansine (IMGN853-0403; NCT02631876) | FRα-positive platinum-resistant ovarian cancer (366 patients) | Mirvetuximab soravtansine did not result in a significant improvement in PFS compared with chemotherapy. | [121] |
5. Targeting EOC via Targeted Antifolates: C1 Metabolism as a Unique Vulnerability for EOC

| Inhibitor | Transporter | Intracellular Target | References |
|---|---|---|---|
| Pemetrexed | PCFT, RFC | Thymidylate synthase, GARFTase, ATIC | [137] |
| AGF17 | FRα, FRβ, PCFT | GARFTase | [144,145] |
| AGF23 | FRα, FRβ, PCFT | GARFTase | [145] |
| AGF94 | FRα, FRβ, PCFT | GARFTase | [14,147] |
| AGF154 | FRα, FRβ, PCFT | GARFTase | [148] |
| AGF278 | FRα, FRβ, PCFT | GARFTase | [149] |
| AGF283 | FRα, FRβ, PCFT | GARFTase | [149] |
| AGF347 | FRα, PCFT, RFC | SHMT1, SHMT2, GARFTase, ATIC | [153,155,156] |
| CT900 | FRα | Thymidylate synthase | [26] |
| (±) SHIN1 | Not determined | SHMT1, SHMT2 | [150] |
| (+) SHIN2 | Not determined | SHMT1, SHMT2 | [151] |
| DS18561882 | Not determined | MTHFD1, MTHFD2 | [152] |
6. Targeting the Tumor Microenvironment in Epithelial Ovarian Cancer
6.1. The Role of the Dynamic TME in EOC Progression
6.2. Targeting the TME in EOC
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef] [Green Version]
- Nero, C.; Ciccarone, F.; Pietragalla, A.; Duranti, S.; Daniele, G.; Salutari, V.; Carbone, M.V.; Scambia, G.; Lorusso, D. Ovarian Cancer Treatments Strategy: Focus on PARP Inhibitors and Immune Check Point Inhibitors. Cancers 2021, 13, 1298. [Google Scholar] [CrossRef] [PubMed]
- Kurnit, K.C.; Fleming, G.F.; Lengyel, E. Updates and New Options in Advanced Epithelial Ovarian Cancer Treatment. Obstet. Gynecol. 2021, 137, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G.; Leamon, C.P.; Reddy, J.A. The folate receptor as a rational therapeutic target for personalized cancer treatment. Drug Resist. Updates 2014, 17, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Elnakat, H.; Ratnam, M. Distribution, functionality and gene regulation of folate receptor isoforms: Implications in targeted therapy. Adv. Drug Deliv. Rev. 2004, 56, 1067–1084. [Google Scholar] [CrossRef]
- Vergote, I.B.; Marth, C.; Coleman, R.L. Role of the folate receptor in ovarian cancer treatment: Evidence, mechanism, and clinical implications. Cancer Metastasis Rev. 2015, 34, 41–52. [Google Scholar] [CrossRef]
- Desmoulin, S.K.; Hou, Z.; Gangjee, A.; Matherly, L.H. The human proton-coupled folate transporter: Biology and therapeutic applications to cancer. Cancer Biol. Ther. 2012, 13, 1355–1373. [Google Scholar] [CrossRef] [Green Version]
- Matherly, L.H.; Hou, Z.; Deng, Y. Human reduced folate carrier: Translation of basic biology to cancer etiology and therapy. Cancer Metastasis Rev. 2007, 26, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Matherly, L.H.; Hou, Z.; Gangjee, A. The promise and challenges of exploiting the proton-coupled folate transporter for selective therapeutic targeting of cancer. Cancer Chemother. Pharmacol. 2018, 81, 1–15. [Google Scholar] [CrossRef]
- Parker, N.; Turk, M.J.; Westrick, E.; Lewis, J.D.; Low, P.S.; Leamon, C.P. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal. Biochem. 2005, 338, 284–293. [Google Scholar] [CrossRef]
- Toffoli, G.; Cernigoi, C.; Russo, A.; Gallo, A.; Bagnoli, M.; Boiocchi, M. Overexpression of folate binding protein in ovarian cancers. Int. J. Cancer 1997, 74, 193–198. [Google Scholar] [CrossRef]
- Weitman, S.D.; Lark, R.H.; Coney, L.R.; Fort, D.W.; Frasca, V.; Zurawski, V.R., Jr.; Kamen, B.A. Distribution of the folate receptor GP38 in normal and malignant cell lines and tissues. Cancer Res. 1992, 52, 3396–3401. [Google Scholar] [PubMed]
- Hou, Z.; Gattoc, L.; O’Connor, C.; Yang, S.; Wallace-Povirk, A.; George, C.; Orr, S.; Polin, L.; White, K.; Kushner, J.; et al. Dual Targeting of Epithelial Ovarian Cancer Via Folate Receptor alpha and the Proton-Coupled Folate Transporter with 6-Substituted Pyrrolo[2,3-d]pyrimidine Antifolates. Mol. Cancer Ther. 2017, 16, 819–830. [Google Scholar] [CrossRef] [Green Version]
- Cresswell, G.M.; Wang, B.; Kischuk, E.M.; Broman, M.M.; Alfar, R.A.; Vickman, R.E.; Dimitrov, D.S.; Kularatne, S.A.; Sundaram, C.P.; Singhal, S.; et al. Folate Receptor Beta Designates Immunosuppressive Tumor-Associated Myeloid Cells That Can Be Reprogrammed with Folate-Targeted Drugs. Cancer Res. 2021, 81, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Puig-Kroger, A.; Sierra-Filardi, E.; Dominguez-Soto, A.; Samaniego, R.; Corcuera, M.T.; Gomez-Aguado, F.; Ratnam, M.; Sanchez-Mateos, P.; Corbi, A.L. Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res. 2009, 69, 9395–9403. [Google Scholar] [CrossRef] [Green Version]
- Nagai, T.; Tanaka, M.; Tsuneyoshi, Y.; Xu, B.; Michie, S.A.; Hasui, K.; Hirano, H.; Arita, K.; Matsuyama, T. Targeting tumor-associated macrophages in an experimental glioma model with a recombinant immunotoxin to folate receptor beta. Cancer Immunol. Immunother. 2009, 58, 1577–1586. [Google Scholar] [CrossRef]
- Tie, Y.; Zheng, H.; He, Z.; Yang, J.; Shao, B.; Liu, L.; Luo, M.; Yuan, X.; Liu, Y.; Zhang, X.; et al. Targeting folate receptor beta positive tumor-associated macrophages in lung cancer with a folate-modified liposomal complex. Signal Transduct. Target. Ther. 2020, 5, 6. [Google Scholar] [CrossRef]
- Matherly, L.H.; Wilson, M.R.; Hou, Z. The major facilitative folate transporters solute carrier 19A1 and solute carrier 46A1: Biology and role in antifolate chemotherapy of cancer. Drug Metab. Dispos. 2014, 42, 632–649. [Google Scholar] [CrossRef] [Green Version]
