
CXCL12 in Pancreatic Cancer: Its Function and Potential as a Therapeutic Drug Target



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA Extraction and qPCR
2.2. ELISA
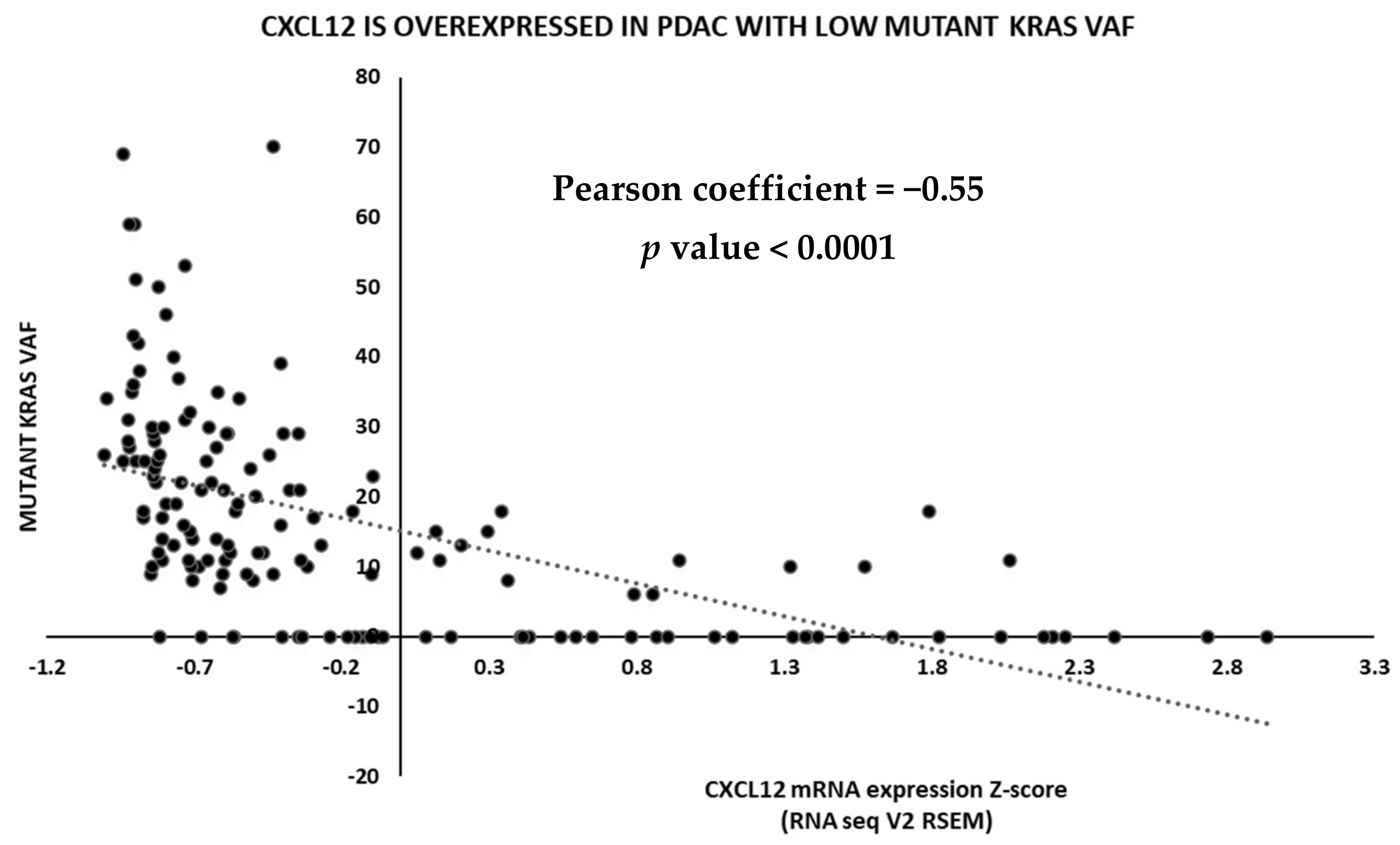
2.3. TCGA Data Analysis
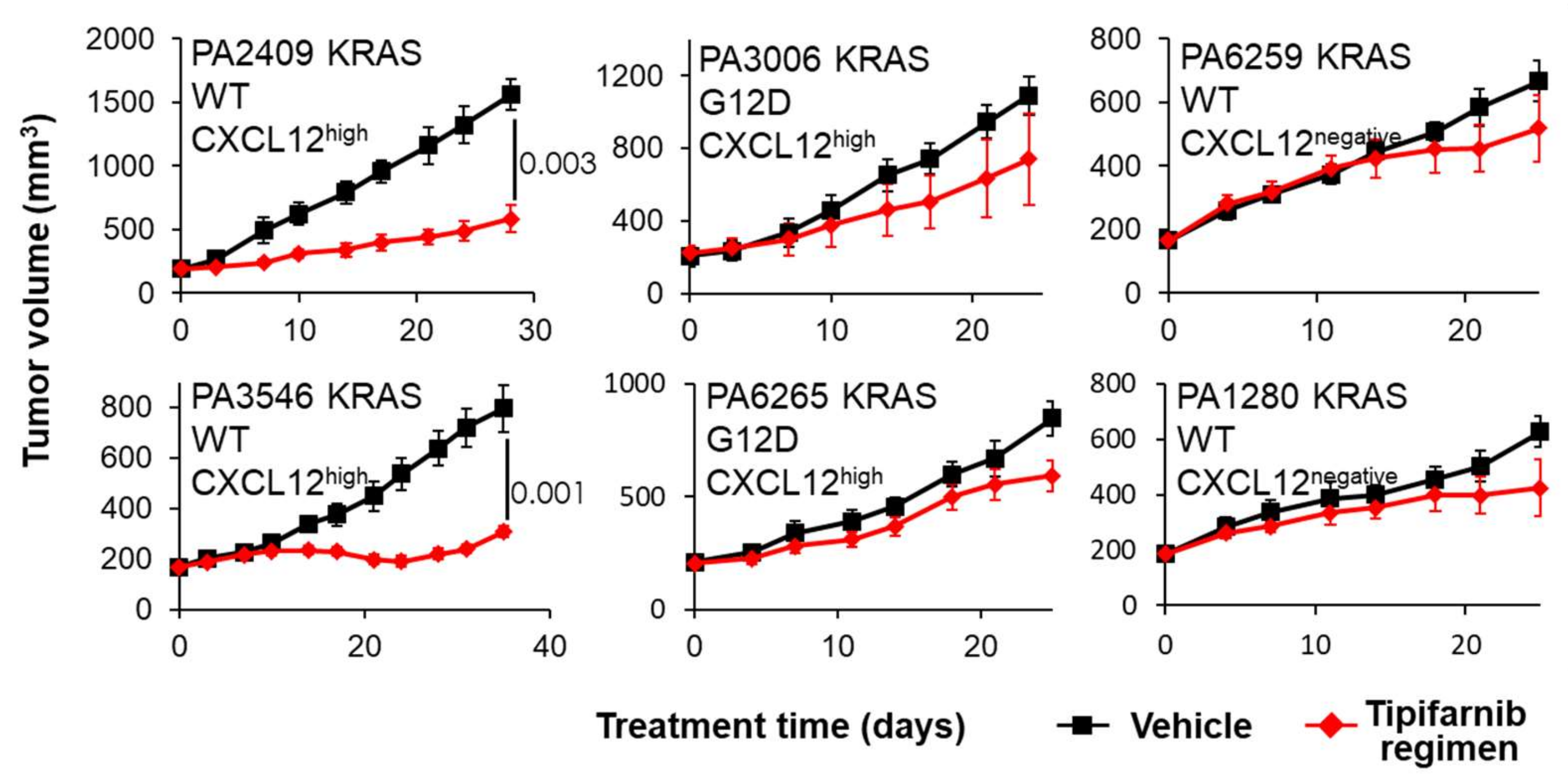
2.4. PDX Mouse Studies
3. CXCL12 Signaling
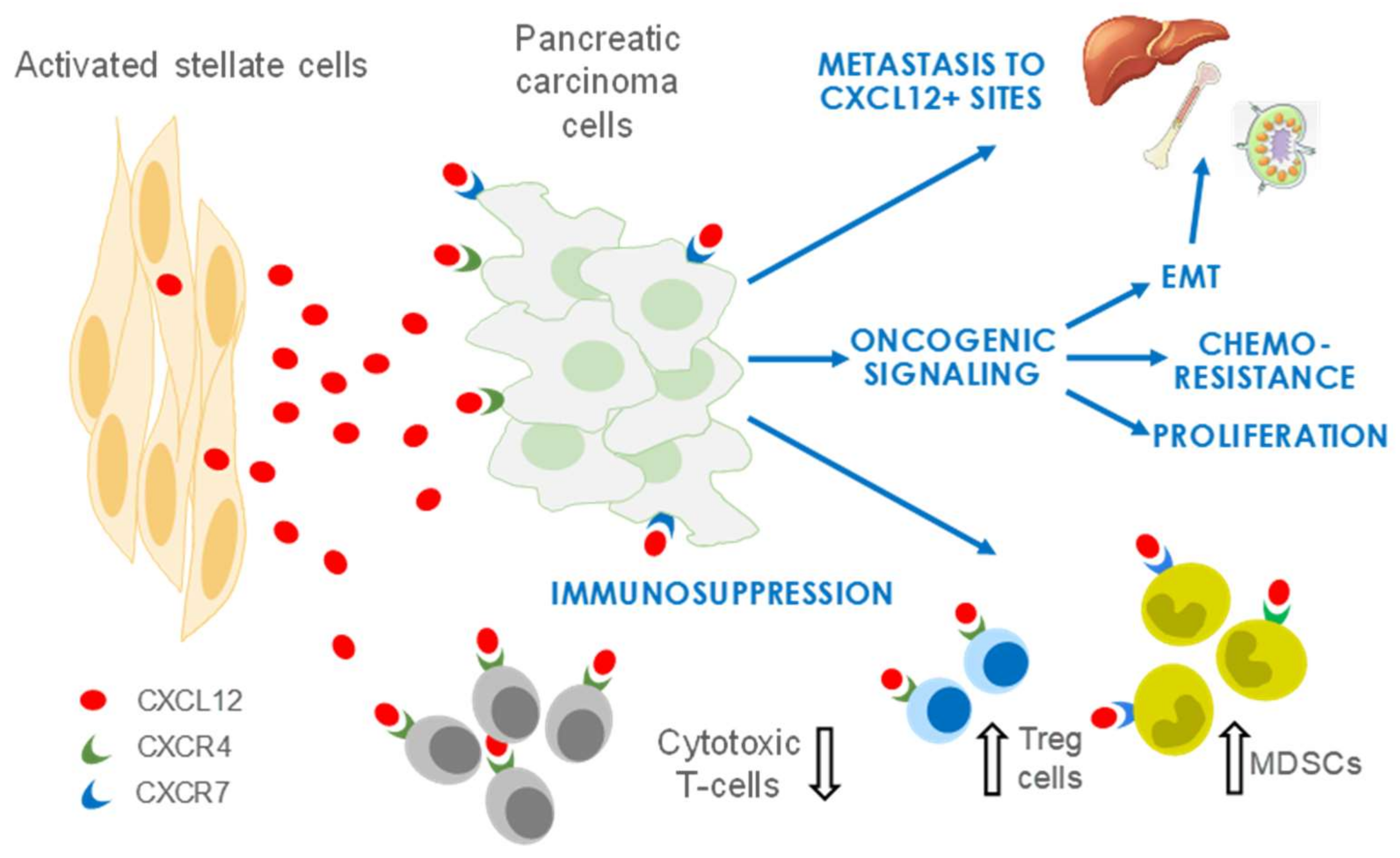
4. CXCL12 Signaling in PDAC
4.1. CXCL12 Promotes PDAC Survival and Proliferation Signaling
4.2. CXCL12 Signaling Promotes Immune Evasion
4.3. CXCL12 Signaling Promotes Angiogenesis
4.4. CXCL12 Signaling Promotes Chemoresistance
5. Therapeutic Targeting of CXCL12/CXCR4 in Pancreatic Cancer
5.1. CXCR4 Antagonists in Pancreatic Cancer
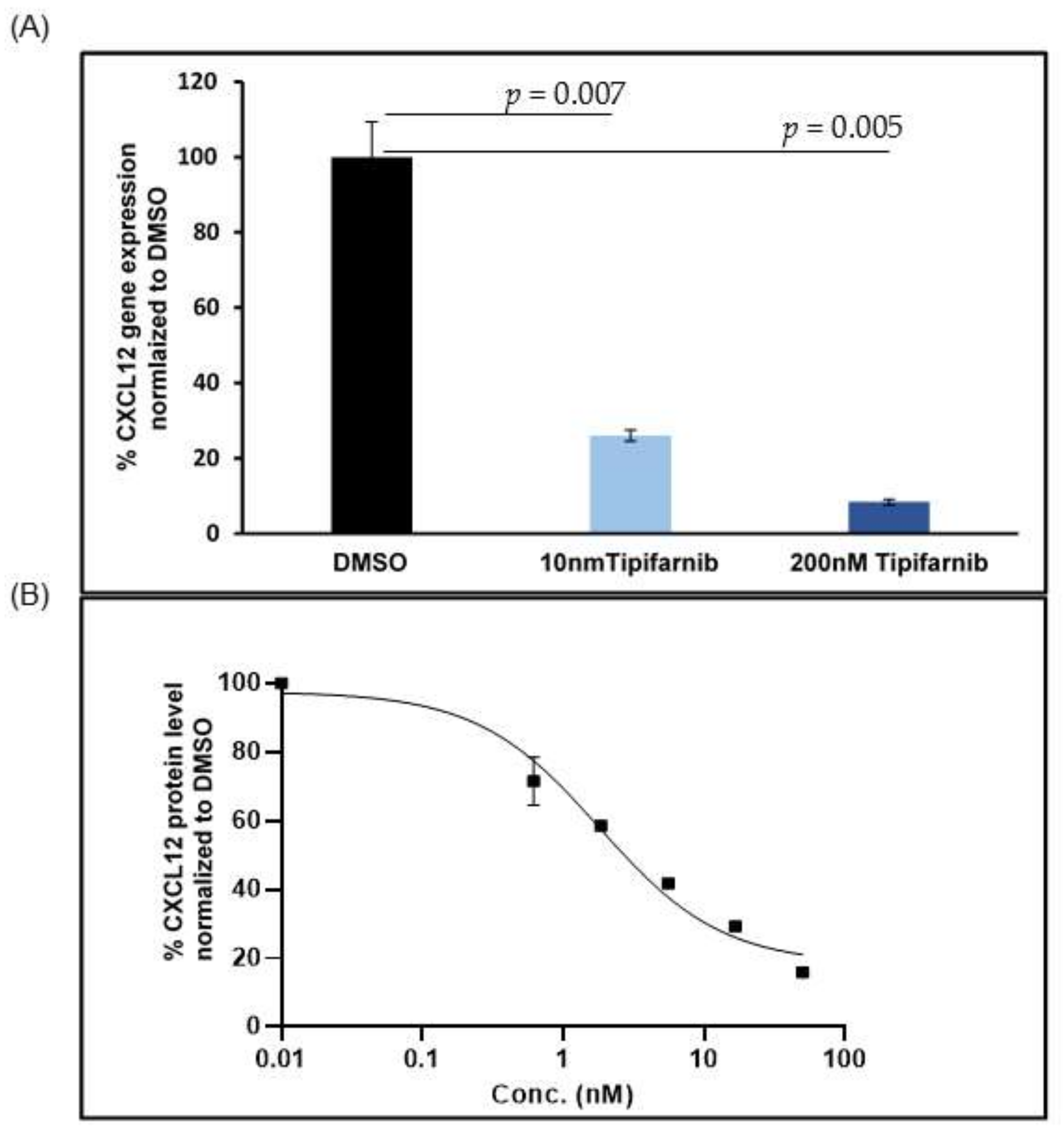
5.2. CXCL12 Antagonists in Pancreatic Cancer
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Rosenberg, P.S.; Jemal, A. Emerging cancer trends among young adults in the USA: Analysis of a population-based cancer registry. Lancet Public Health 2019, 4, e137–e147. [Google Scholar] [CrossRef] [Green Version]
- Sleightholm, R.L.; Neilsen, B.K.; Li, J.; Steele, M.M.; Singh, R.K.; Hollingsworth, M.A.; Oupicky, D. Emerging roles of the CXCL12/CXCR4 axis in pancreatic cancer progression and therapy. Pharm. Ther. 2017, 179, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Hiroshima, Y.; Matsuyama, R.; Homma, Y.; Hoffman, R.M.; Endo, I. Role of the tumor microenvironment in pancreatic cancer. Ann. Gastroenterol. Surg. 2019, 3, 130–137. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Prakash, J. Targeting Pancreatic Stellate Cells in Cancer. Trends Cancer 2019, 5, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma. Mol. Cancer 2018, 17, 62. [Google Scholar] [CrossRef] [Green Version]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Brekken, R.A. Recent advances in understanding cancer-associated fibroblasts in pancreatic cancer. Am. J. Physiol. Cell Physiol. 2020, 319, C233–C243. [Google Scholar] [CrossRef]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Puchert, M.; Engele, J. The peculiarities of the SDF-1/CXCL12 system: In some cells, CXCR4 and CXCR7 sing solos, in others, they sing duets. Cell Tissue Res. 2014, 355, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Azad, B.B.; Nimmagadda, S. The intricate role of CXCR4 in cancer. Adv. Cancer Res. 2014, 124, 31–82. [Google Scholar] [CrossRef] [Green Version]
- Ling, X.; Spaeth, E.; Chen, Y.; Shi, Y.; Zhang, W.; Schober, W.; Hail, N., Jr.; Konopleva, M.; Andreeff, M. The CXCR4 antagonist AMD3465 regulates oncogenic signaling and invasiveness in vitro and prevents breast cancer growth and metastasis in vivo. PLoS ONE 2013, 8, e58426. [Google Scholar] [CrossRef] [Green Version]
