
Glutamine-Derived Aspartate Biosynthesis in Cancer Cells: Role of Mitochondrial Transporters and New Therapeutic Perspectives

 ,
,  , , and
, , and 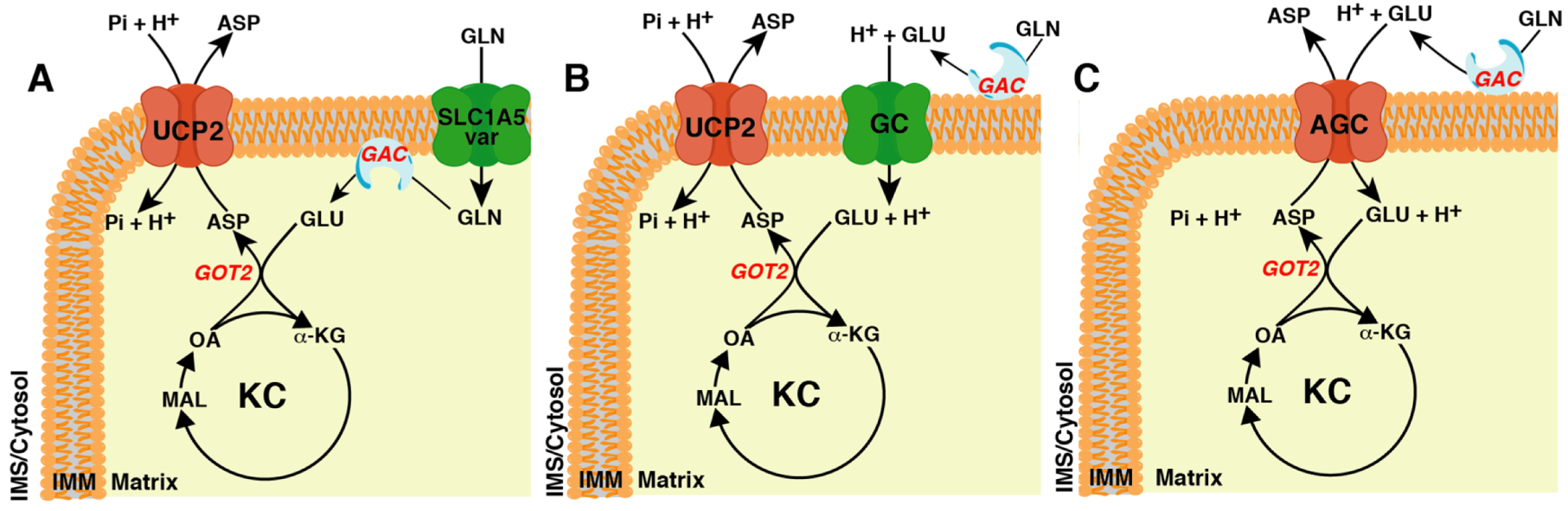{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Glutaminase: A Multifaceted Enzyme in Cancer Cell Metabolism
3. The Mitochondrial Glutamine Carrier: The Last Piece of Glutaminolysis Puzzle
4. The Mitochondrial Glutamate Carrier: The Other Side of the Coin
5. The Mitochondrial Aspartate/Glutamate Carrier: A Key Player of Malate-Aspartate Shuttle
6. Mitochondrial Uncoupling Protein 2: An Aspartate/Pi + H+ Exchanger Belonging to the Mitochondrial Carrier Family
7. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Flier, J.S.; Mueckler, M.M.; Usher, P.; Lodish, H.F. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science 1987, 235, 1492–1495. [Google Scholar] [CrossRef]
- Matoba, S.; Kang, J.-G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 Regulates Mitochondrial Respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor Cell Metabolism: Cancer’s Achilles’ Heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Sayed, N.; Ditsworth, D.; Thompson, C.B. Brick by brick: Metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 2008, 18, 54–61. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.; Wang, Q.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2021, 481, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Fendt, S.-M.; Bell, E.L.; Keibler, M.A.; Olenchock, B.A.; Mayers, J.; Wasylenko, T.M.; Vokes, N.I.; Guarente, L.; Heiden, M.G.V.; Stephanopoulos, G. Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat. Commun. 2013, 4, 2236. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Raho, S.; Capobianco, L.; Malivindi, R.; Vozza, A.; Piazzolla, C.; de Leonardis, F.; Gorgoglione, R.; Scarcia, P.; Pezzuto, F.; Agrimi, G.; et al. KRAS-regulated glutamine metabolism requires UCP2-mediated aspartate transport to support pancreatic cancer growth. Nat. Metab. 2020, 2, 1373–1381. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Gui, D.Y.; Sullivan, L.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Heiden, M.G.V. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016, 24, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 2018, 20, 775–781. [Google Scholar] [CrossRef]
- Kvamme, E.; Tveit, B.; Svenneby, G. Glutaminase from Pig Renal Cortex. J. Biol. Chem. 1970, 245, 1871–1877. [Google Scholar] [CrossRef]
- Elgadi, K.M.; Meguid, R.; Qian, M.; Souba, W.W.; Abcouwer, S.F. Cloning and analysis of unique human glutaminase isoforms generated by tissue-specific alternative splicing. Physiol. Genom. 1999, 1, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Errera, M.; Greenstein, J.P. Phosphate-activated glutaminase in kidney and other tissues. J. Biol. Chem. 1949, 178, 495–502. [Google Scholar] [CrossRef]
- Aoki, C.; Kaneko, T.; Starr, A.; Pickel, V.M. Identification of mitochondrial and non-mitochondrial glutaminase within select neurons and glia of rat forebrain by electron microscopic immunocytochemistry. J. Neurosci. Res. 1991, 28, 531–548. [Google Scholar] [CrossRef]
- Cassago, A.; Ferreira, A.P.S.; Ferreira, I.M.; Fornezari, C.; Gomes, E.R.M.; Greene, K.S.; Pereira, H.M.; Garratt, R.C.; Dias, S.M.G.; Ambrosio, A.L.B. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 1092–1097. [Google Scholar] [CrossRef] [Green Version]
- Kvamme, E.; Torgner, I.; Roberg, B. Evidence indicating that pig renal phosphate-activated glutaminase has a functionally predominant external localization in the inner mitochondrial membrane. J. Biol. Chem. 1991, 266, 13185–13192. [Google Scholar] [CrossRef]
- Roberg, B. The orientation of phosphate activated glutaminase in the inner mitochondrial membrane of synaptic and non-synaptic rat brain mitochondria. Neurochem. Int. 1995, 27, 367–376. [Google Scholar] [CrossRef]
- Shapiro, R.A.; Haser, W.G.; Curthoys, N.P. The orientation of phosphate-dependent glutaminase on the inner membrane of rat renal mitochondria. Arch. Biochem. Biophys. 1985, 243, 1–7. [Google Scholar] [CrossRef]
- Aledo, J.C.; de Pedro, E.; Gómez-Fabre, P.M.; de Castro, I.N.; Márquez, J. Submitochondrial localization and membrane topography of Ehrlich ascitic tumour cell glutaminase. Biochim. Biophys. Acta (BBA)-Biomembr. 1997, 1323, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Fiermonte, G.; Palmieri, L.; Todisco, S.; Agrimi, G.; Palmieri, F.; Walker, J. Identification of the Mitochondrial Glutamate Transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J. Biol. Chem. 2002, 277, 19289–19294. [Google Scholar] [CrossRef] [Green Version]