- Visentin, M.; Zhao, R.; Goldman, I.D. The antifolates. Hematol. Oncol. Clin. N. Am. 2012, 26, 629–648. [Google Scholar] [CrossRef] [Green Version]
- Dekhne, A.S.; Hou, Z.; Gangjee, A.; Matherly, L.H. Therapeutic Targeting of Mitochondrial One-Carbon Metabolism in Cancer. Mol. Cancer Ther. 2020, 19, 2245–2255. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucock, M. Folic acid: Nutritional biochemistry, molecular biology, and role in disease processes. Mol. Genet. Metab. 2000, 71, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Stover, P.J. Physiology of folate and vitamin B12 in health and disease. Nutr. Rev. 2004, 62, S3–S12. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Ann. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.D.; Theti, D.S.; Wood, N.; Green, M.; Raynaud, F.; Valenti, M.; Forster, M.D.; Mitchell, F.; Bavetsias, V.; Henderson, E.; et al. BGC 945, a novel tumor-selective thymidylate synthase inhibitor targeted to alpha-folate receptor-overexpressing tumors. Cancer Res. 2005, 65, 11721–11728. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.L.; Deme, J.C.; Kuteyi, G.; Wu, Z.; Huo, J.; Goldman, I.D.; Owens, R.J.; Biggin, P.C.; Lea, S.M.; Newstead, S. Structural basis of antifolate recognition and transport by PCFT. Nature 2021, 595, 130–134. [Google Scholar] [CrossRef]
- Matherly, L.H.; Goldman, D.I. Membrane transport of folates. Vitam. Horm. 2003, 66, 403–456. [Google Scholar] [PubMed]
- Zhao, R.; Diop-Bove, N.; Visentin, M.; Goldman, I.D. Mechanisms of membrane transport of folates into cells and across epithelia. Annu. Rev. Nutr. 2011, 31, 177–201. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Goldman, I.D. The molecular identity and characterization of a Proton-coupled Folate Transporter--PCFT; biological ramifications and impact on the activity of pemetrexed. Cancer Metastasis Rev. 2007, 26, 129–139. [Google Scholar] [CrossRef]
- Saier, M.H., Jr. Transporter Classification Database. 2012. Available online: http://www.tcdb.org/ (accessed on 1 December 2021).
- Whetstine, J.R.; Flatley, R.M.; Matherly, L.H. The human reduced folate carrier gene is ubiquitously and differentially expressed in normal human tissues: Identification of seven non-coding exons and characterization of a novel promoter. Biochem. J. 2002, 367, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, R.; Russell, R.G.; Goldman, I.D. Localization of the murine reduced folate carrier as assessed by immunohistochemical analysis. Biochim. Biophys. Acta Biomembr. 2001, 1513, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Waes, J.G.-V.; Heller, S.; Bauer, L.K.; Wilberding, J.; Maddox, J.R.; Aleman, F.; Rosenquist, T.H.; Finnell, R.H. Embryonic development in the reduced folate carrier knockout mouse is modulated by maternal folate supplementation. Birth Defects Res. Part A Clin. Mol. Teratol. 2008, 82, 494–507. [Google Scholar] [CrossRef]
- Zhao, R.; Russell, R.G.; Wang, Y.; Liu, L.; Gao, F.; Kneitz, B.; Edelmann, W.; Goldman, I.D. Rescue of embryonic lethality in reduced folate carrier-deficient mice by maternal folic acid supplementation reveals early neonatal failure of hematopoietic organs. J. Biol. Chem. 2001, 276, 10224–10228. [Google Scholar] [CrossRef] [Green Version]
- Siu, M.K.Y.; Kong, D.S.H.; Chan, H.Y.; Wong, E.S.Y.; Philip, P.C.I.; Jiang, L.; Hextan, Y.S.N.; Le, X.F.; Cheung, A.N.Y. Paradoxical Impact of Two Folate Receptors, FRa and RFC, in Ovarian Cancer: Effect on Cell Proliferation, Invasion and Clinical Outcome. PLoS ONE 2012, 7, e47201. [Google Scholar] [CrossRef] [Green Version]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; Sandoval, C.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006, 127, 917–928. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Aluri, S.; Goldman, I.D. The proton-coupled folate transporter (PCFT-SLC46A1) and the syndrome of systemic and cerebral folate deficiency of infancy: Hereditary folate malabsorption. Mol. Asp. Med. 2017, 53, 57–72. [Google Scholar] [CrossRef] [Green Version]
- Qiu, A.; Min, S.H.; Jansen, M.; Malhotra, U.; Tsai, E.; Cabelof, D.C.; Matherly, L.H.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Rodent intestinal folate transporters (SLC46A1): Secondary structure, functional properties, and response to dietary folate restriction. Am. J. Physiol. Cell Physiol. 2007, 293, C1669–C1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Visentin, M.; Suadicani, S.O.; Goldman, I.D. Inhibition of the proton-coupled folate transporter (PCFT-SLC46A1) by bicarbonate and other anions. Mol. Pharmacol. 2013, 84, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Desmoulin, S.K.; Wang, L.; Hales, E.; Polin, L.; White, K.; Kushner, J.; Stout, M.; Hou, Z.; Cherian, C.; Gangjee, A.; et al. Therapeutic targeting of a novel 6-substituted pyrrolo [2,3-d]pyrimidine thienoyl antifolate to human solid tumors based on selective uptake by the proton-coupled folate transporter. Mol. Pharmacol. 2011, 80, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannetti, E.; Zucali, P.A.; Assaraf, Y.G.; Funel, N.; Gemelli, M.; Stark, M.; Thunnissen, E.; Hou, Z.; Muller, I.B.; Struys, E.A.; et al. Role of proton-coupled folate transporter in pemetrexed resistance of mesothelioma: Clinical evidence and new pharmacological tools. Ann. Oncol 2017, 28, 2725–2732. [Google Scholar] [CrossRef]