- Gilardi, M.; Wang, Z.A.-O.; Proietto, M.; Chillà, A.; Calleja-Valera, J.L.; Goto, Y.; Vanoni, M.A.-O.; Janes, M.R.; Mikulski, Z.; Gualberto, A.; et al. Tipifarnib as a Precision Therapy for HRAS-Mutant Head and Neck Squamous Cell Carcinomas. Mol. Cancer Ther. 2020, 19, 1784–1796. [Google Scholar] [CrossRef]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef]
- Tashiro, K.; Tada, H.; Heilker, R.; Shirozu, M.; Nakano, T.; Honjo, T. Signal sequence trap: A cloning strategy for secreted proteins and type I membrane proteins. Science 1993, 261, 600–603. [Google Scholar] [CrossRef]
- Nagasawa, T.; Hirota, S.; Tachibana, K.; Takakura, N.; Nishikawa, S.; Kitamura, Y.; Yoshida, N.; Kikutani, H.; Kishimoto, T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 1996, 382, 635–638. [Google Scholar] [CrossRef]
- Liu, Q.; Li, Z.; Gao, J.L.; Wan, W.; Ganesan, S.; McDermott, D.H.; Murphy, P.M. CXCR4 antagonist AMD3100 redistributes leukocytes from primary immune organs to secondary immune organs, lung, and blood in mice. Eur. J. Immunol. 2015, 45, 1855–1867. [Google Scholar] [CrossRef]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L.; et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Park, H.D.; Lee, Y.; Oh, Y.K.; Jung, J.G.; Park, Y.W.; Myung, K.; Kim, K.H.; Koh, S.S.; Lim, D.S. Pancreatic adenocarcinoma upregulated factor promotes metastasis by regulating TLR/CXCR4 activation. Oncogene 2011, 30, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Saini, V.; Romero, J.; Marchese, A.; Majetschak, M. Ubiquitin receptor binding and signaling in primary human leukocytes. Commun. Integr. Biol. 2010, 3, 608–610. [Google Scholar] [CrossRef] [Green Version]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, X.; Langer, M.; Weber, C.; Koenen, R.R.; von Hundelshausen, P. Touch of chemokines. Front. Immunol. 2012, 3, 175. [Google Scholar] [CrossRef] [Green Version]
- Cojoc, M.; Peitzsch, C.; Trautmann, F.; Polishchuk, L.; Telegeev, G.D.; Dubrovska, A. Emerging targets in cancer management: Role of the CXCL12/CXCR4 axis. Onco Targets Ther. 2013, 6, 1347–1361. [Google Scholar] [CrossRef] [Green Version]
- Walenkamp, A.M.E.; Lapa, C.; Herrmann, K.; Wester, H.J. CXCR4 Ligands: The Next Big Hit? J. Nucl. Med. 2017, 58, 77s–82s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busillo, J.M.; Benovic, J.L. Regulation of CXCR4 signaling. Biochim. Biophys. Acta 2007, 1768, 952–963. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.J.; Zhao, J.; Sun, Y.; Hu, W.; Wu, Y.L.; Cen, B.; Wu, G.X.; Pei, G. β-arrestin differentially regulates the chemokine receptor CXCR4-mediated signaling and receptor internalization, and this implicates multiple interaction sites between beta-arrestin and CXCR4. J. Biol. Chem. 2000, 275, 2479–2485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Cheng, Z.; Ma, L.; Pei, G. Beta-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J. Biol. Chem. 2002, 277, 49212–49219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumann, U.; Cameroni, E.; Pruenster, M.; Mahabaleshwar, H.; Raz, E.; Zerwes, H.G.; Rot, A.; Thelen, M. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE 2010, 5, e9175. [Google Scholar] [CrossRef] [Green Version]