- Lunetti, P.; Cappello, A.R.; Marsano, R.M.; Pierri, C.L.; Carrisi, C.; Martello, E.; Caggese, C.; Dolce, V.; Capobianco, L. Mitochondrial glutamate carriers from Drosophila melanogaster: Biochemical, evolutionary and modeling studies. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 1245–1255. [Google Scholar] [CrossRef] [Green Version]
- Porcelli, V.; Vozza, A.; Calcagnile, V.; Gorgoglione, R.; Arrigoni, R.; Fontanesi, F.; Marobbio, C.M.; Castegna, A.; Palmieri, F.; Palmieri, L. Molecular identification and functional characterization of a novel glutamate transporter in yeast and plant mitochondria. Biochim. Biophys. Acta 2018, 1859, 1249–1258. [Google Scholar] [CrossRef]
- Aledo, J.C.; Gómez-Fabre, P.M.; Olalla, L.; Márquez, J. Identification of two human glutaminase loci and tissue-specific expression of the two related genes. Mamm. Genome 2000, 11, 1107–1110. [Google Scholar] [CrossRef]
- Katt, W.P.; Lukey, M.J.; Cerione, R.A. A tale of two glutaminases: Homologous enzymes with distinct roles in tumorigenesis. Future Med. Chem. 2017, 9, 223–243. [Google Scholar] [CrossRef] [Green Version]
- Szeliga, M.; Obara-Michlewska, M. Glutamine in neoplastic cells: Focus on the expression and roles of glutaminases. Neurochem. Int. 2009, 55, 71–75. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; de Marzo, A.M.; van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [Green Version]
- Martín-Rufián, M.; Nascimento-Gomes, R.; Higuero, A.; Crisma, A.R.; Campos-Sandoval, J.A.; Gomez, F.J.M.; Cardona, C.; Cheng, T.; Lobo, C.; Segura, J.A.; et al. Both GLS silencing and GLS2 overexpression synergize with oxidative stress against proliferation of glioma cells. J. Mol. Med. 2013, 92, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Majewska, E.; Márquez, J.; Albrecht, J.; Szeliga, M. Transfection with GLS2 Glutaminase (GAB) Sensitizes Human Glioblastoma Cell Lines to Oxidative Stress by a Common Mechanism Involving Suppression of the PI3K/AKT Pathway. Cancers 2019, 11, 115. [Google Scholar] [CrossRef] [Green Version]
- Lukey, M.; Cluntun, A.; Katt, W.P.; Lin, M.-C.J.; Druso, J.E.; Ramachandran, S.; Erickson, J.W.; Le, H.; Wang, Z.-E.; Blank, B.; et al. Liver-Type Glutaminase GLS2 Is a Druggable Metabolic Node in Luminal-Subtype Breast Cancer. Cell Rep. 2019, 29, 76–88.e7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Liu, J.; Zhao, Y.; Yue, X.; Zhu, Y.; Wang, X.; Wu, H.; Blanco, F.; Li, S.; Bhanot, G.; et al. Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. eLife 2016, 5, e10727. [Google Scholar] [CrossRef]
- Katunuma, N.; Huzino, A.; Tomino, I. Organ specific control of glutamine metabolism. Adv. Enzym. Regul. 1967, 5, 55–58. [Google Scholar] [CrossRef]
- Kovacevic, Z. Properties and intracellular localization of Ehrlich ascites tumor cell glutaminase. Cancer Res. 1974, 34, 3403–3407. [Google Scholar]
- Roberg, B.; Torgner, I.A.; Laake, J.; Takumi, Y.; Ottersen, O.P.; Kvamme, E. Properties and submitochondrial localization of pig and rat renal phosphate-activated glutaminase. Am. J. Physiol. Physiol. 2000, 279, C648–C657. [Google Scholar] [CrossRef]
- Crompton, M.; Mcgivan, J.D.; Chappell, J.B. The intramitochondrial location of the glutaminase isoenzymes of pig kidney. Biochem. J. 1973, 132, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Laake, J.; Takumi, Y.; Eidet, J.; Torgner, I.; Roberg, B.; Kvamme, E.; Ottersen, O. Postembedding immunogold labelling reveals subcellular localization and pathway-specific enrichment of phosphate activated glutaminase in rat cerebellum. Neuroscience 1999, 88, 1137–1151. [Google Scholar] [CrossRef]
- Kvamme, E.; Roberg, B.; Torgner, I.A. Phosphate-activated glutaminase and mitochondrial glutamine transport in the brain. Neurochem. Res. 2000, 25, 1407–1419. [Google Scholar] [CrossRef]
- Bak, L.K.; Zieminska, E.; Waagepetersen, H.S.; Schousboe, A.; Albrecht, J. Metabolism of [U-13C] Glutamine and [U-13C] Glutamate in Isolated Rat Brain Mitochondria Suggests Functional Phosphate-Activated Glutaminase Activity in Matrix. Neurochem. Res. 2008, 33, 273–278. [Google Scholar] [CrossRef]
