
How Phosphofructokinase-1 Promotes PI3K and YAP/TAZ in Cancer: Therapeutic Perspectives



{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
- (i)
- (ii)
- (iii)
- upregulation of key pro-survival factors, such as anti-apoptotic Bcl-xL and MCL1 proteins [17];
- (iv)
- (v)
- (vi)
- (vii)
- amplification of the WWTR1 (encoding TAZ) and YAP1 genes or mutations and deletions of the FAT1 gene (a tumor suppressor, whose inactivation favors TAZ nuclear translocation), as reported for head and neck squamous cell carcinoma (HNSCC) [23];
- (viii)
- YAP1 gene fusions, as observed in different tumors (for a review see [24]). YAP1 fusion proteins (normally constituted by N-terminal YAP1 and C-terminal part of another protein) can retain a TEAD-dependent YAP activity and can show resistance to inhibition by the Hippo pathway;
- (ix)
- activating point mutations in TAZ gene, which can transform TAZ into an oncogene, as observed in muscle cells [25].
2. The Activation of PI3K/AKT and YAP/TAZ Pathways
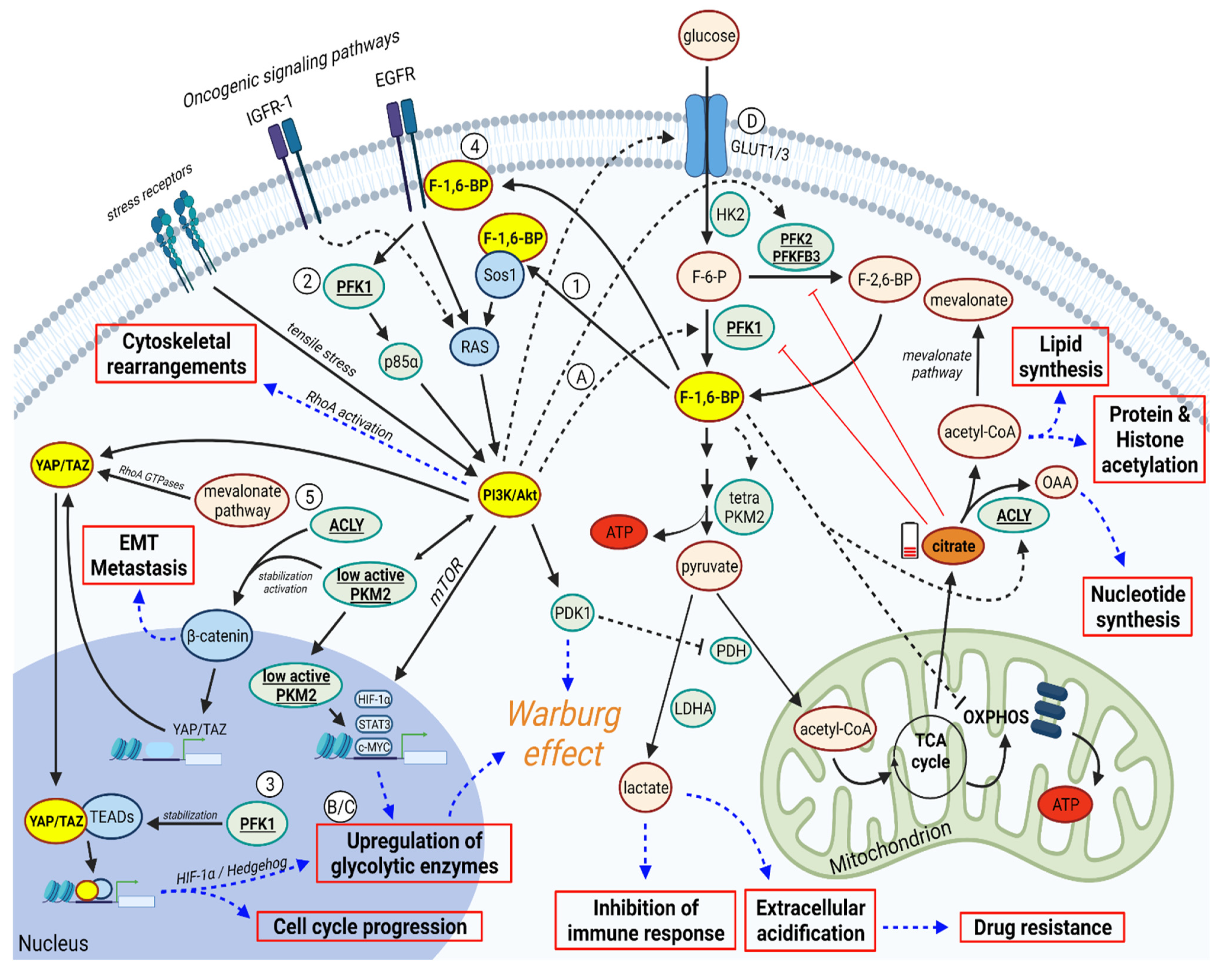
3. PFK1 and F-1,6-BP Activates PI3K/AKT and YAP/TAZ Signaling Pathways
4. In a Feedback Loop, PI3K/AKT and YAP/TAZ can Promote PFK1/F-1,6-BP
5. Discussion
6. Therapeutic Implications
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.L.; Cantley, L.C. PI3K Pathway Alterations in Cancer: Variations on a Theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di-Luoffo, M.; Ben-Meriem, Z.; Lefebvre, P.; Delarue, M.; Guillermet-Guibert, J. PI3K Functions as a Hub in Mechanotransduction. Trends Biochem. Sci. 2021, 46, 878–888. [Google Scholar] [CrossRef]
- Nguyen, C.D.K.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, H.; Li, Y.; Xia, D.; Yang, L.; Ma, Y.; Li, H. The Role of YAP/TAZ Activity in Cancer Metabolic Reprogramming. Molecular Cancer 2018, 17, 134. [Google Scholar] [CrossRef]
- Krajnik, A.; Brazzo, J.A.; Vaidyanathan, K.; Das, T.; Redondo-Muñoz, J.; Bae, Y. Phosphoinositide Signaling and Mechanotransduction in Cardiovascular Biology and Disease. Front. Cell Dev. Biol. 2020, 8, 595849. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in Cancer: Divergent Roles of Isoforms, Modes of Activation and Therapeutic Targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef]
- Rotow, J.; Bivona, T.G. Understanding and Targeting Resistance Mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/MTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef]
- Zeng, R.; Dong, J. The Hippo Signaling Pathway in Drug Resistance in Cancer. Cancers 2021, 13, 318. [Google Scholar] [CrossRef]
- Lien, E.C.; Lyssiotis, C.A.; Cantley, L.C. Metabolic Reprogramming by the PI3K-Akt-MTOR Pathway in Cancer. Recent Results Cancer Res. 2016, 207, 39–72. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Qiu, Y.; Kong, D. Class I Phosphatidylinositol 3-Kinase Inhibitors for Cancer Therapy. Acta Pharm. Sin. B 2017, 7, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Gibault, F.; Sturbaut, M.; Bailly, F.; Melnyk, P.; Cotelle, P. Targeting Transcriptional Enhanced Associate Domains (TEADs). J. Med. Chem. 2018, 61, 5057–5072. [Google Scholar] [CrossRef]