- Hunger, C.; Ödemis, V.; Engele, J. Expression and function of the SDF-1 chemokine receptors CXCR4 and CXCR7 during mouse limb muscle development and regeneration. Exp. Cell. Res. 2012, 318, 2178–2190. [Google Scholar] [CrossRef]
- Sierro, F.; Biben, C.; Martínez-Muñoz, L.; Mellado, M.; Ransohoff, R.M.; Li, M.; Woehl, B.; Leung, H.; Groom, J.; Batten, M.; et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. USA 2007, 104, 14759–14764. [Google Scholar] [CrossRef] [Green Version]
- Levoye, A.; Balabanian, K.; Baleux, F.; Bachelerie, F.; Lagane, B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 2009, 113, 6085–6093. [Google Scholar] [CrossRef] [Green Version]
- Decaillot, F.M.; Kazmi, M.A.; Lin, Y.; Ray-Saha, S.; Sakmar, T.P.; Sachdev, P. CXCR7/CXCR4 heterodimer constitutively recruits beta-arrestin to enhance cell migration. J. Biol. Chem. 2011, 286, 32188–32197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, G.; Stanco, A.; Long, J.E.; Crawford, D.; Potter, G.B.; Pleasure, S.J.; Behrens, T.; Rubenstein, J.L. CXCR4 and CXCR7 have distinct functions in regulating interneuron migration. Neuron 2011, 69, 61–76. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Han, J.; Kim, K.; Choi, H.; Cho, E.G.; Lee, T.R. CXCR7 mediates SDF1-induced melanocyte migration. Pigment. Cell Melanoma. Res. 2013, 26, 58–66. [Google Scholar] [CrossRef]
- Heinrich, E.L.; Lee, W.; Lu, J.; Lowy, A.M.; Kim, J. Chemokine CXCL12 activates dual CXCR4 and CXCR7-mediated signaling pathways in pancreatic cancer cells. J. Transl. Med. 2012, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.; Li, G.X.; Kong, Y.Y.; Shen, X.X.; Qu, Y.Y.; Jia, Z.W.; Wang, Y.; Dai, B.; Ye, D.W. Chemokine Receptors CXCR4 and CXCR7 are Associated with Tumor Aggressiveness and Prognosis in Extramammary Paget Disease. J. Cancer 2017, 8, 2471–2477. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Marchesi, F.; Monti, P.; Leone, B.E.; Zerbi, A.; Vecchi, A.; Piemonti, L.; Mantovani, A.; Allavena, P. Increased survival, proliferation, and migration in metastatic human pancreatic tumor cells expressing functional CXCR4. Cancer Res. 2004, 64, 8420–8427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachem, M.G.; Schunemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Ma, Q.; Liu, Q.; Yu, H.; Zhao, L.; Shen, S.; Yao, J. Blockade of SDF-1/CXCR4 signalling inhibits pancreatic cancer progression in vitro via inactivation of canonical Wnt pathway. Br. J. Cancer 2008, 99, 1695–1703. [Google Scholar] [CrossRef]