- Li, B.; Cao, Y.; Meng, G.; Qian, L.; Xu, T.; Yan, C.; Luo, O.; Wang, S.; Wei, J.; Ding, Y.; et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine 2019, 39, 239–254. [Google Scholar] [CrossRef] [Green Version]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.-K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2019, 31, 267–283.e12. [Google Scholar] [CrossRef]
- Kekuda, R.; Prasad, P.D.; Fei, Y.-J.; Torres-Zamorano, V.; Sinha, S.; Yang-Feng, T.L.; Leibach, F.H.; Ganapathy, V. Cloning of the Sodium-dependent, Broad-scope, Neutral Amino Acid Transporter Bo from a Human Placental Choriocarcinoma Cell Line. J. Biol. Chem. 1996, 271, 18657–18661. [Google Scholar] [CrossRef] [Green Version]
- Gyimesi, G.; Hediger, M. Sequence Features of Mitochondrial Transporter Protein Families. Biomolecules 2020, 10, 1611. [Google Scholar] [CrossRef]
- Bricker, D.K.; Taylor, E.B.; Schell, J.C.; Orsak, T.; Boutron, A.; Chen, Y.-C.; Cox, J.E.; Cardon, C.M.; Van Vranken, J.G.; Dephoure, N.; et al. A Mitochondrial Pyruvate Carrier Required for Pyruvate Uptake in Yeast, Drosophila, and Humans. Science 2012, 337, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Raemy, E.; Montessuit, S.; Veuthey, J.-L.; Zamboni, N.; Westermann, B.; Kunji, E.R.S.; Martinou, J.-C. Identification and Functional Expression of the Mitochondrial Pyruvate Carrier. Science 2012, 337, 93–96. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Asp. Med. 2013, 34, 465–484. [Google Scholar] [CrossRef]
- Curcio, R.; Lunetti, P.; Zara, V.; Ferramosca, A.; Marra, F.; Fiermonte, G.; Cappello, A.R.; de Leonardis, F.; Capobianco, L.; Dolce, V. Drosophila melanogaster Mitochondrial Carriers: Similarities and Differences with the Human Carriers. Int. J. Mol. Sci. 2020, 21, 6052. [Google Scholar] [CrossRef]
- Vozza, A.; de Leonardis, F.; Paradies, E.; de Grassi, A.; Pierri, C.L.; Parisi, G.; Marobbio, C.M.T.; Lasorsa, F.M.; Muto, L.; Capobianco, L.; et al. Biochemical characterization of a new mitochondrial transporter of dephosphocoenzyme A in Drosophila melanogaster. Biochim. Biophys. Acta (BBA)-Bioenerg. 2017, 1858, 137–146. [Google Scholar] [CrossRef]
- Cormerais, Y.; Massard, P.A.; Vucetic, M.; Giuliano, S.; Tambutté, E.; Durivault, J.; Vial, V.; Endou, H.; Wempe, M.F.; Parks, S.K.; et al. The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J. Biol. Chem. 2018, 293, 2877–2887. [Google Scholar] [CrossRef] [Green Version]
- Scalise, M.; Pochini, L.; Console, L.; Losso, M.A.; Indiveri, C. The Human SLC1A5 (ASCT2) Amino Acid Transporter: From Function to Structure and Role in Cell Biology. Front. Cell Dev. Biol. 2018, 6, 96. [Google Scholar] [CrossRef]
- Zhang, H.; Cui, K.; Yao, S.; Yin, Y.; Liu, D.; Huang, Z. Comprehensive molecular and clinical characterization of SLC1A5 in human cancers. Pathol.-Res. Pr. 2021, 224, 153525. [Google Scholar] [CrossRef]
- Csanadi, A.; Oser, A.; Aumann, K.; Gumpp, V.; Rawluk, J.; Nestle, U.; Kayser, C.; Wiesemann, S.; Werner, M.; Kayser, G. Overexpression of SLC1a5 in lymph node metastases outperforms assessment in the primary as a negative prognosticator in non-small cell lung cancer. Pathology 2018, 50, 269–275. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, Z.; Tu, M.; Meng, W.; Gao, H.; Li, M.D.; Li, L. Correlation between Prognostic Biomarker SLC1A5 and Immune Infiltrates in Various Types of Cancers Including Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 608641. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, R.; Shuai, Y.; Huang, Y.; Jin, R.; Wang, X.; Luo, J. ASCT2 (SLC1A5)-dependent glutamine uptake is involved in the progression of head and neck squamous cell carcinoma. Br. J. Cancer 2020, 122, 82–93. [Google Scholar] [CrossRef]
- Ma, H.; Wu, Z.; Peng, J.; Li, Y.; Huang, H.; Liao, Y.; Zhou, M.; Sun, L.; Huang, N.; Shi, M.; et al. Inhibition of SLC1A5 sensitizes colorectal cancer to cetuximab. Int. J. Cancer 2018, 142, 2578–2588. [Google Scholar] [CrossRef] [Green Version]
- Pozza, E.D.; Fiorini, C.; Dando, I.; Menegazzi, M.; Sgarbossa, A.; Costanzo, C.; Palmieri, M.; Donadelli, M. Role of mitochondrial uncoupling protein 2 in cancer cell resistance to gemcitabine. Biochim. Biophys. Acta (BBA)-Bioenerg. 2012, 1823, 1856–1863. [Google Scholar] [CrossRef] [Green Version]
- Donadelli, M.; Dando, I.; Fiorini, C.; Palmieri, M. UCP2, a mitochondrial protein regulated at multiple levels. Cell. Mol. Life Sci. 2014, 71, 1171–1190. [Google Scholar] [CrossRef]