- García-Escudero, R.; Segrelles, C.; Dueñas, M.; Pombo, M.; Ballestín, C.; Alonso-Riaño, M.; Nenclares, P.; Álvarez-Rodríguez, R.; Sánchez-Aniceto, G.; Ruíz-Alonso, A.; et al. Overexpression of PIK3CA in Head and Neck Squamous Cell Carcinoma Is Associated with Poor Outcome and Activation of the YAP Pathway. Oral. Oncol. 2018, 79, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Britschgi, A.; Andraos, R.; Brinkhaus, H.; Klebba, I.; Romanet, V.; Müller, U.; Murakami, M.; Radimerski, T.; Bentires-Alj, M. JAK2/STAT5 Inhibition Circumvents Resistance to PI3K/MTOR Blockade: A Rationale for Cotargeting These Pathways in Metastatic Breast Cancer. Cancer Cell 2012, 22, 796–811. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.-H.; Kim, H.-B.; Kim, M.-C.; Lee, J.-M.; Lee, J.H.; Kim, J.-H.; Kim, J.-W.; Park, W.-Y.; Kim, S.-Y.; Kim, J.B.; et al. Hippo-Mediated Suppression of IRS2/AKT Signaling Prevents Hepatic Steatosis and Liver Cancer. J. Clin. Investig. 2018, 128, 1010–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmani, M.; Aust, M.M.; Attkisson, E.; Williams, D.C.; Ferreira-Gonzalez, A.; Grant, S. Dual Inhibition of Bcl-2 and Bcl-XL Strikingly Enhances PI3K Inhibition-Induced Apoptosis in Human Myeloid Leukemia Cells through a GSK3- and Bim-Dependent Mechanism. Cancer Res. 2013, 73, 1340–1351. [Google Scholar] [CrossRef] [Green Version]
- Serra, V.; Scaltriti, M.; Prudkin, L.; Eichhorn, P.J.A.; Ibrahim, Y.H.; Chandarlapaty, S.; Markman, B.; Rodriguez, O.; Guzman, M.; Rodriguez, S.; et al. PI3K Inhibition Results in Enhanced HER Signaling and Acquired ERK Dependency in HER2-Overexpressing Breast Cancer. Oncogene 2011, 30, 2547–2557. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Shen, W.H. PTEN: A New Guardian of the Genome. Oncogene 2008, 27, 5443–5453. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo Effector YAP Promotes Resistance to RAF- and MEK-Targeted Cancer Therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Faraji, F.; Ramirez, S.I.; Anguiano Quiroz, P.Y.; Mendez-Molina, A.N.; Gutkind, J.S. Genomic Hippo Pathway Alterations and Persistent YAP/TAZ Activation: New Hallmarks in Head and Neck Cancer. Cells 2022, 11, 1370. [Google Scholar] [CrossRef] [PubMed]
- Szulzewsky, F.; Holland, E.C.; Vasioukhin, V. YAP1 and Its Fusion Proteins in Cancer Initiation, Progression and Therapeutic Resistance. Developmental. Biol. 2021, 475, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Sun, C.; De Mello, V.; Selfe, J.; Missiaglia, E.; Shipley, J.; Murray, G.I.; Zammit, P.S.; Wackerhage, H. The Hippo Effector TAZ (WWTR1) Transforms Myoblasts and TAZ Abundance Is Associated with Reduced Survival in Embryonal Rhabdomyosarcoma. J. Pathol. 2016, 240, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Liu, R.; Li, J.; Zhang, C.; Wang, Y.; Cai, Q.; Qian, X.; Xia, Y.; Zheng, Y.; Piao, Y.; et al. Stabilization of Phosphofructokinase 1 Platelet Isoform by AKT Promotes Tumorigenesis. Nat. Commun. 2017, 8, 949. [Google Scholar] [CrossRef] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s Contributions to Current Concepts of Cancer Metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Icard, P.; Shulman, S.; Farhat, D.; Steyaert, J.-M.; Alifano, M.; Lincet, H. How the Warburg Effect Supports Aggressiveness and Drug Resistance of Cancer Cells? Drug Resist. Updates 2018, 38, 1–11. [Google Scholar] [CrossRef]