- Shen, B.; Zheng, M.Q.; Lu, J.W.; Jiang, Q.; Wang, T.H.; Huang, X.E. CXCL12-CXCR4 promotes proliferation and invasion of pancreatic cancer cells. Asian Pac. J. Cancer Prev. 2013, 14, 5403–5408. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ma, Q.; Xu, Q.; Liu, H.; Lei, J.; Duan, W.; Bhat, K.; Wang, F.; Wu, E.; Wang, Z. SDF-1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial-mesenchymal transition in vitro through non-canonical activation of Hedgehog pathway. Cancer Lett. 2012, 322, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billadeau, D.D.; Chatterjee, S.; Bramati, P.; Sreekumar, R.; Shah, V.; Hedin, K.; Urrutia, R. Characterization of the CXCR4 signaling in pancreatic cancer cells. Int. J. Gastrointest. Cancer 2006, 37, 110–119. [Google Scholar] [CrossRef]
- Singh, A.P.; Arora, S.; Bhardwaj, A.; Srivastava, S.K.; Kadakia, M.P.; Wang, B.; Grizzle, W.E.; Owen, L.B.; Singh, S. CXCL12/CXCR4 protein signaling axis induces sonic hedgehog expression in pancreatic cancer cells via extracellular regulated kinase- and Akt kinase-mediated activation of nuclear factor kappaB: Implications for bidirectional tumor-stromal interactions. J. Biol. Chem. 2012, 287, 39115–39124. [Google Scholar] [CrossRef] [Green Version]
- Weekes, C.D.; Song, D.; Arcaroli, J.; Wilson, L.A.; Rubio-Viqueira, B.; Cusatis, G.; Garrett-Mayer, E.; Messersmith, W.A.; Winn, R.A.; Hidalgo, M. Stromal cell-derived factor 1alpha mediates resistance to mTOR-directed therapy in pancreatic cancer. Neoplasia 2012, 14, 690–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Ohuchida, K.; Fei, S.; Zheng, B.; Guan, W.; Feng, H.; Kibe, S.; Ando, Y.; Koikawa, K.; Abe, T.; et al. Inhibition of ERK1/2 in cancer-associated pancreatic stellate cells suppresses cancer-stromal interaction and metastasis. J. Exp. Clin. Cancer Res. 2019, 38, 221. [Google Scholar] [CrossRef]
- Zhou, W.; Guo, S.; Liu, M.; Burow, M.E.; Wang, G. Targeting CXCL12/CXCR4 Axis in Tumor Immunotherapy. Curr. Med. Chem. 2019, 26, 3026–3041. [Google Scholar] [CrossRef]
- Ma, J.; Su, H.; Yu, B.; Guo, T.; Gong, Z.; Qi, J.; Zhao, X.; Du, J. CXCL12 gene silencing down-regulates metastatic potential via blockage of MAPK/PI3K/AP-1 signaling pathway in colon cancer. Clin. Transl. Oncol. 2018, 20, 1035–1045. [Google Scholar] [CrossRef] [Green Version]
- Samara, G.J.; Lawrence, D.M.; Chiarelli, C.J.; Valentino, M.D.; Lyubsky, S.; Zucker, S.; Vaday, G.G. CXCR4-mediated adhesion and MMP-9 secretion in head and neck squamous cell carcinoma. Cancer Lett. 2004, 214, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, Y.; Zhang, S.; Chen, Z.; Fan, L.; Shen, X.; Zhou, S.; Chen, D. Stem cell autocrine CXCL12/CXCR4 stimulates invasion and metastasis of esophageal cancer. Oncotarget 2017, 8, 36149–36160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, H.L.; Atkins, M.B.; Subedi, P.; Wu, J.; Chambers, J.; Joseph Mattingly, T., II; Campbell, J.D.; Allen, J.; Ferris, A.E.; Schilsky, R.L.; et al. The promise of Immuno-oncology: Implications for defining the value of cancer treatment. J. Immunother. Cancer 2019, 7, 129. [Google Scholar] [CrossRef] [Green Version]