- Wong, C.C.; Qian, Y.; Li, X.; Xu, J.; Kang, W.; Tong, J.H.; To, K.-F.; Jin, Y.; Li, W.; Chen, H.; et al. SLC25A22 Promotes Proliferation and Survival of Colorectal Cancer Cells with KRAS Mutations and Xenograft Tumor Progression in Mice via Intracellular Synthesis of Aspartate. Gastroenterology 2016, 151, 945–960.e6. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chung, A.C.; Li, S.; Wu, L.; Xu, J.; Yu, J.; Wong, C.; Cai, Z. LC-MS-based metabolomics revealed SLC25A22 as an essential regulator of aspartate-derived amino acids and polyamines in KRAS-mutant colorectal cancer. Oncotarget 2017, 8, 101333–101344. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Chen, Y.; Yu, Y.; Pan, W.; Cui, Y.; Xu, X.; Peng, K.; Liu, M.; Rashid, K.; Hou, Y.; et al. SLC25A18 has prognostic value in colorectal cancer and represses Warburg effect and cell proliferation via Wnt signaling. Am. J. Cancer Res. 2020, 10, 1548–1567. [Google Scholar]
- Chen, M.-W.; Wu, X.-J. SLC25A22 Promotes Proliferation and Metastasis of Osteosarcoma Cells via the PTEN Signaling Pathway. Technol. Cancer Res. Treat. 2018, 17, 1533033818811143. [Google Scholar] [CrossRef]
- Du, P.; Liang, H.; Fu, X.; Wu, P.; Wang, C.; Chen, H.; Zheng, B.; Zhang, J.; Hu, S.; Zeng, R.; et al. SLC25A22 promotes proliferation and metastasis by activating MAPK/ERK pathway in gallbladder cancer. Cancer Cell Int. 2019, 19, 33. [Google Scholar] [CrossRef]
- Palmieri, L.; Pardo, B.; Lasorsa, F.; del Arco, A.; Kobayashi, K.; Iijima, M.; Runswick, M.; Walker, J.; Saheki, T.; Satrústegui, J. Citrin and aralar1 are Ca2+-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001, 20, 5060–5069. [Google Scholar] [CrossRef]
- del Arco, A.; Satrústegui, J. Molecular Cloning of Aralar, a New Member of the Mitochondrial Carrier Superfamily That Binds Calcium and Is Present in Human Muscle and Brain. J. Biol. Chem. 1998, 273, 23327–23334. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Sinasac, D.; Iijima, M.; Boright, A.P.; Begum, L.; Lee, J.R.; Yasuda, T.; Ikeda, S.; Hirano, R.; Terazono, H.; et al. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat. Genet. 1999, 22, 159–163. [Google Scholar] [CrossRef]
- Lunetti, P.; Marsano, R.M.; Curcio, R.; Dolce, V.; Fiermonte, G.; Cappello, A.R.; Marra, F.; Moschetti, R.; Li, Y.; Aiello, D.; et al. The mitochondrial aspartate/glutamate carrier (AGC or Aralar1) isoforms in D. melanogaster: Biochemical characterization, gene structure, and evolutionary analysis. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2021, 1865, 129854. [Google Scholar] [CrossRef]
- Fiermonte, G.; Walker, J.; Palmieri, F. Abundant bacterial expression and reconstitution of an intrinsic membrane-transport protein from bovine mitochondria. Biochem. J. 1993, 294, 293–299. [Google Scholar] [CrossRef]
- Monné, M.; Miniero, D.V.; Bisaccia, F.; Fiermonte, G. The mitochondrial oxoglutarate carrier: From identification to mechanism. J. Bioenerg. Biomembr. 2012, 45, 1–13. [Google Scholar] [CrossRef]
- LaNoue, K.F.; Tischler, M.E. Electrogenic Characteristics of the Mitochondrial Glutamate-Aspartate Antiporter. J. Biol. Chem. 1974, 249, 7522–7528. [Google Scholar] [CrossRef]
- Lanoue, K.F.; Meijer, A.J.; Brouwer, A. Evidence for electrogenic aspartate transport in rat liver mitochondria. Arch. Biochem. Biophys. 1974, 161, 544–550. [Google Scholar] [CrossRef]
- Lasorsa, F.; Pinton, P.; Palmieri, L.; Fiermonte, G.; Rizzuto, R.; Palmieri, F. Recombinant Expression of the Ca2+-sensitive Aspartate/Glutamate Carrier Increases Mitochondrial ATP Production in Agonist-stimulated Chinese Hamster Ovary Cells. J. Biol. Chem. 2003, 278, 38686–38692. [Google Scholar] [CrossRef] [Green Version]
- Profilo, E.; Peña-Altamira, L.E.; Corricelli, M.; Castegna, A.; Danese, A.; Agrimi, G.; Petralla, S.; Giannuzzi, G.; Porcelli, V.; Sbano, L.; et al. Down-regulation of the mitochondrial aspartate-glutamate carrier isoform 1 AGC1 inhibits proliferation and N-acetylaspartate synthesis in Neuro2A cells. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1422–1435. [Google Scholar] [CrossRef]
- Cavero, S.; Vozza, A.; del Arco, A.; Palmieri, L.; Villa, A.; Blanco, E.; Runswick, M.J.; Walker, J.; Cerdan, S.; Satrústegui, J. Identification and metabolic role of the mitochondrial aspartate-glutamate transporter in Saccharomyces cerevisiae. Mol. Microbiol. 2003, 50, 1257–1269. [Google Scholar] [CrossRef]
