
Targeted Disruption of E6/p53 Binding Exerts Broad Activity and Synergism with Paclitaxel and Topotecan against HPV-Transformed Cancer Cells

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Cell Lines
2.3. Cell Viability Assays
2.4. Two-Dimensional Clonogenic Assays
2.5. Tumor Spheroid Formation Assays
2.6. Western Blot Analysis
2.7. Gene Expression Analysis by Quantitative Real-Time PCR (qPCR)
2.8. Cytofluorimetric and Immunofluorescence Analysis for Cell-Cycle Phase Determination
2.9. In Situ Staining of Senescence-Associated β-Galactosidase Activity, Autofluorescence Analysis, and Detection of Apoptosis
2.10. Cell Migration Assays
2.11. Drug Combination Studies
2.12. Statistical Analysis
3. Results
3.1. Cpd12 Selectively Affects the Viability and Proliferation of HPV-Transformed Cells of Both Cervical and Head-and-Neck Origin and Rescues p53 Levels and Transcriptional Activity
3.2. The Cell-Cycle Arrest Induced by Cpd12 Occurs in the G1 Phase
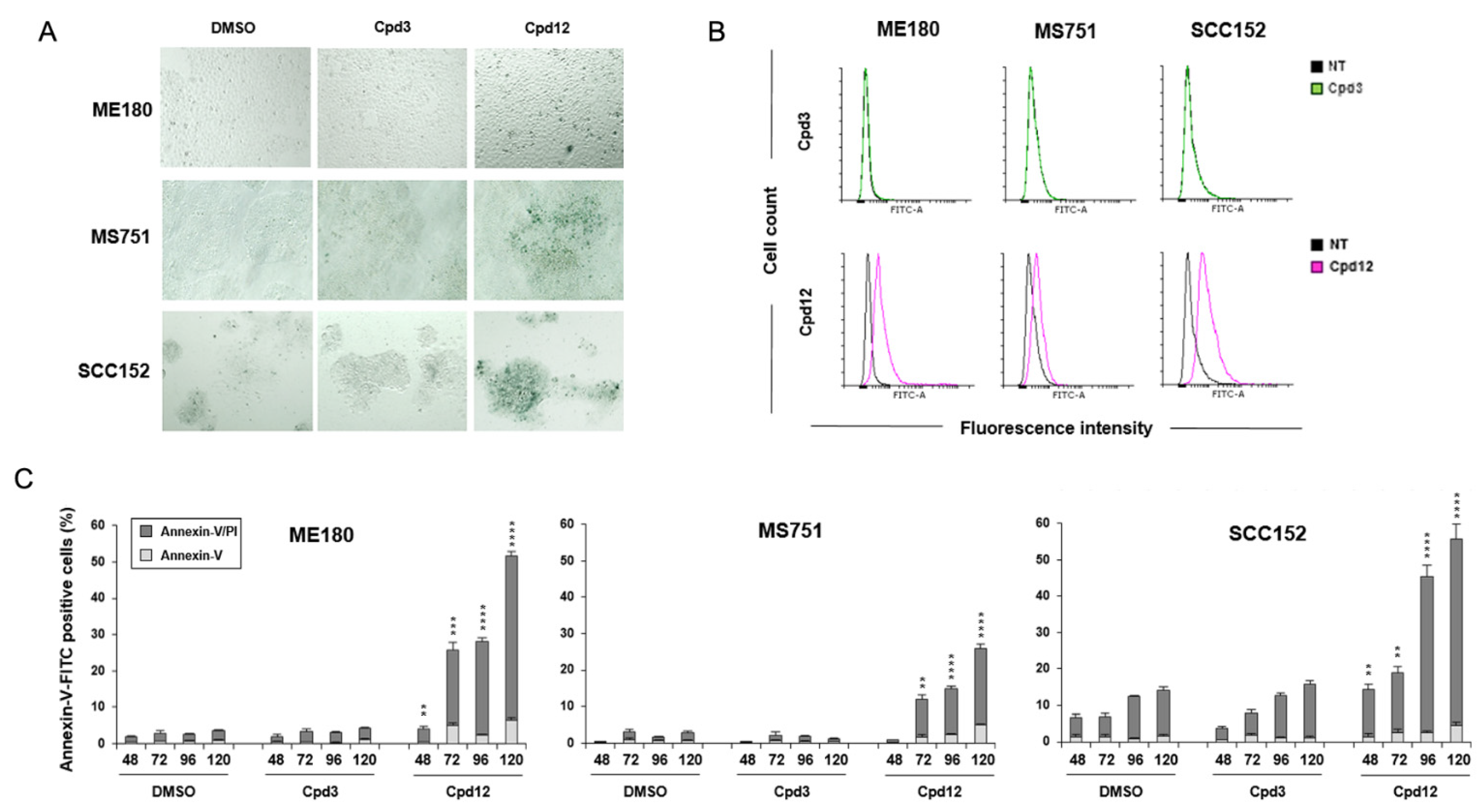
3.3. Cpd12 Treatment Induces Senescent and Apoptotic Responses in Both Cervical and Head-and-Neck Cancer Cells
3.4. The Cpd12-Induced Rescue of p53 Blocks the Migration of HPV-Transformed Cells
3.5. Cpd12 Enhances the Cytotoxic Effect of Clinically-Approved Anticancer Drugs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tommasino, M. The human papillomavirus family and its role in carcinogenesis. Semin. Cancer Biol. 2014, 26, 13–21. [Google Scholar] [CrossRef]
- de Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [Green Version]
- Tumban, E. A Current Update on Human Papillomavirus-Associated Head and Neck Cancers. Viruses 2019, 11, 922. [Google Scholar] [CrossRef] [Green Version]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Khan, M.J.; Castle, P.E.; Lorincz, A.T.; Wacholder, S.; Sherman, M.; Scott, D.R.; Rush, B.B.; Glass, A.G.; Schiffman, M. The elevated 10-year risk of cervical precancer and cancer in women with human papillomavirus (HPV) type 16 or 18 and the possible utility of type-specific HPV testing in clinical practice. J. Natl. Cancer Inst. 2005, 97, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Clifford, G.; Buonaguro, F.M. Classification of weakly carcinogenic human papillomavirus types: Addressing the limits of epidemiology at the borderline. Infect. Agents Cancer 2009, 4, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcaro, M.; Castanon, A.; Ndlela, B.; Checchi, M.; Soldan, K.; Lopez-Bernal, J.; Elliss-Brookes, L.; Sasieni, P. The effects of the national HPV vaccination programme in England, UK, on cervical cancer and grade 3 cervical intraepithelial neoplasia incidence: A register-based observational study. Lancet 2021, 398, 2084–2092. [Google Scholar] [CrossRef]