- Ren, J.-G.; Seth, P.; Ye, H.; Guo, K.; Hanai, J.; Husain, Z.; Sukhatme, V.P. Citrate Suppresses Tumor Growth in Multiple Models through Inhibition of Glycolysis, the Tricarboxylic Acid Cycle and the IGF-1R Pathway. Sci. Rep. 2017, 7, 4537. [Google Scholar] [CrossRef] [Green Version]
- Icard, P.; Coquerel, A.; Wu, Z.; Gligorov, J.; Fuks, D.; Fournel, L.; Lincet, H.; Simula, L. Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. Int. J. Mol. Sci. 2021, 22, 6587. [Google Scholar] [CrossRef]
- Abe, K.; Yamamoto, N.; Hayashi, K.; Takeuchi, A.; Tsuchiya, H. Caffeine Citrate Enhanced Cisplatin Antitumor Effects in Osteosarcoma and Fibrosarcoma in Vitro and in Vivo. BMC Cancer 2019, 19, 689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, H.; Eshima, K.; Ishida, T. Neutralization of Acidic Tumor Microenvironment (TME) with Daily Oral Dosing of Sodium Potassium Citrate (K/Na Citrate) Increases Therapeutic Effect of Anti-Cancer Agent in Pancreatic Cancer Xenograft Mice Model. Biol. Pharm. Bull. 2021, 44, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Pilon-Thomas, S.; Kodumudi, K.N.; El-Kenawi, A.E.; Russell, S.; Weber, A.M.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.W.; Mulé, J.J.; Ibrahim-Hashim, A.; et al. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res. 2016, 76, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- Faes, S.; Duval, A.P.; Planche, A.; Uldry, E.; Santoro, T.; Pythoud, C.; Stehle, J.-C.; Horlbeck, J.; Letovanec, I.; Riggi, N.; et al. Acidic Tumor Microenvironment Abrogates the Efficacy of MTORC1 Inhibitors. Mol. Cancer 2016, 15, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.Y.; Sabatini, D.M. MTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.B.; Hay, N. Is Akt the “Warburg Kinase”?-Akt-Energy Metabolism Interactions and Oncogenesis. Semin. Cancer Biol. 2009, 19, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.C.; Vaira, V.; Caino, M.C.; Tang, H.-Y.Y.; Seo, J.H.; Kossenkov, A.V.; Ottobrini, L.; Martelli, C.; Lucignani, G.; Bertolini, I.; et al. Mitochondrial Akt Regulation of Hypoxic Tumor Reprogramming. Cancer Cell 2016, 30, 257–272. [Google Scholar] [CrossRef] [Green Version]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-Derived Lactate Modifies Antitumor Immune Response: Effect on Myeloid-Derived Suppressor Cells and NK Cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Taylor, S.; Spugnini, E.P.; Assaraf, Y.G.; Azzarito, T.; Rauch, C.; Fais, S. Microenvironment Acidity as a Major Determinant of Tumor Chemoresistance: Proton Pump Inhibitors (PPIs) as a Novel Therapeutic Approach. Drug Resist. Updat 2015, 23, 69–78. [Google Scholar] [CrossRef]