- Saheki, T.; Iijima, M.; Li, M.X.; Kobayashi, K.; Horiuchi, M.; Ushikai, M.; Okumura, F.; Meng, X.J.; Inoue, I.; Tajima, A.; et al. Citrin/Mitochondrial Glycerol-3-phosphate Dehydrogenase Double Knock-out Mice Recapitulate Features of Human Citrin Deficiency. J. Biol. Chem. 2007, 282, 25041–25052. [Google Scholar] [CrossRef] [Green Version]
- Fiermonte, G.; Soon, D.; Chaudhuri, A.; Paradies, E.; Lee, P.J.; Krywawych, S.; Palmieri, F.; Lachmann, R.H. An Adult with Type 2 Citrullinemia Presenting in Europe. N. Engl. J. Med. 2008, 358, 1408–1409. [Google Scholar] [CrossRef] [Green Version]
- Dimmock, D.; Maranda, B.; Dionisi-Vici, C.; Wang, J.; Kleppe, S.; Fiermonte, G.; Bai, R.; Hainline, B.; Hamosh, A.; O’Brien, W.E.; et al. Citrin deficiency, a perplexing global disorder. Mol. Genet. Metab. 2009, 96, 44–49. [Google Scholar] [CrossRef]
- Wibom, R.; Lasorsa, F.; Töhönen, V.; Barbaro, M.; Sterky, F.H.; Kucinski, T.; Naess, K.; Jonsson, M.; Pierri, C.L.; Palmieri, F.; et al. AGC1 Deficiency Associated with Global Cerebral Hypomyelination. N. Engl. J. Med. 2009, 361, 489–495. [Google Scholar] [CrossRef] [Green Version]
- Alkan, H.F.; Walter, K.E.; Luengo, A.; Madreiter-Sokolowski, C.T.; Stryeck, S.; Lau, A.N.; Al-Zoughbi, W.; Lewis, C.A.; Thomas, C.J.; Hoefler, G.; et al. Cytosolic Aspartate Availability Determines Cell Survival When Glutamine Is Limiting. Cell Metab. 2018, 28, 706–720.e6. [Google Scholar] [CrossRef] [Green Version]
- Alkan, H.F.; Bogner-Strauss, J.G. Maintaining cytosolic aspartate levels is a major function of the TCA cycle in proliferating cells. Mol. Cell. Oncol. 2019, 6, e1536843. [Google Scholar] [CrossRef] [Green Version]
- Vozza, A.; Parisi, G.; de Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef] [Green Version]
- Arsenijevic, D.; Onuma, H.; Pecqueur, C.; Raimbault, S.; Manning, B.S.; Miroux, B.; Couplan, E.; Alves-Guerra, M.-C.; Goubern, M.; Surwit, R.; et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 2000, 26, 435–439. [Google Scholar] [CrossRef]
- Pecqueur, C.; Alves-Guerra, M.-C.; Gelly, C.; Lévi-Meyrueis, C.; Couplan, E.; Collins, S.; Ricquier, D.; Bouillaud, F.; Miroux, B. Uncoupling Protein 2, In Vivo Distribution, Induction upon Oxidative Stress, and Evidence for Translational Regulation. J. Biol. Chem. 2001, 276, 8705–8712. [Google Scholar] [CrossRef] [Green Version]
- Baffy, G. Uncoupling protein-2 and cancer. Mitochondrion 2010, 10, 243–252. [Google Scholar] [CrossRef]
- Fleury, C.; Neverova, M.; Collins, S.; Raimbault, S.; Champigny, O.; Levi-Meyrueis, C.; Bouillaud, F.; Seldin, M.F.; Surwit, R.S.; Ricquier, D.; et al. Uncoupling protein-2: A novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 1997, 15, 269–272. [Google Scholar] [CrossRef]
- Nübel, T.; Emre, Y.; Rabier, D.; Chadefaux, B.; Ricquier, D.; Bouillaud, F. Modified glutamine catabolism in macrophages of Ucp2 knock-out mice. Biochim. Biophys. Acta (BBA)-Bioenerg. 2008, 1777, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Owen, O.E.; Kalhan, S.; Hanson, R.W. The Key Role of Anaplerosis and Cataplerosis for Citric Acid Cycle Function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F. Mitochondrial transporters of the SLC25 family and associated diseases: A review. J. Inherit. Metab. Dis. 2014, 37, 565–575. [Google Scholar] [CrossRef]
- Sayeed, A.; Meng, Z.; Luciani, G.; Chen, L.-C.; Bennington, J.L.; Dairkee, S.H. Negative regulation of UCP2 by TGFβ signaling characterizes low and intermediate-grade primary breast cancer. Cell Death Dis. 2010, 1, e53. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Bradshaw, A.-D.; Gera, S.; Dewan, M.Z.; Xu, R. The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J. Clin. Med. 2017, 6, 5. [Google Scholar] [CrossRef]
- Rupprecht, A.; Moldzio, R.; Mödl, B.; Pohl, E.E. Glutamine regulates mitochondrial uncoupling protein 2 to promote glutaminolysis in neuroblastoma cells. Biochim. Biophys. Acta (BBA)-Bioenerg. 2019, 1860, 391–401. [Google Scholar] [CrossRef]
- Azzu, V.; Affourtit, C.; Breen, E.P.; Parker, N.; Brand, M.D. Dynamic regulation of uncoupling protein 2 content in INS-1E insulinoma cells. Biochim. Biophys. Acta (BBA)-Bioenerg. 2008, 1777, 1378–1383. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, K.; Chen, J.; Guo, J.; Yin, Y.; Cai, X.; Guo, X.; Wang, G.; Yang, R.; Zhu, L.; et al. In Vitro Evidence Suggests That miR-133a-mediated Regulation of Uncoupling Protein 2 (UCP2) Is an Indispensable Step in Myogenic Differentiation. J. Biol. Chem. 2009, 284, 5362–5369. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.-L.; Jiang, B.-G.; Li, W.-T.; Zou, J.-J.; Shi, Y.-Q.; Liu, Z.-M. MicroRNA-15a positively regulates insulin synthesis by inhibiting uncoupling protein-2 expression. Diabetes Res. Clin. Pr. 2011, 91, 94–100. [Google Scholar] [CrossRef]