- Bruni, L.; Saura-Lazaro, A.; Montoliu, A.; Brotons, M.; Alemany, L.; Diallo, M.S.; Afsar, O.Z.; LaMontagne, D.S.; Mosina, L.; Contreras, M.; et al. HPV vaccination introduction worldwide and WHO and UNICEF estimates of national HPV immunization coverage 2010–2019. Prev. Med. 2021, 144, 106399. [Google Scholar] [CrossRef] [PubMed]
- Enerly, E.; Flingtorp, R.; Christiansen, I.K.; Campbell, S.; Hansen, M.; Myklebust, T.Å.; Weiderpass, E.; Nygård, M. An observational study comparing HPV prevalence and type distribution between HPV-vaccinated and -unvaccinated girls after introduction of school-based HPV vaccination in Norway. PLoS ONE 2019, 14, e0223612. [Google Scholar] [CrossRef]
- Lynge, E.; Thamsborg, L.; Larsen, L.G.; Christensen, J.; Johansen, T.; Hariri, J.; Christiansen, S.; Rygaard, C.; Andersen, B. Prevalence of high-risk human papillomavirus after HPV-vaccination in Denmark. Int. J. Cancer 2020, 147, 3446–3452. [Google Scholar] [CrossRef]
- Messa, L.; Loregian, A. HPV-induced cancers: Preclinical therapeutic advancements. Expert Opin. Investig. Drugs 2021. [Google Scholar] [CrossRef] [PubMed]
- Stern, P.L.; van der Burg, S.H.; Hampson, I.N.; Broker, T.R.; Fiander, A.; Lacey, C.J.; Kitchener, H.C.; Einstein, M.H. Therapy of human papillomavirus-related disease. Vaccine 2012, 30 (Suppl. 5), F71–F82. [Google Scholar] [CrossRef] [Green Version]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; de Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086. [Google Scholar] [CrossRef]
- Celegato, M.; Messa, L.; Goracci, L.; Mercorelli, B.; Bertagnin, C.; Spyrakis, F.; Suarez, I.; Cousido-Siah, A.; Travé, G.; Banks, L.; et al. A novel small-molecule inhibitor of the human papillomavirus E6-p53 interaction that reactivates p53 function and blocks cancer cells growth. Cancer Lett. 2020, 470, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Messa, L.; Celegato, M.; Bertagnin, C.; Mercorelli, B.; Nannetti, G.; Palù, G.; Loregian, A. A quantitative LumiFluo assay to test inhibitory compounds blocking p53 degradation induced by human papillomavirus oncoprotein E6 in living cells. Sci. Rep. 2018, 8, 6020. [Google Scholar] [CrossRef] [Green Version]
- Messa, L.; Celegato, M.; Bertagnin, C.; Mercorelli, B.; Alvisi, G.; Banks, L.; Palù, G.; Loregian, A. The Dimeric Form of HPV16 E6 Is Crucial to Drive YAP/TAZ Upregulation through the Targeting of hScrib. Cancers 2021, 13, 4083. [Google Scholar] [CrossRef]