- Hilmi, M.; Bartholin, L.; Neuzillet, C. Immune therapies in pancreatic ductal adenocarcinoma: Where are we now? World J. Gastroenterol. 2018, 24, 2137–2151. [Google Scholar] [CrossRef]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [Green Version]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winograd, R.; Byrne, K.T.; Evans, R.A.; Odorizzi, P.M.; Meyer, A.R.; Bajor, D.L.; Clendenin, C.; Stanger, B.Z.; Furth, E.E.; Wherry, E.J.; et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol. Res. 2015, 3, 399–411. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.D.; Jiang, X.; Sullivan, K.M.; Jalikis, F.G.; Smythe, K.S.; Abbasi, A.; Vignali, M.; Park, J.O.; Daniel, S.K.; Pollack, S.M.; et al. Mobilization of CD8(+) T Cells via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 3934–3945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukunaga, A.; Miyamoto, M.; Cho, Y.; Murakami, S.; Kawarada, Y.; Oshikiri, T.; Kato, K.; Kurokawa, T.; Suzuoki, M.; Nakakubo, Y.; et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas 2004, 28, e26–e31. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Watt, J.; Kocher, H.M. The desmoplastic stroma of pancreatic cancer is a barrier to immune cell infiltration. Oncoimmunology 2013, 2, e26788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protti, M.P.; De Monte, L. Immune infiltrates as predictive markers of survival in pancreatic cancer patients. Front. Physiol. 2013, 4, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 2018, 33, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yan, R.; Li, J.; Gao, Y.; Moresco, P.; Yao, M.; Hechtman, J.F.; Weiss, M.J.; Janowitz, T.; Fearon, D.T. Pancreatic cancer cells assemble a CXCL12-keratin 19 coating to resist immunotherapy. bioRXiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, Y.; Ochi, N.; Sawai, H.; Yasuda, A.; Takahashi, H.; Funahashi, H.; Takeyama, H.; Tong, Z.; Guha, S. CXCL8/IL-8 and CXCL12/SDF-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer 2009, 124, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Guleng, B.; Tateishi, K.; Ohta, M.; Kanai, F.; Jazag, A.; Ijichi, H.; Tanaka, Y.; Washida, M.; Morikane, K.; Fukushima, Y.; et al. Blockade of the stromal cell-derived factor-1/CXCR4 axis attenuates in vivo tumor growth by inhibiting angiogenesis in a vascular endothelial growth factor-independent manner. Cancer Res. 2005, 65, 5864–5871. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Srivastava, S.K.; Bhardwaj, A.; Owen, L.B.; Singh, A.P. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: A novel target for therapy. Br. J. Cancer 2010, 103, 1671–1679. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wu, H.; Guan, J.; Wang, L.; Ren, X.; Shi, X.; Liang, Z.; Liu, T. Paracrine SDF-1α signaling mediates the effects of PSCs on GEM chemoresistance through an IL-6 autocrine loop in pancreatic cancer cells. Oncotarget 2015, 6, 3085–3097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, S.; Bhardwaj, A.; Singh, S.; Srivastava, S.K.; McClellan, S.; Nirodi, C.S.; Piazza, G.A.; Grizzle, W.E.; Owen, L.B.; Singh, A.P. An undesired effect of chemotherapy: Gemcitabine promotes pancreatic cancer cell invasiveness through reactive oxygen species-dependent, nuclear factor kappaB- and hypoxia-inducible factor 1alpha-mediated up-regulation of CXCR4. J. Biol. Chem. 2013, 288, 21197–21207. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, M.; Matsuo, Y.; Koide, S.; Tsuboi, K.; Shamoto, T.; Sato, T.; Saito, K.; Takahashi, H.; Takeyama, H. Enhancement of the CXCL12/CXCR4 axis due to acquisition of gemcitabine resistance in pancreatic cancer: Effect of CXCR4 antagonists. BMC Cancer 2016, 16, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Srivastava, S.K.; Zubair, H.; Patel, G.K.; Arora, S.; Khushman, M.; Carter, J.E.; Gorman, G.S.; Singh, S.; Singh, A.P. Co-targeting of CXCR4 and hedgehog pathways disrupts tumor-stromal crosstalk and improves chemotherapeutic efficacy in pancreatic cancer. J. Biol. Chem. 2020, 295, 8413–8424. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Srivastava, S.K.; Singh, S.; Arora, S.; Tyagi, N.; Andrews, J.; McClellan, S.; Carter, J.E.; Singh, A.P. CXCL12/CXCR4 signaling counteracts docetaxel-induced microtubule stabilization via p21-activated kinase 4-dependent activation of LIM domain kinase 1. Oncotarget 2014, 5, 11490–11500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domanska, U.M.; Timmer-Bosscha, H.; Nagengast, W.B.; Oude Munnink, T.H.; Kruizinga, R.C.; Ananias, H.J.; Kliphuis, N.M.; Huls, G.; De Vries, E.G.; de Jong, I.J.; et al. CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy. Neoplasia 2012, 14, 709–718. [Google Scholar] [CrossRef] [Green Version]
- Heckmann, D.; Maier, P.; Laufs, S.; Wenz, F.; Zeller, W.J.; Fruehauf, S.; Allgayer, H. CXCR4 Expression and Treatment with SDF-1α or Plerixafor Modulate Proliferation and Chemosensitivity of Colon Cancer Cells. Transl. Oncol. 2013, 6, 124–132. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, S.; Hoos, J.; Klussmann, S.; Vonhoff, S. RNA aptamers and spiegelmers: Synthesis, purification, and post-synthetic PEG conjugation. Curr. Protoc. Nucleic Acid Chem. 2011. [Google Scholar] [CrossRef]