- Molina, J.R.; Adjei, A.A. The Ras/Raf/MAPK Pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Jancík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical Relevance of KRAS in Human Cancers. J. Biomed. Biotechnol. 2010, 2010, 150960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, K.; Van Leemputte, F.; Fischer, B.; Bonini, B.M.; Quezada, H.; Tsytlonok, M.; Haesen, D.; Vanthienen, W.; Bernardes, N.; Gonzalez-Blas, C.B.; et al. Fructose-1,6-Bisphosphate Couples Glycolytic Flux to Activation of Ras. Nat. Commun. 2017, 8, 922. [Google Scholar] [CrossRef] [PubMed]
- Han, Y. Analysis of the Role of the Hippo Pathway in Cancer. J. Transl. Med. 2019, 17, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borreguero-Muñoz, N.; Fletcher, G.C.; Aguilar-Aragon, M.; Elbediwy, A.; Vincent-Mistiaen, Z.I.; Thompson, B.J. The Hippo Pathway Integrates PI3K-Akt Signals with Mechanical and Polarity Cues to Control Tissue Growth. PLoS Biol. 2019, 17, e3000509. [Google Scholar] [CrossRef]
- Ibar, C.; Irvine, K.D. Integration of Hippo-YAP Signaling with Metabolism. Dev. Cell 2020, 54, 256–267. [Google Scholar] [CrossRef]
- Gomez, M.; Gomez, V.; Hergovich, A. The Hippo Pathway in Disease and Therapy: Cancer and Beyond. Clin. Transl. Med. 2014, 3, 22. [Google Scholar] [CrossRef]
- Fan, R.; Kim, N.-G.; Gumbiner, B.M. Regulation of Hippo Pathway by Mitogenic Growth Factors via Phosphoinositide 3-Kinase and Phosphoinositide-Dependent Kinase-1. Proc. Natl. Acad. Sci. USA 2013, 110, 2569–2574. [Google Scholar] [CrossRef] [Green Version]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ Functions and Their Regulation at a Glance. J. Cell Sci. 2020, 133, jcs230425. [Google Scholar] [CrossRef]
- Tumaneng, K.; Schlegelmilch, K.; Russell, R.C.; Yimlamai, D.; Basnet, H.; Mahadevan, N.; Fitamant, J.; Bardeesy, N.; Camargo, F.D.; Guan, K.-L. YAP Mediates Crosstalk between the Hippo and PI(3)K–TOR Pathways by Suppressing PTEN via MiR-29. Nat. Cell Biol. 2012, 14, 1322–1329. [Google Scholar] [CrossRef]
- Qian, X.; He, L.; Hao, M.; Li, Y.; Li, X.; Liu, Y.; Jiang, H.; Xu, L.; Li, C.; Wu, W.; et al. YAP Mediates the Interaction between the Hippo and PI3K/Akt Pathways in Mesangial Cell Proliferation in Diabetic Nephropathy. Acta Diabetol. 2021, 58, 47–62. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. β-Catenin-Driven Cancers Require a YAP1 Transcriptional Complex for Survival and Tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-Wide Association between YAP/TAZ/TEAD and AP-1 at Enhancers Drives Oncogenic Growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo Transducer TAZ Confers Cancer Stem Cell-Related Traits on Breast Cancer Cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Janse van Rensburg, H.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Liu, R.; Li, J.; Wang, Y.; Tan, L.; Li, X.J.; Qian, X.; Zhang, C.; Xia, Y.; Xu, D.; et al. EGFR-Phosphorylated Platelet Isoform of Phosphofructokinase 1 Promotes PI3K Activation. Molecular. Cell 2018, 70, 197–210.e7. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Jin, Z.; Lv, H.; Jin, K.; Jonas, K.; Zhu, C.; Chen, B. PFKP Is Highly Expressed in Lung Cancer and Regulates Glucose Metabolism. Cell Oncol. 2020, 43, 617–629. [Google Scholar] [CrossRef]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic Glycolysis Tunes YAP/ TAZ Transcriptional Activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef]