- Miller, I.; Min, M.; Yang, C.; Tian, C.; Gookin, S.; Carter, D.; Spencer, S.L. Ki67 is a Graded Rather than a Binary Marker of Proliferation versus Quiescence. Cell Rep. 2018, 24, 1105–1112.e1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Bulk, S.; Berkhof, J.; Bulkmans, N.W.; Zielinski, G.D.; Rozendaal, L.; van Kemenade, F.J.; Snijders, P.J.F.; Meijer, C.J.L.M. Preferential risk of HPV16 for squamous cell carcinoma and of HPV18 for adenocarcinoma of the cervix compared to women with normal cytology in The Netherlands. Br. J. Cancer 2006, 94, 171–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.S.; Weissfeld, J.L.; Ragin, C.C.; Rossie, K.M.; Martin, C.L.; Shuster, M.; Ishwad, C.S.; Law, J.C.; Myers, E.N.; Johnson, J.T.; et al. The influence of clinical and demographic risk factors on the establishment of head and neck squamous cell carcinoma cell lines. Oral Oncol. 2007, 43, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.Y.; Szekely, L.; Bao, W.; Selivanova, G. Rescue of p53 function by small molecule RITA in cervical carcinoma by blocking E6-mediated degradation. Cancer Res. 2012, 70, 3372–3381. [Google Scholar] [CrossRef] [Green Version]
- Wanzel, M.; Vischedyk, J.B.; Gittler, M.P.; Gremke, N.; Seiz, J.R.; Hefter, M.; Noack, M.; Savai, R.; Mernberger, M.; Charles, J.P.; et al. CRISPR-Cas9-based target validation for p53-reactivating model compounds. Nat. Chem. Biol. 2016, 12, 22–28. [Google Scholar] [CrossRef] [Green Version]
- de Magalhaes, J.P.; Passos, J.F. Stress, cell senescence and organismal ageing. Mech. Ageing Dev. 2018, 170, 2–9. [Google Scholar] [CrossRef]
- Lawless, C.; Wang, C.; Jurk, D.; Merz, A.; von Zglinicki, T.; Passos, J.F. Quantitative assessment of markers for cell senescence. Exp. Gerontol. 2010, 45, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Galvis, D.; Walsh, D.; Harries, L.W.; Latorre, E.; Rankin, J. A dynamical systems model for the measurement of cellular senescence. J. R. Soc. Interface 2019, 16, 20190311. [Google Scholar] [CrossRef]
- Bertolo, A.; Baur, M.; Guerrero, J.; Pötzel, T.; Stoyanov, J. Autofluorescence is a Reliable in vitro Marker of Cellular Senescence in Human Mesenchymal Stromal Cells. Sci. Rep. 2019, 9, 2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budczies, J.; von Winterfeld, M.; Klauschen, F.; Bockmayr, M.; Lennerz, J.K.; Denkert, C.; Wolf, T.; Warth, A.; Dietel, M.; Anagnostopoulos, I.; et al. The landscape of metastatic progression patterns across major human cancers. Oncotarget 2015, 6, 570–583. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef]
- Xu, J.; Wang, H.; Wang, H.; Chen, Q.; Zhang, L.; Song, C.; Zhou, Q.; Hong, Y. The inhibition of miR-126 in cell migration and invasion of cervical cancer through regulating ZEB1. Hereditas 2019, 156, 11. [Google Scholar] [CrossRef] [Green Version]