- Wang, H.; An, H.; Wang, B.; Liao, Q.; Li, W.; Jin, X.; Cui, S.; Zhang, Y.; Ding, Y.; Zhao, L. miR-133a represses tumour growth and metastasis in colorectal cancer by targeting LIM and SH3 protein 1 and inhibiting the MAPK pathway. Eur. J. Cancer 2013, 49, 3924–3935. [Google Scholar] [CrossRef]
- Dong, Y.; Zhao, J.; Wu, C.-W.; Zhang, L.; Liu, X.; Kang, W.; Leung, W.-W.; Zhang, N.; Chan, F.K.; Sung, J.J.Y.; et al. Tumor Suppressor Functions of miR-133a in Colorectal Cancer. Mol. Cancer Res. 2013, 11, 1051–1060. [Google Scholar] [CrossRef] [Green Version]
- Nohata, N. microRNA-1/133a and microRNA-206/133b clusters: Dysregulation and functional roles in human cancers. Oncotarget 2012, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Kojima, S.; Chiyomaru, T.; Kawakami, K.; Yoshino, H.; Enokida, H.; Nohata, N.; Fuse, M.; Ichikawa, T.; Naya, Y.; Nakagawa, M.; et al. Tumour suppressors miR-1 and miR-133a target the oncogenic function of purine nucleoside phosphorylase (PNP) in prostate cancer. Br. J. Cancer 2011, 106, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.-T.; Xu, W.-X.; Li, H.; Xia, M. Emerging roles of MiR-133a in human cancers. J. Cancer 2021, 12, 198–206. [Google Scholar] [CrossRef]
- Liu, W.; Shi, X.; Wang, B. microRNA-133a exerts tumor suppressive role in oral squamous cell carcinoma through the Notch signaling pathway via downregulation of CTBP2. Cancer Gene Ther. 2021, 1–11. [Google Scholar] [CrossRef]
- Mataki, H.; Enokida, H.; Chiyomaru, T.; Mizuno, K.; Matsushita, R.; Goto, Y.; Nishikawa, R.; Higashimoto, I.; Samukawa, T.; Nakagawa, M.; et al. Downregulation of the microRNA-1/133a cluster enhances cancer cell migration and invasion in lung-squamous cell carcinoma via regulation of Coronin1C. J. Hum. Genet. 2014, 60, 53–61. [Google Scholar] [CrossRef]
- Vandewalle, V.; Essaghir, A.; Bollaert, E.; Lenglez, S.; Graux, C.; Schoemans, H.; Saussoy, P.; Michaux, L.; Valk, P.J.M.; Demoulin, J.; et al. miR-15a-5p and miR-21-5p contribute to chemoresistance in cytogenetically normal acute myeloid leukaemia by targeting PDCD4, ARL2 and BTG2. J. Cell. Mol. Med. 2020, 25, 575–585. [Google Scholar] [CrossRef]
- Lovat, F.; Nigita, G.; Distefano, R.; Nakamura, T.; Gasparini, P.; Tomasello, L.; Fadda, P.; Ibrahimova, N.; Catricalà, S.; Palamarchuk, A.; et al. Combined loss of function of two different loci of miR-15/16 drives the pathogenesis of acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2020, 117, 12332–12340. [Google Scholar] [CrossRef]
- Guo, S.; Fesler, A.; Huang, W.; Wang, Y.; Yang, J.; Wang, X.; Zheng, Y.; Hwang, G.-R.; Ju, J. Functional Significance and Therapeutic Potential of miR-15a Mimic in Pancreatic Ductal Adenocarcinoma. Mol. Ther.-Nucleic Acids 2020, 19, 228–239. [Google Scholar] [CrossRef]
- Liu, T.; Xu, Z.; Ou, D.; Liu, J.; Zhang, J. The miR-15a/16 gene cluster in human cancer: A systematic review. J. Cell. Physiol. 2019, 234, 5496–5506. [Google Scholar] [CrossRef]
- Guo, S.; Xu, X.; Tang, Y.; Zhang, C.; Li, J.; Ouyang, Y.; Ju, J.; Bie, P.; Wang, H. miR-15a inhibits cell proliferation and epithelial to mesenchymal transition in pancreatic ductal adenocarcinoma by down-regulating Bmi-1 expression. Cancer Lett. 2014, 344, 40–46. [Google Scholar] [CrossRef]
- Aqeilan, R.I.; Calin, G.; Croce, C.M. miR-15a and miR-16-1 in cancer: Discovery, function and future perspectives. Cell Death Differ. 2009, 17, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J.; Fu, A.; Robson-Doucette, C.; Allister, E.M.; Wheeler, M.B.; Screaton, R.; Harper, M.-E. Glutathionylation State of Uncoupling Protein-2 and the Control of Glucose-stimulated Insulin Secretion. J. Biol. Chem. 2012, 287, 39673–39685. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.; Seifert, E.L.; Bouillaud, F.; Aguer, C.; Collins, S.; Harper, M.-E. Glutathionylation Acts as a Control Switch for Uncoupling Proteins UCP2 and UCP3. J. Biol. Chem. 2011, 286, 21865–21875. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, E.; Esteves, P.; Sancerni, T.; Lenoir, V.; Aparicio, T.; Bouillaud, F.; Dentin, R.; Prip-Buus, C.; Ricquier, D.; Pecqueur, C.; et al. UCP2 Deficiency Increases Colon Tumorigenesis by Promoting Lipid Synthesis and Depleting NADPH for Antioxidant Defenses. Cell Rep. 2019, 28, 2306–2316.e5. [Google Scholar] [CrossRef] [Green Version]