- Lim, S.-O.; Li, C.-W.; Xia, W.; Lee, H.-H.; Chang, S.-S.; Shen, J.; Hsu, J.L.; Raftery, D.; Djukovic, D.; Gu, H.; et al. EGFR Signaling Enhances Aerobic Glycolysis in Triple-Negative Breast Cancer Cells to Promote Tumor Growth and Immune Escape. Cancer Res. 2016, 76, 1284–1296. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 Regulates β-Catenin Transactivation upon EGFR Activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. The Key Role of the WNT/β-Catenin Pathway in Metabolic Reprogramming in Cancers under Normoxic Conditions. Cancers 2021, 13, 5557. [Google Scholar] [CrossRef]
- Konsavage, W.M.; Kyler, S.L.; Rennoll, S.A.; Jin, G.; Yochum, G.S. Wnt/β-Catenin Signaling Regulates Yes-Associated Protein (YAP) Gene Expression in Colorectal Carcinoma Cells. J. Biol. Chem. 2012, 287, 11730–11739. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; Poli, V. PKM2, STAT3 and HIF-1α: The Warburg’s Vicious Circle. JAK-STAT 2012, 1, 194–196. [Google Scholar] [CrossRef] [Green Version]
- Marín-Hernández, A.; Gallardo-Pérez, J.C.; Ralph, S.J.; Rodríguez-Enríquez, S.; Moreno-Sánchez, R. HIF-1alpha Modulates Energy Metabolism in Cancer Cells by Inducing over-Expression of Specific Glycolytic Isoforms. Mini Rev. Med. Chem. 2009, 9, 1084–1101. [Google Scholar] [CrossRef] [Green Version]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.-S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.-X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [Green Version]
- Hanai, J.I.; Doro, N.; Sasaki, A.T.; Kobayashi, S.; Cantley, L.C.; Seth, P.; Sukhatme, V.P. Inhibition of Lung Cancer Growth: ATP Citrate Lyase Knockdown and Statin Treatment Leads to Dual Blockade of Mitogen-Activated Protein Kinase (MAPK) and Phosphatidylinositol-3-Kinase (PI3K)/AKT Pathways. J. Cell Physiol. 2012, 227, 1709–1720. [Google Scholar] [CrossRef] [Green Version]
- Potapova, I.A.; El-Maghrabi, M.R.; Doronin, S.V.; Benjamin, W.B. Phosphorylation of Recombinant Human ATP:Citrate Lyase by CAMP-Dependent Protein Kinase Abolishes Homotropic Allosteric Regulation of the Enzyme by Citrate and Increases the Enzyme Activity. Allosteric Activation of ATP:Citrate Lyase by Phosphorylated Sugars. Biochemistry 2000, 39, 1169–1179. [Google Scholar] [CrossRef]
- Han, Q.; Chen, C.-A.; Yang, W.; Liang, D.; Lv, H.-W.; Lv, G.-S.; Zong, Q.-N.; Wang, H.-Y. ATP-Citrate Lyase Regulates Stemness and Metastasis in Hepatocellular Carcinoma via the Wnt/β-Catenin Signaling Pathway. Hepatobiliary Pancreat. Dis. Int. 2021, 20, 251–261. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic Control of YAP and TAZ by the Mevalonate Pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Zhou, Y.; Tao, J.; Calvisi, D.F.; Chen, X. Role of Lipogenesis Rewiring in Hepatocellular Carcinoma. Semin. Liver Dis. 2022, 42, 77–86. [Google Scholar] [CrossRef]