- American National Cancer Institute. Available online: www.cancer.gov/about-cancer/treatment/drugs/cervical (accessed on 17 November 2021).
- American National Cancer Institute. Available online: www.cancer.gov/about-cancer/treatment/drugs/tpf (accessed on 17 November 2021).
- Markowitz, L.E.; Hariri, S.; Lin, C.; Dunne, E.F.; Steinau, M.; McQuillan, G.; Unger, E.R. Reduction in human papillomavirus (HPV) prevalence among young women following HPV vaccine introduction in the United States, National Health and Nutrition Examination Surveys, 2003–2010. J. Infect. Dis. 2013, 208, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, E.M.; Ravicz, J.R.; Liu, S.; Chawla, S.P.; Hall, F.L. Cell cycle checkpoint control: The cyclin G1/Mdm2/p53 axis emerges as a strategic target for broad-spectrum cancer gene therapy—A review of molecular mechanisms for oncologists. Mol. Clin. Oncol. 2018, 9, 115–134. [Google Scholar] [CrossRef] [Green Version]
- Kho, E.Y.; Wang, H.K.; Banerjee, N.S.; Broker, T.R.; Chow, L.T. HPV-18 E6 mutants reveal p53 modulation of viral DNA amplification in organotypic cultures. Proc. Natl. Acad. Sci. USA 2013, 110, 7542–7549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.T.; Hubert, W.G.; Ruesch, M.N.; Laimins, L.A. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 8449–8454. [Google Scholar] [CrossRef] [Green Version]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012, 30, 679–692. [Google Scholar] [CrossRef]
- Lanni, J.S.; Lowe, S.W.; Licitra, E.J.; Liu, J.O.; Jacks, T. p53-independent apoptosis induced by paclitaxel through an indirect mechanism. Proc. Natl. Acad. Sci. USA 1997, 94, 9679–9683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, A.F.; Donaldson, K.L.; Fairchild, C.; Lee, F.Y.; Foster, S.A.; Demers, G.W.; Galloway, D.A. Loss of normal p53 function confers sensitization to Taxol by increasing G2/M arrest and apoptosis. Nat. Med. 1996, 2, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Feeney, G.P.; Errington, R.J.; Wiltshire, M.; Marquez, N.; Chappell, S.C.; Smith, P.J. Tracking the cell cycle origins for escape from topotecan action by breast cancer cells. Br. J. Cancer 2003, 88, 1310–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, A.C.; Brown, R. Induction of p53-dependent and p53-independent cellular responses by topoisomerase 1 inhibitors. Br. J. Cancer 1998, 78, 745–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taron, M.; Plasencia, C.; Abad, A.; Martin, C.; Guillot, M. Cytotoxic effects of topotecan combined with various active G2/M-phase anticancer drugs in human tumor-derived cell lines. Investig. New Drugs 2000, 18, 139–147. [Google Scholar] [CrossRef]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Combination Index (CI) a | ||||