- Wilson, M.R.; Hou, Z.; Yang, S.; Polin, L.; Kushner, J.; White, K.; Huang, J.; Ratnam, M.; Gangjee, A.; Matherly, L.H. Targeting Nonsquamous Nonsmall Cell Lung Cancer via the Proton-Coupled Folate Transporter with 6-Substituted Pyrrolo[2,3-d]Pyrimidine Thienoyl Antifolates. Mol. Pharmacol. 2016, 89, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Diop-Bove, N.K.; Wu, J.; Zhao, R.; Locker, J.; Goldman, I.D. Hypermethylation of the human proton-coupled folate transporter (SLC46A1) minimal transcriptional regulatory region in an antifolate-resistant HeLa cell line. Mol. Cancer Ther. 2009, 8, 2424–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonen, N.; Bram, E.E.; Assaraf, Y.G. PCFT/SLC46A1 promoter methylation and restoration of gene expression in human leukemia cells. Biochem. Biophys. Res. Commun. 2008, 376, 787–792. [Google Scholar] [CrossRef]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Nishimura, K.; Al Hosseini, H.S.; Bianchi, E.; Wright, G.J.; Jovine, L. Divergent evolution of vitamin B9 binding underlies Juno-mediated adhesion of mammalian gametes. Curr. Biol. 2016, 26, R100–R101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christoph, D.C.; Asuncion, B.R.; Hassan, B.; Tran, C.; Maltzman, J.D.; O’Shannessy, D.J.; Wynes, M.W.; Gauler, T.C.; Wohlschlaeger, J.; Hoiczyk, M.; et al. Significance of folate receptor alpha and thymidylate synthase protein expression in patients with non-small-cell lung cancer treated with pemetrexed. J. Thorac. Oncol. 2013, 8, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Nunez, M.I.; Behrens, C.; Woods, D.M.; Lin, H.; Suraokar, M.; Kadara, H.; Hofstetter, W.; Kalhor, N.; Lee, J.J.; Franklin, W.; et al. High expression of folate receptor alpha in lung cancer correlates with adenocarcinoma histology and EGFR [corrected] mutation. J. Thorac. Oncol. 2012, 7, 833–840. [Google Scholar] [CrossRef] [Green Version]
- Czeizel, A.E.; Dudas, I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N. Engl. J. Med. 1992, 327, 1832–1835. [Google Scholar] [CrossRef] [PubMed]
- Nakashima-Matsushita, N.; Homma, T.; Yu, S.; Matsuda, T.; Sunahara, N.; Nakamura, T.; Tsukano, M.; Ratnam, M.; Matsuyama, T. Selective expression of folate receptor beta and its possible role in methotrexate transport in synovial macrophages from patients with rheumatoid arthritis. Arthritis Rheum. 1999, 42, 1609–1616. [Google Scholar] [CrossRef]
- Ratnam, M.; Marquardt, H.; Duhring, J.L.; Freisheim, J.H. Homologous membrane folate binding proteins in human placenta: Cloning and sequence of a cDNA. Biochemistry 1989, 28, 8249–8254. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.F.; Wang, H.; Behm, F.G.; Mathew, P.; Wu, M.; Booth, R.; Ratnam, M. Folate receptor type beta is a neutrophilic lineage marker and is differentially expressed in myeloid leukemia. Cancer 1999, 85, 348–357. [Google Scholar] [CrossRef]
- Shen, F.; Wu, M.; Ross, J.F.; Miller, D.; Ratnam, M. Folate receptor type gamma is primarily a secretory protein due to lack of an efficient signal for glycosylphosphatidylinositol modification: Protein characterization and cell type specificity. Biochemistry 1995, 34, 5660–5665. [Google Scholar] [CrossRef] [PubMed]
- Kamen, B.A.; Wang, M.T.; Streckfuss, A.J.; Peryea, X.; Anderson, R.G. Delivery of folates to the cytoplasm of MA104 cells is mediated by a surface membrane receptor that recycles. J. Biol. Chem. 1988, 263, 13602–13609. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chang, M.C.; Huang, C.Y.; Chiang, Y.C.; Lin, H.W.; Chen, C.A.; Hsieh, C.Y.; Cheng, W.F. Serous ovarian carcinoma patients with high alpha-folate receptor had reducing survival and cytotoxic chemo-response. Mol. Oncol. 2012, 6, 360–369. [Google Scholar] [CrossRef] [Green Version]
- Elwood, P.C.; Nachmanoff, K.; Saikawa, Y.; Page, S.T.; Pacheco, P.; Roberts, S.; Chung, K.N. The divergent 5’ termini of the alpha human folate receptor (hFR) mRNAs originate from two tissue-specific promoters and alternative splicing: Characterization of the alpha hFR gene structure. Biochemistry 1997, 36, 1467–1478. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.J.; Chung, K.N.; Nachmanoff, K.; Elwood, P.C. Tissue-specific promoters of the alpha human folate receptor gene yield transcripts with divergent 5’ leader sequences and different translational efficiencies. Biochem. J. 1997, 326, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Page, S.T.; Owen, W.C.; Price, K.; Elwood, P.C. Expression of the human placental folate receptor transcript is regulated in human tissues. Organization and full nucleotide sequence of the gene. J. Mol. Biol. 1993, 229, 1175–1183. [Google Scholar] [CrossRef] [Green Version]
- Kelley, K.M.; Rowan, B.G.; Ratnam, M. Modulation of the folate receptor alpha gene by the estrogen receptor: Mechanism and implications in tumor targeting. Cancer Res. 2003, 63, 2820–2828. [Google Scholar]
- Tran, T.; Shatnawi, A.; Zheng, X.; Kelley, K.M.; Ratnam, M. Enhancement of folate receptor alpha expression in tumor cells through the glucocorticoid receptor: A promising means to improved tumor detection and targeting. Cancer Res. 2005, 65, 4431–4441. [Google Scholar] [CrossRef] [Green Version]
- Campbell, I.G.; Jones, T.A.; Foulkes, W.D.; Trowsdale, J. Folate-binding protein is a marker for ovarian cancer. Cancer Res. 1991, 51, 5329–5338. [Google Scholar]
- Lawrence, S.A.; Hackett, J.C.; Moran, R.G. Tetrahydrofolate Recognition by the Mitochondrial Folate Transporter. J. Biol. Chem. 2011, 286, 31480–31489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, E.A.; Titus, S.A.; Taylor, S.M.; Jackson-Cook, C.; Moran, R.G. A mutation inactivating the mitochondrial inner membrane folate transporter creates a glycine requirement for survival of chinese hamster cells. J. Biol. Chem. 2004, 279, 33829–33836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, S.A.; Titus, S.A.; Ferguson, J.; Heineman, A.L.; Taylor, S.M.; Moran, R.G. Mammalian mitochondrial and cytosolic folylpolyglutamate synthetase maintain the subcellular compartmentalization of folates. J. Biol. Chem. 2014, 289, 29386–29396. [Google Scholar] [CrossRef] [Green Version]