- Liu, L.; Yan, H.; Ruan, M.; Yang, H.; Wang, L.; Lei, B.; Sun, X.; Chang, C.; Huang, G.; Xie, W. An AKT/PRMT5/SREBP1 Axis in Lung Adenocarcinoma Regulates de Novo Lipogenesis and Tumor Growth. Cancer Sci. 2021, 112, 3083–3098. [Google Scholar] [CrossRef]
- Sato, R.; Okamoto, A.; Inoue, J.; Miyamoto, W.; Sakai, Y.; Emoto, N.; Shimano, H.; Maeda, M. Transcriptional Regulation of the ATP Citrate-Lyase Gene by Sterol Regulatory Element-Binding Proteins. J. Biol. Chem. 2000, 275, 12497–12502. [Google Scholar] [CrossRef] [Green Version]
- Shu, Z.; Gao, Y.; Zhang, G.; Zhou, Y.; Cao, J.; Wan, D.; Zhu, X.; Xiong, W. A Functional Interaction between Hippo-YAP Signalling and SREBPs Mediates Hepatic Steatosis in Diabetic Mice. J. Cell Mol. Med. 2019, 23, 3616–3628. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, Y.; Ma, Y.; Yang, L.; Wang, T.; Meng, X.; Zong, Z.; Sun, X.; Hua, X.; Li, H. Yes-Associated Protein (YAP) Binds to HIF-1α and Sustains HIF-1α Protein Stability to Promote Hepatocellular Carcinoma Cell Glycolysis under Hypoxic Stress. J. Exp. Clin. Cancer Res. 2018, 37, 216. [Google Scholar] [CrossRef]
- Kilic-Eren, M.; Boylu, T.; Tabor, V. Targeting PI3K/Akt Represses Hypoxia Inducible Factor-1α Activation and Sensitizes Rhabdomyosarcoma and Ewing’s Sarcoma Cells for Apoptosis. Cancer Cell Int. 2013, 13, 36. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Han, H.; Liu, G.-P.; Ma, Y.-X.; Pan, R.-L.; Sang, L.-J.; Li, R.-H.; Yang, L.-J.; Marks, J.R.; Wang, W.; et al. LncRNA Wires up Hippo and Hedgehog Signaling to Reprogramme Glucose Metabolism. EMBO J. 2017, 36, 3325–3335. [Google Scholar] [CrossRef]
- Wang, W.; Xiao, Z.-D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK Modulates Hippo Pathway Activity to Regulate Energy Homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.G.; Tsomides, A.; Yimlamai, D.; Hwang, K.L.; Miesfeld, J.; Galli, G.G.; Fowl, B.H.; Fort, M.; Ma, K.Y.; Sullivan, M.R.; et al. Yap Regulates Glucose Utilization and Sustains Nucleotide Synthesis to Enable Organ Growth. EMBO J. 2018, 37, e100294. [Google Scholar] [CrossRef]
- Actis Dato, V.; Sánchez, M.C.; Chiabrando, G.A. LRP1 Mediates the IGF-1-Induced GLUT1 Expression on the Cell Surface and Glucose Uptake in Müller Glial Cells. Sci. Rep. 2021, 11, 4742. [Google Scholar] [CrossRef]
- Fang, J.; Zhou, S.-H.; Fan, J.; Yan, S.-X. Roles of Glucose Transporter-1 and the Phosphatidylinositol 3-kinase/Protein Kinase B Pathway in Cancer Radioresistance. Mol. Med. Rep. 2015, 11, 1573–1581. [Google Scholar] [CrossRef]
- Wang, F.; Li, L.; Zhang, Z. Platelet Isoform of Phosphofructokinase Promotes Aerobic Glycolysis and the Progression of Non-small Cell Lung Cancer. Mol. Med. Rep. 2021, 23, 74. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Li, X.; Liu, J.; Qian, L.; Ke, H.; Yao, C.; Tian, W.; Liu, Y.; Zhang, J. Expression of PFKFB3 and Ki67 in Lung Adenocarcinomas and Targeting PFKFB3 as a Therapeutic Strategy. Mol. Cell Biochem. 2018, 445, 123–134. [Google Scholar] [CrossRef]