|---|---|---|---|---|
| Drug Combination at Equipotent Ratio (Fold of IC50 b) | Cisplatin + Cpd12 | Paclitaxel + Cpd12 | Topotecan + Cpd12 | |
| HeLa | 0.125× | 0.457 ± 0.091 | 0.196 ± 0.095 | 0.494 ± 0.084 |
| 0.25× | 0.797 ± 0.103 | 0.329 ± 0.104 | 0.520 ± 0.169 | |
| 0.5× | 0.677 ± 0.116 | 0.355 ± 0.085 | 0.442 ± 0.102 | |
| 1× | 0.901 ± 0.117 | 0.482 ± 0.107 | 0.663 ± 0.134 | |
| CaSki | 0.125× | 0.895 ± 0.271 | 0.104 ± 0.060 | 0.348 ± 0.057 |
| 0.25× | 0.705 ± 0.087 | 0.180 ± 0.079 | 0.398 ± 0.102 | |
| 0.5× | 0.577 ± 0.125 | 0.268 ± 0.108 | 0.386 ± 0.021 | |
| 1× | 0.914 ± 0.162 | 0.516 ± 0.187 | 0.624 ± 0.026 | |
| MS751 | 0.125× | 0.832 ± 0.092 | 0.161 ± 0.072 | 0.649 ± 0.221 |
| 0.25× | 0.930 ± 0.035 | 0.197 ± 0.037 | 0.785 ± 0.071 | |
| 0.5× | 0.814 ± 0.219 | 0.274 ± 0.052 | 0.472 ± 0.165 | |
| 1× | 0.653 ± 0.103 | 0.279 ± 0.132 | 0.372 ± 0.136 | |
| Cisplatin + Cpd12 | Paclitaxel + Cpd12 | 5-Fluorouracil + Cpd12 | ||
| SCC152 | 0.125× | 0.484 ± 0.124 | 0.135 ± 0.021 | 0.636 ± 0.074 |
| 0.25× | 0.598 ± 0.097 | 0.242 ± 0.035 | 0.635 ± 0.081 | |
| 0.5× | 0.796 ± 0.235 | 0.438 ± 0.026 | 0.643 ± 0.037 | |
| 1× | 0.691 ± 0.179 | 0.355 ± 0.051 | 0.431 ± 0.062 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Celegato, M.; Messa, L.; Bertagnin, C.; Mercorelli, B.; Loregian, A. Targeted Disruption of E6/p53 Binding Exerts Broad Activity and Synergism with Paclitaxel and Topotecan against HPV-Transformed Cancer Cells. Cancers 2022, 14, 193. https://doi.org/10.3390/cancers14010193
Celegato M, Messa L, Bertagnin C, Mercorelli B, Loregian A. Targeted Disruption of E6/p53 Binding Exerts Broad Activity and Synergism with Paclitaxel and Topotecan against HPV-Transformed Cancer Cells. Cancers. 2022; 14(1):193. https://doi.org/10.3390/cancers14010193
Chicago/Turabian StyleCelegato, Marta, Lorenzo Messa, Chiara Bertagnin, Beatrice Mercorelli, and Arianna Loregian. 2022. "Targeted Disruption of E6/p53 Binding Exerts Broad Activity and Synergism with Paclitaxel and Topotecan against HPV-Transformed Cancer Cells" Cancers 14, no. 1: 193. https://doi.org/10.3390/cancers14010193
APA StyleCelegato, M., Messa, L., Bertagnin, C., Mercorelli, B., & Loregian, A. (2022). Targeted Disruption of E6/p53 Binding Exerts Broad Activity and Synergism with Paclitaxel and Topotecan against HPV-Transformed Cancer Cells. Cancers, 14(1), 193. https://doi.org/10.3390/cancers14010193