- Shane, B. Folylpolyglutamate synthesis and role in the regulation of one-carbon metabolism. Vitam. Horm. 1989, 45, 263–335. [Google Scholar] [CrossRef] [PubMed]
- Kory, N.; Wyant, G.A.; Prakash, G.; Uit de Bos, J.; Bottanelli, F.; Pacold, M.E.; Chan, S.H.; Lewis, C.A.; Wang, T.; Keys, H.R.; et al. SFXN1 is a mitochondrial serine transporter required for one-carbon metabolism. Science 2018, 362, eaat9528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locasale, J.W. Serine, glycine and the one-carbon cycle: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- Ye, J.; Fan, J.; Venneti, S.; Wan, Y.W.; Pawel, B.R.; Zhang, J.; Finley, L.W.; Lu, C.; Lindsten, T.; Cross, J.R.; et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014, 4, 1406–1417. [Google Scholar] [CrossRef] [Green Version]
- Krupenko, S.A.; Krupenko, N.I. ALDH1L1 and ALDH1L2 Folate Regulatory Enzymes in Cancer. Adv. Exp. Med. Biol. 2018, 1032, 127–143. [Google Scholar] [CrossRef]
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pareek, V.; Pedley, A.M.; Benkovic, S.J. Human de novo purine biosynthesis. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 1–16. [Google Scholar] [CrossRef]
- Lu, S.C. S-Adenosylmethionine. Int. J. Biochem. Cell Biol. 2000, 32, 391–395. [Google Scholar] [CrossRef]
- Matthews, B.G.; Bowden, N.A.; Wong-Brown, M.W. Epigenetic Mechanisms and Therapeutic Targets in Chemoresistant High-Grade Serous Ovarian Cancer. Cancers 2021, 13, 5993. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Lin, T.Y.; Lee, G.; Paddock, M.N.; Momb, J.; Cheng, Z.; Li, Q.; Fei, D.L.; Stein, B.D.; Ramsamooj, S.; et al. Mitochondrial One-Carbon Pathway Supports Cytosolic Folate Integrity in Cancer Cells. Cell 2018, 175, 1546–1560. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5, 4128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badar, T.; Patel, K.P.; Thompson, P.A.; DiNardo, C.; Takahashi, K.; Cabrero, M.; Borthakur, G.; Cortes, J.; Konopleva, M.; Kadia, T.; et al. Detectable FLT3-ITD or RAS mutation at the time of transformation from MDS to AML predicts for very poor outcomes. Leuk. Res. 2015, 39, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Nikiforov, M.A.; Chandriani, S.; O’Connell, B.; Petrenko, O.; Kotenko, I.; Beavis, A.; Sedivy, J.M.; Cole, M.D. A functional screen for Myc-responsive genes reveals serine hydroxymethyltransferase, a major source of the one-carbon unit for cell metabolism. Mol. Cell. Biol. 2002, 22, 5793–5800. [Google Scholar] [CrossRef] [Green Version]
- Shin, M.; Momb, J.; Appling, D.R. Human mitochondrial MTHFD2 is a dual redox cofactor-specific methylenetetrahydrofolate dehydrogenase/methenyltetrahydrofolate cyclohydrolase. Cancer Metab. 2017, 5, 11. [Google Scholar] [CrossRef]
- Nilsson, R.; Nicolaidou, V.; Koufaris, C. Mitochondrial MTHFD isozymes display distinct expression, regulation, and association with cancer. Gene 2019, 716, 144032. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://seer.cancer.gov/statfacts/html/ovary.html (accessed on 1 December 2021).
- Worzfeld, T.; Strandmann, E.P.V.; Huber, M.; Adhikary, T.; Wagner, U.; Reinartz, S.; Muller, R. The Unique Molecular and Cellular Microenvironment of Ovarian Cancer. Front. Oncol. 2017, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- Lokadasan, R.; James, F.V.; Narayanan, G.; Prabhakaran, P.K. Targeted agents in epithelial ovarian cancer: Review on emerging therapies and future developments. Ecancermedicalscience 2016, 10, 626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.; Ueda, Y.; Naka, T.; Enomoto, T. Therapeutic strategies in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 2012, 31, 14. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.A.; Raja, F.A.; Fotopoulou, C.; Gonzalez-Martin, A.; Colombo, N.; Sessa, C. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24 (Suppl. S6), vi24–vi32. [Google Scholar] [CrossRef]
- Boussios, S.; Zarkavelis, G.; Seraj, E.; Zerdes, I.; Tatsi, K.; Pentheroudakis, G. Non-epithelial Ovarian Cancer: Elucidating Uncommon Gynaecological Malignancies. Anticancer Res. 2016, 36, 5031–5042. [Google Scholar] [CrossRef] [Green Version]
- Devouassoux-Shisheboran, M.; Genestie, C. Pathobiology of ovarian carcinomas. Chin. J. Cancer 2015, 34, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Neff, R.T.; Senter, L.; Salani, R. BRCA mutation in ovarian cancer: Testing, implications and treatment considerations. Ther. Adv. Med. Oncol. 2017, 9, 519–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramus, S.J.; Gayther, S.A. The contribution of BRCA1 and BRCA2 to ovarian cancer. Mol. Oncol. 2009, 3, 138–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [Green Version]
- Pal, T.; Permuth-Wey, J.; Betts, J.A.; Krischer, J.P.; Fiorica, J.; Arango, H.; LaPolla, J.; Hoffman, M.; Martino, M.A.; Wakeley, K.; et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 2005, 104, 2807–2816. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Timms, K.M.; Carey, M.S.; Gutin, A.; Meyer, L.A.; Flake, D.D., 2nd; Abkevich, V.; Potter, J.; Pruss, D.; Glenn, P.; et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J. Clin. Oncol. 2010, 28, 3570–3576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, R.L.; Nemeth, E.; Tran, H.; Shvartsman, H.; Cass, I.; Narod, S.; Karlan, B.Y. BRCA1 promoter region hypermethylation in ovarian carcinoma: A population-based study. Cancer Res. 2000, 60, 5329–5333. [Google Scholar] [PubMed]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [Green Version]