- Gupta, S.; Ramjaun, A.R.; Haiko, P.; Wang, Y.; Warne, P.H.; Nicke, B.; Nye, E.; Stamp, G.; Alitalo, K.; Downward, J. Binding of Ras to Phosphoinositide 3-Kinase P110alpha Is Required for Ras-Driven Tumorigenesis in Mice. Cell 2007, 129, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Medarde, A.; Santos, E. Ras in Cancer and Developmental Diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.; Roy, D.; Sarkar Bhattacharya, S.; Jin, L.; Jung, D.; Zhang, S.; Kalogera, E.; Staub, J.; Wang, Y.; Xuyang, W.; et al. Therapeutic Targeting of PFKFB3 with a Novel Glycolytic Inhibitor PFK158 Promotes Lipophagy and Chemosensitivity in Gynecologic Cancers. Int. J. Cancer 2019, 144, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Lypova, N.; Dougherty, S.M.; Lanceta, L.; Chesney, J.; Imbert-Fernandez, Y. PFKFB3 Inhibition Impairs Erlotinib-Induced Autophagy in NSCLCs. Cells 2021, 10, 1679. [Google Scholar] [CrossRef]
- Nissler, K.; Petermann, H.; Wenz, I.; Brox, D. Fructose 2,6-Bisphosphate Metabolism in Ehrlich Ascites Tumour Cells. J. Cancer Res. Clin. Oncol. 1995, 121, 739–745. [Google Scholar] [CrossRef]
- Nelson, D.L.; Cox, M.M. Lehninger Principles of Biochemistry, 8th ed.; W.H. Freeman: New York, NY, USA, 2021. [Google Scholar]
- Mosaoa, R.; Kasprzyk-Pawelec, A.; Fernandez, H.R.; Avantaggiati, M.L. The Mitochondrial Citrate Carrier SLC25A1/CIC and the Fundamental Role of Citrate in Cancer, Inflammation and Beyond. Biomolecules 2021, 11, 141. [Google Scholar] [CrossRef]
- Icard, P.; Fournel, L.; Coquerel, A.; Gligorov, J.; Alifano, M.; Lincet, H. Citrate Targets FBPase and Constitutes an Emerging Novel Approach for Cancer Therapy. Cancer Cell Int. 2018, 18, 175. [Google Scholar] [CrossRef]
- Díaz-Ruiz, R.; Avéret, N.; Araiza, D.; Pinson, B.; Uribe-Carvajal, S.; Devin, A.; Rigoulet, M. Mitochondrial Oxidative Phosphorylation Is Regulated by Fructose 1,6-Bisphosphate. A Possible Role in Crabtree Effect Induction? J. Biol. Chem. 2008, 283, 26948–26955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Chen, R.; Yu, Z.; Li, R.; Li, J.; Zhao, X.; Song, S.; Liu, J.; Huang, G. Dichloroacetate Restores Drug Sensitivity in Paclitaxel-Resistant Cells by Inducing Citric Acid Accumulation. Mol. Cancer 2015, 14, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.-A.; Zhang, X.-D.; Guo, X.-Y.; Xian, S.-L.; Lu, Y.-F. 3-Bromopyruvate and Sodium Citrate Target Glycolysis, Suppress Survivin, and Induce Mitochondrial-Mediated Apoptosis in Gastric Cancer Cells and Inhibit Gastric Orthotopic Transplantation Tumor Growth. Oncol. Rep. 2016, 35, 1287–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Zhou, J.; Yan, X.; Bi, X.; Liang, J.; Lu, S.; Luo, L.; Zhou, D.; Yin, Z. Citrate Activates Autophagic Death of Prostate Cancer Cells via Downregulation CaMKII/AKT/MTOR Pathway. Life Sci. 2021, 275, 119355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Varin, E.; Allouche, S.; Lu, Y.; Poulain, L.; Icard, P. Effect of Citrate on Malignant Pleural Mesothelioma Cells: A Synergistic Effect with Cisplatin. Anticancer. Res. 2009, 29, 1249–1254. [Google Scholar]