- Hoellenriegel, J.; Zboralski, D.; Maasch, C.; Rosin, N.Y.; Wierda, W.G.; Keating, M.J.; Kruschinski, A.; Burger, J.A. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood 2014, 123, 1032–1039. [Google Scholar] [CrossRef]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef] [Green Version]
- Saada-Bouzid, E.; Le Tourneau, C. Beyond EGFR Targeting in SCCHN: Angiogenesis, PI3K, and Other Molecular Targets. Front. Oncol. 2019, 9, 74. [Google Scholar] [CrossRef]
- Choi, W.T.; Yang, Y.; Xu, Y.; An, J. Targeting chemokine receptor CXCR4 for treatment of HIV-1 infection, tumor progression, and metastasis. Curr. Top. Med. Chem. 2014, 14, 1574–1589. [Google Scholar] [CrossRef] [Green Version]
- Uy, G.L.; Rettig, M.P.; Motabi, I.H.; McFarland, K.; Trinkaus, K.M.; Hladnik, L.M.; Kulkarni, S.; Abboud, C.N.; Cashen, A.F.; Stockerl-Goldstein, K.E.; et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood 2012, 119, 3917–3924. [Google Scholar] [CrossRef]
- Gao, Z.; Wang, X.; Wu, K.; Zhao, Y.; Hu, G. Pancreatic stellate cells increase the invasion of human pancreatic cancer cells through the stromal cell-derived factor-1/CXCR4 axis. Pancreatology 2010, 10, 186–193. [Google Scholar] [CrossRef]
- Biasci, D.; Smoragiewicz, M.; Connell, C.M.; Wang, Z.; Gao, Y.; Thaventhiran, J.E.D.; Basu, B.; Magiera, L.; Johnson, T.I.; Bax, L.; et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc. Natl. Acad. Sci. USA 2020, 117, 28960–28970. [Google Scholar] [CrossRef] [PubMed]
- Fearon, D.T.; Janowitz, T. AMD3100/Plerixafor overcomes immune inhibition by the CXCL12-KRT19 coating on pancreatic and colorectal cancer cells. Br. J. Cancer 2021, 125, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Williams, A.; Schreiber, J.; Hohmann, N.; Pruefer, U.; Krauss, J.; Jäger, D.; Frömming, A.; Beyer, D.; Eulberg, D.; et al. Combined inhibition of CXCL12 and PD-1 in MSS colorectal and pancreatic cancer: Modulation of the microenvironment and clinical effects. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Zboralski, D.; Hoehlig, K.; Eulberg, D.; Frömming, A.; Vater, A. Increasing Tumor-Infiltrating T Cells through Inhibition of CXCL12 with NOX-A12 Synergizes with PD-1 Blockade. Cancer Immunol. Res. 2017, 5, 950–956. [Google Scholar] [CrossRef] [Green Version]
- Demir, I.E.; Kujundzic, K.; Pfitzinger, P.L.; Saricaoglu, Ö.C.; Teller, S.; Kehl, T.; Reyes, C.M.; Ertl, L.S.; Miao, Z.; Schall, T.J.; et al. Early pancreatic cancer lesions suppress pain through CXCL12-mediated chemoattraction of Schwann cells. Proc. Natl. Acad. Sci. USA 2017, 114, E85–E94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualberto, A.; Scholz, C.R.; Cutsem, E.V. Patient reported abdominal pain as a surrogate of the clinical benefit of tipifarnib in pancreatic cancer patients. J. Clin. Oncol. 2019, 37, 275. [Google Scholar] [CrossRef] [Green Version]