- Corney, D.C.; Flesken-Nikitin, A.; Choi, J.; Nikitin, A.Y. Role of p53 and Rb in ovarian cancer. Adv. Exp. Med. Biol. 2008, 622, 99–117. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- du Bois, A.; Reuss, A.; Pujade-Lauraine, E.; Harter, P.; Ray-Coquard, I.; Pfisterer, J. Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: A combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: By the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d’Investigateurs Nationaux Pour les Etudes des Cancers de l’Ovaire (GINECO). Cancer 2009, 115, 1234–1244. [Google Scholar] [CrossRef]
- van der Burg, M.E.; van Lent, M.; Buyse, M.; Kobierska, A.; Colombo, N.; Favalli, G.; Lacave, A.J.; Nardi, M.; Renard, J.; Pecorelli, S. The effect of debulking surgery after induction chemotherapy on the prognosis in advanced epithelial ovarian cancer. Gynecological Cancer Cooperative Group of the European Organization for Research and Treatment of Cancer. N. Engl. J. Med. 1995, 332, 629–634. [Google Scholar] [CrossRef]
- Vergote, I.B.; De Wever, I.; Decloedt, J.; Tjalma, W.; Van Gramberen, M.; van Dam, P. Neoadjuvant chemotherapy versus primary debulking surgery in advanced ovarian cancer. Semin. Oncol. 2000, 27, 31–36. [Google Scholar] [CrossRef]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.S.; DeGeest, K.; Hartenbach, E.M.; Baergen, R. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: A Gynecologic Oncology Group study. J. Clin. Oncol. 2003, 21, 3194–3200. [Google Scholar] [CrossRef]
- Sato, S.; Itamochi, H. Neoadjuvant chemotherapy in advanced ovarian cancer: Latest results and place in therapy. Ther. Adv. Med. Oncol. 2014, 6, 293–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Nag, S.; Aggarwal, S.; Rauthan, A.; Warrier, N. Maintenance therapy for recurrent epithelial ovarian cancer: Current therapies and future perspectives—A review. J. Ovarian Res. 2019, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Scandurra, G.; Lombardo, V.; Gattuso, G.; Lavoro, A.; Distefano, A.B.; Scibilia, G.; Scollo, P. A multidisciplinary approach remains the best strategy to improve and strengthen the management of ovarian cancer (Review). Int. J. Oncol. 2021, 59, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Vernooij, F.; Heintz, A.P.; Coebergh, J.W.; Massuger, L.F.; Witteveen, P.O.; van der Graaf, Y. Specialized and high-volume care leads to better outcomes of ovarian cancer treatment in the Netherlands. Gynecol. Oncol. 2009, 112, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Cortez, A.J.; Tudrej, P.; Kujawa, K.A.; Lisowska, K.M. Advances in ovarian cancer therapy. Cancer Chemother. Pharmacol. 2018, 81, 17–38. [Google Scholar] [CrossRef] [Green Version]
- Evans, T.; Matulonis, U. PARP inhibitors in ovarian cancer: Evidence, experience and clinical potential. Ther. Adv. Med. Oncol. 2017, 9, 253–267. [Google Scholar] [CrossRef]
- Kobel, M.; Madore, J.; Ramus, S.J.; Clarke, B.A.; Pharoah, P.D.; Deen, S.; Bowtell, D.D.; Odunsi, K.; Menon, U.; Morrison, C.; et al. Evidence for a time-dependent association between FOLR1 expression and survival from ovarian carcinoma: Implications for clinical testing. An Ovarian Tumour Tissue Analysis consortium study. Br. J. Cancer 2014, 111, 2297–2307. [Google Scholar] [CrossRef] [Green Version]
- Scaranti, M.; Cojocaru, E.; Banerjee, S.; Banerji, U. Exploiting the folate receptor alpha in oncology. Nat. Rev. Clin. Oncol. 2020, 17, 349–359. [Google Scholar] [CrossRef]
- Ebel, W.; Routhier, E.L.; Foley, B.; Jacob, S.; McDonough, J.M.; Patel, R.K.; Turchin, H.A.; Chao, Q.; Kline, J.B.; Old, L.J.; et al. Preclinical evaluation of MORAb-003, a humanized monoclonal antibody antagonizing folate receptor-alpha. Cancer Immun. 2007, 7, 6. [Google Scholar]
- Ledermann, J.A.; Canevari, S.; Thigpen, T. Targeting the folate receptor: Diagnostic and therapeutic approaches to personalize cancer treatments. Ann. Oncol. 2015, 26, 2034–2043. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Spidel, J.L.; Maddage, C.J.; Rybinski, K.A.; Kennedy, R.P.; Krauthauser, C.L.; Park, Y.C.; Albone, E.F.; Jacob, S.; Goserud, M.T.; et al. The antitumor activity of the human FOLR1-specific monoclonal antibody, farletuzumab, in an ovarian cancer mouse model is mediated by antibody-dependent cellular cytotoxicity. Cancer Biol. Ther. 2013, 14, 1032–1038. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Graybill, W.S.; Previs, R.A.; Hu, W.; Ivan, C.; Mangala, L.S.; Zand, B.; Nick, A.M.; Jennings, N.B.; Dalton, H.J.; et al. Immunotherapy targeting folate receptor induces cell death associated with autophagy in ovarian cancer. Clin. Cancer Res. 2015, 21, 448–459. [Google Scholar] [CrossRef] [Green Version]
- Konner, J.A.; Bell-McGuinn, K.M.; Sabbatini, P.; Hensley, M.L.; Tew, W.P.; Pandit-Taskar, N.; Vander Els, N.; Phillips, M.D.; Schweizer, C.; Weil, S.C.; et al. Farletuzumab, a humanized monoclonal antibody against folate receptor alpha, in epithelial ovarian cancer: A phase I study. Clin. Cancer Res. 2010, 16, 5288–5295. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, D.K.; White, A.J.; Weil, S.C.; Phillips, M.; Coleman, R.L. Farletuzumab (a monoclonal antibody against folate receptor alpha) in relapsed platinum-sensitive ovarian cancer. Gynecol. Oncol. 2013, 129, 452–458. [Google Scholar] [CrossRef]