- Imai, K.; Fukuda, T.; Wada, T.; Kawanishi, M.; Tasaka, R.; Yasui, T.; Sumi, T. UCP2 expression may represent a predictive marker of neoadjuvant chemotherapy effectiveness for locally advanced uterine cervical cancer. Oncol. Lett. 2017, 14, 951–957. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhang, C.; Jackson, K.; Shen, X.; Chunjing, Z.; Li, G.; Kevil, C.; Xingui, S.; Shi, R.; Zhao, Y. UCP2 Knockout Suppresses Mouse Skin Carcinogenesis. Cancer Prev. Res. 2015, 8, 487–491. [Google Scholar] [CrossRef] [Green Version]
- Donadelli, M.; Dando, I.; Pozza, E.D.; Palmieri, M. Mitochondrial uncoupling protein 2 and pancreatic cancer: A new potential target therapy. World J. Gastroenterol. 2015, 21, 3232–3238. [Google Scholar] [CrossRef]
- Yu, G.; Liu, J.; Xu, K.; Dong, J. Uncoupling protein 2 mediates resistance to gemcitabine-induced apoptosis in hepatocellular carcinoma cell lines. Biosci. Rep. 2015, 35, e00231. [Google Scholar] [CrossRef]
- Li, W.; Nichols, K.; Nathan, C.-A.; Zhao, Y. Mitochondrial uncoupling protein 2 is up-regulated in human head and neck, skin, pancreatic, and prostate tumors. Cancer Biomark. 2013, 13, 377–383. [Google Scholar] [CrossRef]
- Dando, I.; Fiorini, C.; Pozza, E.D.; Padroni, C.; Costanzo, C.; Palmieri, M.; Donadelli, M. UCP2 inhibition triggers ROS-dependent nuclear translocation of GAPDH and autophagic cell death in pancreatic adenocarcinoma cells. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1833, 672–679. [Google Scholar] [CrossRef] [Green Version]
- Pfefferle, A.; Mailloux, R.; Adjeitey, C.N.-K.; Harper, M.-E. Glutathionylation of UCP2 sensitizes drug resistant leukemia cells to chemotherapeutics. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1833, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Yang, Y.; Han, Y.; Li, X.; Wang, X.; Li, X.; Zhang, Z.; Wang, Y. UCP2 Inhibits ROS-Mediated Apoptosis in A549 under Hypoxic Conditions. PLoS ONE 2012, 7, e30714. [Google Scholar] [CrossRef] [Green Version]
- Bertholet, A.M.; Chouchani, E.T.; Kazak, L.; Angelin, A.; Fedorenko, A.; Long, J.Z.; Vidoni, S.; Garrity, R.; Cho, J.; Terada, N.; et al. H+ transport is an integral function of the mitochondrial ADP/ATP carrier. Nature 2019, 571, 515–520. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Encarnacion-Rosado, J.; Kimmelman, A.C. Harnessing metabolic dependencies in pancreatic cancers. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 482–492. [Google Scholar] [CrossRef]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef] [Green Version]
- Meléndez-Rodríguez, F.; Urrutia, A.A.; Lorendeau, D.; Rinaldi, G.; Roche, O.; Böğürcü-Seidel, N.; Muelas, M.O.; Ciller, C.M.; Turiel, G.; Bouthelier, A.; et al. HIF1α Suppresses Tumor Cell Proliferation through Inhibition of Aspartate Biosynthesis. Cell Rep. 2019, 26, 2257–2265.e4. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Luengo, A.; Danai, L.V.; Bush, L.N.; Diehl, F.F.; Hosios, A.M.; Lau, A.N.; Elmiligy, S.; Malstrom, S.; Lewis, C.A.; et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 2018, 20, 782–788. [Google Scholar] [CrossRef]
- Cheng, C.-T.; Qi, Y.; Wang, Y.-C.; Chi, K.; Chung, Y.; Ouyang, C.; Chen, Y.-R.; Oh, M.E.; Sheng, X.; Tang, Y.; et al. Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction. Commun. Biol. 2018, 1, 178. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.; Menon, D.; Bernfeld, E.; Mroz, V.; Kalan, S.; Loayza, D.; Foster, D.A. Aspartate Rescues S-phase Arrest Caused by Suppression of Glutamine Utilization in KRas-driven Cancer Cells. J. Biol. Chem. 2016, 291, 9322–9329. [Google Scholar] [CrossRef] [Green Version]
- Matés, J.M.; Campos-Sandoval, J.A.; Santos-Jiménez, J.D.L.; Segura, J.A.; Alonso, F.J.; Márquez, J. Metabolic Reprogramming of Cancer by Chemicals that Target Glutaminase Isoenzymes. Curr. Med. Chem. 2020, 27, 5317–5339. [Google Scholar] [CrossRef]
- Masisi, B.K.; El Ansari, R.; Alfarsi, L.; Rakha, E.A.; Green, A.R.; Craze, M.L. The role of glutaminase in cancer. Histopathology 2020, 76, 498–508. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef]