- Poznansky, M.C.; Olszak, I.T.; Foxall, R.; Evans, R.H.; Luster, A.D.; Scadden, D.T. Active movement of T cells away from a chemokine. Nat. Med. 2000, 6, 543–548. [Google Scholar] [CrossRef]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e411. [Google Scholar] [CrossRef] [Green Version]
- Daniel, S.K.; Seo, Y.D.; Pillarisetty, V.G. The CXCL12-CXCR4/CXCR7 axis as a mechanism of immune resistance in gastrointestinal malignancies. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kalatskaya, I.; Berchiche, Y.A.; Gravel, S.; Limberg, B.J.; Rosenbaum, J.S.; Heveker, N. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol. Pharmacol. 2009, 75, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Albert, S.; Riveiro, M.E.; Halimi, C.; Hourseau, M.; Couvelard, A.; Serova, M.; Barry, B.; Raymond, E.; Faivre, S. Focus on the role of the CXCL12/CXCR4 chemokine axis in head and neck squamous cell carcinoma. Head Neck 2013, 35, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wu, C.; Zhang, Z.; Yan, K.; Li, C.; Li, Y.; Li, L. CXCL12 is associated with FoxP3(+) tumor-infiltrating lymphocytes and affects the survival of patients with oral squamous cell carcinoma. Oncol. Lett. 2019, 18, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Nazari, A.; Khorramdelazad, H.; Hassanshahi, G. Biological/pathological functions of the CXCL12/CXCR4/CXCR7 axes in the pathogenesis of bladder cancer. Int. J. Clin. Oncol. 2017, 22, 991–1000. [Google Scholar] [CrossRef]
- Kajiyama, H.; Shibata, K.; Terauchi, M.; Ino, K.; Nawa, A.; Kikkawa, F. Involvement of SDF-1alpha/CXCR4 axis in the enhanced peritoneal metastasis of epithelial ovarian carcinoma. Int. J. Cancer 2008, 122, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Lefort, S.; Thuleau, A.; Kieffer, Y.; Sirven, P.; Bieche, I.; Marangoni, E.; Vincent-Salomon, A.; Mechta-Grigoriou, F. CXCR4 inhibitors could benefit to HER2 but not to triple-negative breast cancer patients. Oncogene 2017, 36, 1211–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malik, S.; Westcott, J.M.; Brekken, R.A.; Burrows, F.J. CXCL12 in Pancreatic Cancer: Its Function and Potential as a Therapeutic Drug Target. Cancers 2022, 14, 86. https://doi.org/10.3390/cancers14010086
Malik S, Westcott JM, Brekken RA, Burrows FJ. CXCL12 in Pancreatic Cancer: Its Function and Potential as a Therapeutic Drug Target. Cancers. 2022; 14(1):86. https://doi.org/10.3390/cancers14010086
Chicago/Turabian StyleMalik, Shivani, Jill M. Westcott, Rolf A. Brekken, and Francis J. Burrows. 2022. "CXCL12 in Pancreatic Cancer: Its Function and Potential as a Therapeutic Drug Target" Cancers 14, no. 1: 86. https://doi.org/10.3390/cancers14010086
APA StyleMalik, S., Westcott, J. M., Brekken, R. A., & Burrows, F. J. (2022). CXCL12 in Pancreatic Cancer: Its Function and Potential as a Therapeutic Drug Target. Cancers, 14(1), 86. https://doi.org/10.3390/cancers14010086