- Sato, S.; Itamochi, H. Profile of farletuzumab and its potential in the treatment of solid tumors. OncoTargets Ther. 2016, 9, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Vergote, I.; Armstrong, D.; Scambia, G.; Teneriello, M.; Sehouli, J.; Schweizer, C.; Weil, S.C.; Bamias, A.; Fujiwara, K.; Ochiai, K.; et al. A Randomized, Double-Blind, Placebo-Controlled, Phase III Study to Assess Efficacy and Safety of Weekly Farletuzumab in Combination with Carboplatin and Taxane in Patients with Ovarian Cancer in First Platinum-Sensitive Relapse. J. Clin. Oncol. 2016, 34, 2271–2278. [Google Scholar] [CrossRef]
- Ab, O.; Whiteman, K.R.; Bartle, L.M.; Sun, X.; Singh, R.; Tavares, D.; LaBelle, A.; Payne, G.; Lutz, R.J.; Pinkas, J.; et al. IMGN853, a Folate Receptor-alpha (FRalpha)-Targeting Antibody-Drug Conjugate, Exhibits Potent Targeted Antitumor Activity against FRalpha-Expressing Tumors. Mol. Cancer Ther. 2015, 14, 1605–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.N.; Borghaei, H.; O’Malley, D.M.; Jeong, W.; Seward, S.M.; Bauer, T.M.; Perez, R.P.; Matulonis, U.A.; Running, K.L.; Zhang, X.; et al. Phase 1 dose-escalation study of mirvetuximab soravtansine (IMGN853), a folate receptor alpha-targeting antibody-drug conjugate, in patients with solid tumors. Cancer 2017, 123, 3080–3087. [Google Scholar] [CrossRef]
- Moore, K.N.; Oza, A.M.; Colombo, N.; Oaknin, A.; Scambia, G.; Lorusso, D.; Konecny, G.E.; Banerjee, S.; Murphy, C.G.; Tanyi, J.L.; et al. Phase III, randomized trial of mirvetuximab soravtansine versus chemotherapy in patients with platinum-resistant ovarian cancer: Primary analysis of FORWARD I. Ann. Oncol. 2021, 32, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.A.; Dorton, R.; Westrick, E.; Dawson, A.; Smith, T.; Xu, L.C.; Vetzel, M.; Kleindl, P.; Vlahov, I.R.; Leamon, C.P. Preclinical evaluation of EC145, a folate-vinca alkaloid conjugate. Cancer Res. 2007, 67, 4434–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorusso, P.M.; Edelman, M.J.; Bever, S.L.; Forman, K.M.; Pilat, M.; Quinn, M.F.; Li, J.; Heath, E.I.; Malburg, L.M.; Klein, P.J.; et al. Phase I study of folate conjugate EC145 (Vintafolide) in patients with refractory solid tumors. J. Clin. Oncol. 2012, 30, 4011–4016. [Google Scholar] [CrossRef]
- Naumann, R.W.; Coleman, R.L.; Burger, R.A.; Sausville, E.A.; Kutarska, E.; Ghamande, S.A.; Gabrail, N.Y.; Depasquale, S.E.; Nowara, E.; Gilbert, L.; et al. PRECEDENT: A randomized phase II trial comparing vintafolide (EC145) and pegylated liposomal doxorubicin (PLD) in combination versus PLD alone in patients with platinum-resistant ovarian cancer. J. Clin. Oncol. 2013, 31, 4400–4406. [Google Scholar] [CrossRef] [PubMed]
- Vergote, I.; Leamon, C.P. Vintafolide: A novel targeted therapy for the treatment of folate receptor expressing tumors. Ther. Adv. Med. Oncol. 2015, 7, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Banerji, U.; Garces, A.H.I.; Michalarea, V.; Ruddle, R.; Raynaud, F.I.; Riisnaes, R.; Rodrigues, D.N.; Tunariu, N.; Porter, J.C.; Ward, S.E.; et al. An investigator-initiated phase I study of ONX-0801 a first-in-class alpha folate receptor targeted, small molecule thymidylate synthase inhibitor in solid tumors. J. Clin. Oncol. 2017, 35, 2503. [Google Scholar] [CrossRef]
- Konda, S.D.; Aref, M.; Brechbiel, M.; Wiener, E.C. Development of a tumor-targeting MR contrast agent using the high-affinity folate receptor: Work in progress. Investig. Radiol. 2000, 35, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Meier, R.; Henning, T.D.; Boddington, S.; Tavri, S.; Arora, S.; Piontek, G.; Rudelius, M.; Corot, C.; Daldrup-Link, H.E. Breast cancers: MR imaging of folate-receptor expression with the folate-specific nanoparticle P1133. Radiology 2010, 255, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Siegel, B.A.; Dehdashti, F.; Mutch, D.G.; Podoloff, D.A.; Wendt, R.; Sutton, G.P.; Burt, R.W.; Ellis, P.R.; Mathias, C.J.; Green, M.A.; et al. Evaluation of 111In-DTPA-folate as a receptor-targeted diagnostic agent for ovarian cancer: Initial clinical results. J. Nucl. Med. 2003, 44, 700–707. [Google Scholar]
- Fisher, R.E.; Siegel, B.A.; Edell, S.L.; Oyesiku, N.M.; Morgenstern, D.E.; Messmann, R.A.; Amato, R.J. Exploratory study of 99mTc-EC20 imaging for identifying patients with folate receptor-positive solid tumors. J. Nucl. Med. 2008, 49, 899–906. [Google Scholar] [CrossRef] [Green Version]
- Brand, C.; Sadique, A.; Houghton, J.L.; Gangangari, K.; Ponte, J.F.; Lewis, J.S.; Pillarsetty, N.V.K.; Konner, J.A.; Reiner, T. Leveraging PET to image folate receptor alpha therapy of an antibody-drug conjugate. EJNMMI Res. 2018, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.D.; Jallad, K.N.; Thompson, D.H.; Ben-Amotz, D.; Low, P.S. Optical imaging of metastatic tumors using a folate-targeted fluorescent probe. J. Biomed. Opt. 2003, 8, 636–641. [Google Scholar] [CrossRef]
- Tummers, Q.R.; Hoogstins, C.E.; Gaarenstroom, K.N.; de Kroon, C.D.; van Poelgeest, M.I.; Vuyk, J.; Bosse, T.; Smit, V.T.; van de Velde, C.J.; Cohen, A.F.; et al. Intraoperative imaging of folate receptor alpha positive ovarian and breast cancer using the tumor specific agent EC17. Oncotarget 2016, 7, 32144–32155. [Google Scholar] [CrossRef] [Green Version]
- van Dam, G.M.; Themelis, G.; Crane, L.M.; Harlaar, N.J.; Pleijhuis, R.G.; Kelder, W.; Sarantopoulos, A.; de Jong, J.S.; Arts, H.J.; van der Zee, A.G.; et al. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-alpha targeting: First in-human results. Nat. Med. 2011, 17, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-imaging-drug-help-identify-ovarian-cancer-lesions (accessed on 1 December 2021).