- Koch, K.; Hartmann, R.; Tsiampali, J.; Uhlmann, C.; Nickel, A.-C.; He, X.; Kamp, M.A.; Sabel, M.; Barker, R.A.; Steiger, H.-J.; et al. A comparative pharmaco-metabolomic study of glutaminase inhibitors in glioma stem-like cells confirms biological effectiveness but reveals differences in target-specificity. Cell Death Discov. 2020, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F.; Monné, M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1863, 2362–2378. [Google Scholar] [CrossRef]
- Lytovchenko, O.; Kunji, E.R. Expression and putative role of mitochondrial transport proteins in cancer. Biochim. Biophys. Acta (BBA)-Bioenerg. 2017, 1858, 641–654. [Google Scholar] [CrossRef]
- Kolukula, V.K.; Sahu, G.; Wellstein, A.; Rodriguez, O.C.; Preet, A.; Iacobazzi, V.; D’Orazi, G.; Albanese, C.; Palmieri, F.; Avantaggiati, M.L. SLC25A1, or CIC, is a novel transcriptional target of mutant p53 and a negative tumor prognostic marker. Oncotarget 2014, 5, 1212–1225. [Google Scholar] [CrossRef] [Green Version]
- Mosaoa, R.; Kasprzyk-Pawelec, A.; Fernandez, H.; Avantaggiati, M. The Mitochondrial Citrate Carrier SLC25A1/CIC and the Fundamental Role of Citrate in Cancer, Inflammation and Beyond. Biomolecules 2021, 11, 141. [Google Scholar] [CrossRef]
- Shi, W.; Tang, T.; Li, X.; Deng, S.; Li, R.; Wang, Y.; Wang, Y.; Xia, T.; Zhang, Y.; Zen, K.; et al. Methylation-mediated silencing of miR-133a-3p promotes breast cancer cell migration and stemness via miR-133a-3p/MAML1/DNMT3A positive feedback loop. J. Exp. Clin. Cancer Res. 2018, 38, 429. [Google Scholar] [CrossRef] [Green Version]
- Sui, Y.; Zhang, X.; Yang, H.; Wei, W.; Wang, M. MicroRNA-133a acts as a tumour suppressor in breast cancer through targeting LASP1. Oncol. Rep. 2017, 39, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Yao, Y.F.; Hu, S.N.; Gao, J.; Zhang, L.-L. MiR-133a Is Functionally Involved in Doxorubicin-Resistance in Breast Cancer Cells MCF-7 via Its Regulation of the Expression of Uncoupling Protein 2. PLoS ONE 2015, 10, e0129843. [Google Scholar] [CrossRef] [Green Version]
- Dieter, C.; Assmann, T.S.; Costa, A.R.; Canani, L.H.; De Souza, B.M.; Bauer, A.C.; Crispim, D. MiR-30e-5p and MiR-15a-5p Expressions in Plasma and Urine of Type 1 Diabetic Patients with Diabetic Kidney Disease. Front. Genet. 2019, 10, 563. [Google Scholar] [CrossRef]
- de Groen, F.L.M.; Timmer, L.M.; Menezes, R.X.; Diosdado, B.; Hooijberg, E.; Meijer, G.A.; Steenbergen, R.D.M.; Carvalho, B. Oncogenic Role of miR-15a-3p in 13q Amplicon-Driven Colorectal Adenoma-to-Carcinoma Progression. PLoS ONE 2015, 10, e0132495. [Google Scholar] [CrossRef]
- Jin, X.; Chen, N.; Zheng, R.-H.; Zhang, H.; Chen, Y.-P.; Xiang, Z. miRNA-133a-UCP2 pathway regulates inflammatory bowel disease progress by influencing inflammation, oxidative stress and energy metabolism. World J. Gastroenterol. 2017, 23, 76–86. [Google Scholar] [CrossRef]
- Ferrara, C.T.; Boodhansingh, K.E.; Paradies, E.; Giuseppe, F.; Steinkrauss, L.J.; Topor, L.S.; Quintos, J.B.; Ganguly, A.; de Leon, D.; Palmieri, F.; et al. Novel Hypoglycemia Phenotype in Congenital Hyperinsulinism Due to Dominant Mutations of Uncoupling Protein 2. J. Clin. Endocrinol. Metab. 2017, 102, 942–949. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorgoglione, R.; Impedovo, V.; Riley, C.L.; Fratantonio, D.; Tiziani, S.; Palmieri, L.; Dolce, V.; Fiermonte, G. Glutamine-Derived Aspartate Biosynthesis in Cancer Cells: Role of Mitochondrial Transporters and New Therapeutic Perspectives. Cancers 2022, 14, 245. https://doi.org/10.3390/cancers14010245
Gorgoglione R, Impedovo V, Riley CL, Fratantonio D, Tiziani S, Palmieri L, Dolce V, Fiermonte G. Glutamine-Derived Aspartate Biosynthesis in Cancer Cells: Role of Mitochondrial Transporters and New Therapeutic Perspectives. Cancers. 2022; 14(1):245. https://doi.org/10.3390/cancers14010245
Chicago/Turabian StyleGorgoglione, Ruggiero, Valeria Impedovo, Christopher L. Riley, Deborah Fratantonio, Stefano Tiziani, Luigi Palmieri, Vincenza Dolce, and Giuseppe Fiermonte. 2022. "Glutamine-Derived Aspartate Biosynthesis in Cancer Cells: Role of Mitochondrial Transporters and New Therapeutic Perspectives" Cancers 14, no. 1: 245. https://doi.org/10.3390/cancers14010245
APA StyleGorgoglione, R., Impedovo, V., Riley, C. L., Fratantonio, D., Tiziani, S., Palmieri, L., Dolce, V., & Fiermonte, G. (2022). Glutamine-Derived Aspartate Biosynthesis in Cancer Cells: Role of Mitochondrial Transporters and New Therapeutic Perspectives. Cancers, 14(1), 245. https://doi.org/10.3390/cancers14010245