- Lincet, H.; Kafara, P.; Giffard, F.; Abeilard-Lemoisson, E.; Duval, M.; Louis, M.-H.; Poulain, L.; Icard, P. Inhibition of Mcl-1 Expression by Citrate Enhances the Effect of Bcl-XL Inhibitors on Human Ovarian Carcinoma Cells. J. Ovarian Res. 2013, 6, 72. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Zhang, X.; Wang, T.; Xian, S.; Lu, Y. 3-Bromopyruvate and Sodium Citrate Induce Apoptosis in Human Gastric Cancer Cell Line MGC-803 by Inhibiting Glycolysis and Promoting Mitochondria-Regulated Apoptosis Pathway. Biochem. Biophys. Res. Commun. 2016, 475, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Sakamoto, K. Citric Acid Promoted Melanin Synthesis in B16F10 Mouse Melanoma Cells, but Inhibited It in Human Epidermal Melanocytes and HMV-II Melanoma Cells via the GSK3β/β-Catenin Signaling Pathway. PLoS ONE 2020, 15, e0243565. [Google Scholar] [CrossRef]
- Xu, X.; LI, B.; Huang, P.; Wan, X.; QIN, Y.; Zhou, L.; Liu, H.; BAI, H.; GAO, Y.; Wang, C.; et al. Citrate Induces Apoptosis of the Acute Monocytic Leukemia U937 Cell Line through Regulation of HIF-1α Signaling. Mol. Med. Rep. 2013, 8, 1379–1384. [Google Scholar] [CrossRef] [Green Version]
- Hung, K.-C.; Wang, S.-G.; Lin, M.-L.; Chen, S.-S. Citrate-Induced P85α–PTEN Complex Formation Causes G2/M Phase Arrest in Human Pharyngeal Squamous Carcinoma Cell Lines. Int. J. Mol. Sci. 2019, 20, 2105. [Google Scholar] [CrossRef] [Green Version]
- Hanai, J.I.; Doro, N.; Seth, P.; Sukhatme, V.P. ATP Citrate Lyase Knockdown Impacts Cancer Stem Cells in Vitro. Cell Death Dis. 2013, 4, e696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Shi, J.; Lin, Q.; Ma, X.; Pang, Y.; Mao, H.; Li, R.; Lu, W.; Wang, Y.; Liu, P. Corrigendum: Targeting ACLY Attenuates Tumor Growth and Acquired Cisplatin Resistance in Ovarian Cancer by Inhibiting the PI3K-AKT Pathway and Activating the AMPK-ROS Pathway. Front. Oncol. 2021, 11, 742374. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simula, L.; Alifano, M.; Icard, P. How Phosphofructokinase-1 Promotes PI3K and YAP/TAZ in Cancer: Therapeutic Perspectives. Cancers 2022, 14, 2478. https://doi.org/10.3390/cancers14102478
Simula L, Alifano M, Icard P. How Phosphofructokinase-1 Promotes PI3K and YAP/TAZ in Cancer: Therapeutic Perspectives. Cancers. 2022; 14(10):2478. https://doi.org/10.3390/cancers14102478
Chicago/Turabian StyleSimula, Luca, Marco Alifano, and Philippe Icard. 2022. "How Phosphofructokinase-1 Promotes PI3K and YAP/TAZ in Cancer: Therapeutic Perspectives" Cancers 14, no. 10: 2478. https://doi.org/10.3390/cancers14102478
APA StyleSimula, L., Alifano, M., & Icard, P. (2022). How Phosphofructokinase-1 Promotes PI3K and YAP/TAZ in Cancer: Therapeutic Perspectives. Cancers, 14(10), 2478. https://doi.org/10.3390/cancers14102478