- Roche, M.; Parisi, L.; Li, L.; Knehans, A.; Phaeton, R.; Kesterson, J.P. The role of pemetrexed in recurrent epithelial ovarian cancer: A scoping review. Oncol. Rev. 2018, 12, 346. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Moran, R.G.; Goldman, I.D. Pemetrexed: Biochemical and cellular pharmacology, mechanisms, and clinical applications. Mol. Cancer Ther. 2007, 6, 404–417. [Google Scholar] [CrossRef] [Green Version]
- Issaeva, N.; Thomas, H.D.; Djureinovic, T.; Jaspers, J.E.; Stoimenov, I.; Kyle, S.; Pedley, N.; Gottipati, P.; Zur, R.; Sleeth, K.; et al. 6-thioguanine selectively kills BRCA2-defective tumors and overcomes PARP inhibitor resistance. Cancer Res. 2010, 70, 6268–6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronder, J.L.; Moran, R.G. A defect in the p53 response pathway induced by de novo purine synthesis inhibition. J. Biol. Chem. 2003, 278, 48861–48871. [Google Scholar] [CrossRef] [Green Version]
- Bertino, J.R.; Waud, W.R.; Parker, W.B.; Lubin, M. Targeting tumors that lack methylthioadenosine phosphorylase (MTAP) activity: Current strategies. Cancer Biol. Ther. 2011, 11, 627–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, H.; Tran, P.; Carrera, C.J.; Hori, Y.; Rosenbach, M.D.; Carson, D.A.; Nobori, T. Methylthioadenosine phosphorylase cDNA transfection alters sensitivity to depletion of purine and methionine in A549 lung cancer cells. Cancer Res. 1996, 56, 5653–5658. [Google Scholar]
- Hoxhaj, G.; Hughes-Hallett, J.; Timson, R.C.; Ilagan, E.; Yuan, M.; Asara, J.M.; Ben-Sahra, I.; Manning, B.D. The mTORC1 Signaling Network Senses Changes in Cellular Purine Nucleotide Levels. Cell Rep. 2017, 21, 1331–1346. [Google Scholar] [CrossRef] [Green Version]
- Rothbart, S.B.; Racanelli, A.C.; Moran, R.G. Pemetrexed indirectly activates the metabolic kinase AMPK in human carcinomas. Cancer Res. 2010, 70, 10299–10309. [Google Scholar] [CrossRef] [Green Version]
- Desmoulin, S.K.; Wang, Y.; Wu, J.; Stout, M.; Hou, Z.; Fulterer, A.; Chang, M.H.; Romero, M.F.; Cherian, C.; Gangjee, A.; et al. Targeting the proton-coupled folate transporter for selective delivery of 6-substituted pyrrolo[2,3-d]pyrimidine antifolate inhibitors of de novo purine biosynthesis in the chemotherapy of solid tumors. Mol. Pharmacol. 2010, 78, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Wang, Y.; Cherian, C.; Hou, Z.; Buck, S.A.; Matherly, L.H.; Gangjee, A. Synthesis and discovery of high affinity folate receptor-specific glycinamide ribonucleotide formyltransferase inhibitors with antitumor activity. J. Med. Chem. 2008, 51, 5052–5063. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Cherian, C.; Desmoulin, S.K.; Polin, L.; Deng, Y.; Wu, J.; Hou, Z.; White, K.; Kushner, J.; Matherly, L.H.; et al. Synthesis and antitumor activity of a novel series of 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolate inhibitors of purine biosynthesis with selectivity for high affinity folate receptors and the proton-coupled folate transporter over the reduced folate carrier for cellular entry. J. Med. Chem. 2010, 53, 1306–1318. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Desmoulin, S.K.; Cherian, C.; Polin, L.; White, K.; Kushner, J.; Fulterer, A.; Chang, M.H.; Mitchell-Ryan, S.; Stout, M.; et al. Synthesis, biological, and antitumor activity of a highly potent 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolate inhibitor with proton-coupled folate transporter and folate receptor selectivity over the reduced folate carrier that inhibits beta-glycinamide ribonucleotide formyltransferase. J. Med. Chem. 2011, 54, 7150–7164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wallace, A.; Raghavan, S.; Deis, S.M.; Wilson, M.R.; Yang, S.; Polin, L.; White, K.; Kushner, J.; Orr, S.; et al. 6-Substituted Pyrrolo[2,3-d]pyrimidine Thienoyl Regioisomers as Targeted Antifolates for Folate Receptor alpha and the Proton-Coupled Folate Transporter in Human Tumors. J. Med. Chem. 2015, 58, 6938–6959. [Google Scholar] [CrossRef] [Green Version]
- Ravindra, M.; Wilson, M.R.; Tong, N.; O’Connor, C.; Karim, M.; Polin, L.; Wallace-Povirk, A.; White, K.; Kushner, J.; Hou, Z.; et al. Fluorine-Substituted Pyrrolo[2,3-d]Pyrimidine Analogues with Tumor Targeting via Cellular Uptake by Folate Receptor alpha and the Proton-Coupled Folate Transporter and Inhibition of de Novo Purine Nucleotide Biosynthesis. J. Med. Chem. 2018, 61, 4228–4248. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Ghergurovich, J.M.; Mainolfi, N.; Suri, V.; Jeong, S.K.; Hsin-Jung Li, S.; Friedman, A.; Manfredi, M.G.; Gitai, Z.; Kim, H.; et al. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2017, 114, 11404–11409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Canaveras, J.C.; Lancho, O.; Ducker, G.S.; Ghergurovich, J.M.; Xu, X.; da Silva-Diz, V.; Minuzzo, S.; Indraccolo, S.; Kim, H.; Herranz, D.; et al. SHMT inhibition is effective and synergizes with methotrexate in T-cell acute lymphoblastic leukemia. Leukemia 2021, 35, 377–388. [Google Scholar] [CrossRef]
- Kawai, J.; Toki, T.; Ota, M.; Inoue, H.; Takata, Y.; Asahi, T.; Suzuki, M.; Shimada, T.; Ono, K.; Suzuki, K.; et al. Discovery of a Potent, Selective, and Orally Available MTHFD2 Inhibitor (DS18561882) with in Vivo Antitumor Activity. J. Med. Chem. 2019, 62, 10204–10220. [Google Scholar] [CrossRef]
- Dekhne, A.S.; Shah, K.; Ducker, G.S.; Katinas, J.M.; Wong-Roushar, J.; Nayeen, M.J.; Doshi, A.; Ning, C.; Bao, X.; Fruhauf, J.; et al. Novel pyrrolo[3,2-d]pyrimidine compounds target mitochondrial and cytosolic one-carbon metabolism with broad-spectrum antitumor efficacy. Mol. Cancer Ther. 2019, 18, 1787–1799. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, C.; Wallace-Povirk, A.; Ning, C.; Fruhauf, J.; Tong, N.; Gangjee, A.; Matherly, L.H.; Hou, Z. Folate transporter dynamics and therapy with classic and tumor-targeted antifolates. Sci Rep. 2021, 11, 6389. [Google Scholar] [CrossRef] [PubMed]
- Dekhne, A.S.; Ning, C.; Nayeen, M.J.; Shah, K.; Kalpage, H.; Frühauf, J.; Wallace-Povirk, A.; O’Connor, C.; Hou, Z.; Kim, S.; et al. Cellular Pharmacodynamics of a Novel Pyrrolo[3,2-d]pyrimidine Inhibitor Targeting Mitochondrial and Cytosolic One-Carbon Metabolism. Mol. Pharmacol. 2020, 97, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Wallace-Povirk, A.; O’Connor, C.; Bao, X.; Katinas, J.; Wong-Roushar, J.; Dekhne, A.; Hou, Z.; Nayeen, M.J.; Shah, K.; Nunez, J.; et al. Abstract 2348: Targeting mitochondrial and cytosolic one-carbon metabolism in epithelial ovarian cancer via folate receptor alpha. In Proceedings of the AACR Annual Meeting, Philadelphia, PA, USA, 10–15 April 2021; p. 2348. [Google Scholar]
- Li, Q.; Yang, F.; Shi, X.; Bian, S.; Shen, F.; Wu, Y.; Zhu, C.; Fu, F.; Wang, J.; Zhou, J.; et al. MTHFD2 promotes ovarian cancer growth and metastasis via activation of the STAT3 signaling pathway. FEBS Open Bio 2021, 11, 2845–2857. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, R.; Jemth, A.S.; Gustafsson, N.M.; Farnegardh, K.; Loseva, O.; Wiita, E.; Bonagas, N.; Dahllund, L.; Llona-Minguez, S.; Haggblad, M.; et al. Crystal Structure of the Emerging Cancer Target MTHFD2 in Complex with a Substrate-Based Inhibitor. Cancer Res. 2017, 77, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Ju, H.Q.; Lu, Y.X.; Chen, D.L.; Zuo, Z.X.; Liu, Z.X.; Wu, Q.N.; Mo, H.Y.; Wang, Z.X.; Wang, D.S.; Pu, H.Y.; et al. Modulation of Redox Homeostasis by Inhibition of MTHFD2 in Colorectal Cancer: Mechanisms and Therapeutic Implications. J. Natl. Cancer Inst. 2019, 111, 584–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, J.; Ota, M.; Ohki, H.; Toki, T.; Suzuki, M.; Shimada, T.; Matsui, S.; Inoue, H.; Sugihara, C.; Matsuhashi, N.; et al. Structure-Based Design and Synthesis of an Isozyme-Selective MTHFD2 Inhibitor with a Tricyclic Coumarin Scaffold. ACS Med. Chem. Lett. 2019, 10, 893–898. [Google Scholar] [CrossRef]
- Ducker, G.S.; Chen, L.; Morscher, R.J.; Ghergurovich, J.M.; Esposito, M.; Teng, X.; Kang, Y.; Rabinowitz, J.D. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab. 2016, 23, 1140–1153. [Google Scholar] [CrossRef] [Green Version]
- Bejarano, L.; Jordao, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Ishii, G.; Ochiai, A.; Neri, S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv. Drug Deliv. Rev. 2016, 99, 186–196. [Google Scholar] [CrossRef]
- Gilead, A.; Meir, G.; Neeman, M. The role of angiogenesis, vascular maturation, regression and stroma infiltration in dormancy and growth of implanted MLS ovarian carcinoma spheroids. Int. J. Cancer 2004, 108, 524–531. [Google Scholar] [CrossRef]
- Schauer, I.G.; Sood, A.K.; Mok, S.; Liu, J. Cancer-associated fibroblasts and their putative role in potentiating the initiation and development of epithelial ovarian cancer. Neoplasia 2011, 13, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tang, H.; Cai, J.; Zhang, T.; Guo, J.; Feng, D.; Wang, Z. Ovarian cancer-associated fibroblasts contribute to epithelial ovarian carcinoma metastasis by promoting angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer Lett. 2011, 303, 47–55. [Google Scholar] [CrossRef]
- Givel, A.M.; Kieffer, Y.; Scholer-Dahirel, A.; Sirven, P.; Cardon, M.; Pelon, F.; Magagna, I.; Gentric, G.; Costa, A.; Bonneau, C.; et al. miR200-regulated CXCL12beta promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat. Commun. 2018, 9, 1056. [Google Scholar] [CrossRef]
- Ahmed, Z.; Bicknell, R. Angiogenic signalling pathways. Angiogenesis Protoc. 2009, 467, 3–24. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colvin, E.K. Tumor-associated macrophages contribute to tumor progression in ovarian cancer. Front. Oncol. 2014, 4, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaishi, K.; Komohara, Y.; Tashiro, H.; Ohtake, H.; Nakagawa, T.; Katabuchi, H.; Takeya, M. Involvement of M2-polarized macrophages in the ascites from advanced epithelial ovarian carcinoma in tumor progression via Stat3 activation. Cancer Sci. 2010, 101, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Kornete, M.; Joyce, J.A. Re-education of macrophages as a therapeutic strategy in cancer. Immunotherapy 2019, 11, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.Y.; Dand, S.; Doig, L.; Papenfuss, A.T.; Scott, C.L.; Ho, G.; Ooi, J.D. T-Cell Receptor Therapy in the Treatment of Ovarian Cancer: A Mini Review. Front. Immunol. 2021, 12, 1141. [Google Scholar] [CrossRef] [PubMed]
- Kandalaft, L.E.; Powell, D.J., Jr.; Coukos, G. A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. J. Transl. Med. 2012, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Dong, J.; Yang, N.; Li, S.D.; Yang, Z.Y.; Huang, R.; Li, F.J.; Wang, W.T.; Ren, J.K.; Lei, J.; et al. Tandem CAR-T cells targeting FOLR1 and MSLN enhance the antitumor effects in ovarian cancer. Int. J. Biol. Sci. 2021, 17, 4365–4376. [Google Scholar] [CrossRef]
- Sale, S.; Orsulic, S. Models of ovarian cancer metastasis: Murine models. Drug Discov. Today Dis. Models 2006, 3, 149–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, D.; Orsulic, S. A mouse model for the molecular characterization of brca1-associated ovarian carcinoma. Cancer Res. 2006, 66, 8949–8953. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, A.E.; Ducker, G.S.; Billingham, L.K.; Martinez, C.A.; Mainolfi, N.; Suri, V.; Friedman, A.; Manfredi, M.G.; Weinberg, S.E.; Rabinowitz, J.D.; et al. Serine Metabolism Supports Macrophage IL-1beta Production. Cell Metab. 2019, 29, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Wang, Z.; Zhang, K.; Chi, Z.; Xu, T.; Jiang, D.; Chen, S.; Li, W.; Yang, X.; Zhang, X.; et al. One-Carbon Metabolism Supports S-Adenosylmethionine and Histone Methylation to Drive Inflammatory Macrophages. Mol. Cell 2019, 75, 1147–1160. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wallace-Povirk, A.; Hou, Z.; Nayeen, M.J.; Gangjee, A.; Matherly, L.H. Folate Transport and One-Carbon Metabolism in Targeted Therapies of Epithelial Ovarian Cancer. Cancers 2022, 14, 191. https://doi.org/10.3390/cancers14010191
Wallace-Povirk A, Hou Z, Nayeen MJ, Gangjee A, Matherly LH. Folate Transport and One-Carbon Metabolism in Targeted Therapies of Epithelial Ovarian Cancer. Cancers. 2022; 14(1):191. https://doi.org/10.3390/cancers14010191
Chicago/Turabian StyleWallace-Povirk, Adrianne, Zhanjun Hou, Md. Junayed Nayeen, Aleem Gangjee, and Larry H. Matherly. 2022. "Folate Transport and One-Carbon Metabolism in Targeted Therapies of Epithelial Ovarian Cancer" Cancers 14, no. 1: 191. https://doi.org/10.3390/cancers14010191
APA StyleWallace-Povirk, A., Hou, Z., Nayeen, M. J., Gangjee, A., & Matherly, L. H. (2022). Folate Transport and One-Carbon Metabolism in Targeted Therapies of Epithelial Ovarian Cancer. Cancers, 14(1), 191. https://doi.org/10.3390/cancers14010191