Whole-Exome Sequencing Reveals Clinical Potential of Circulating Tumor DNA from Peritoneal Fluid and Plasma in Endometrial Cancer

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
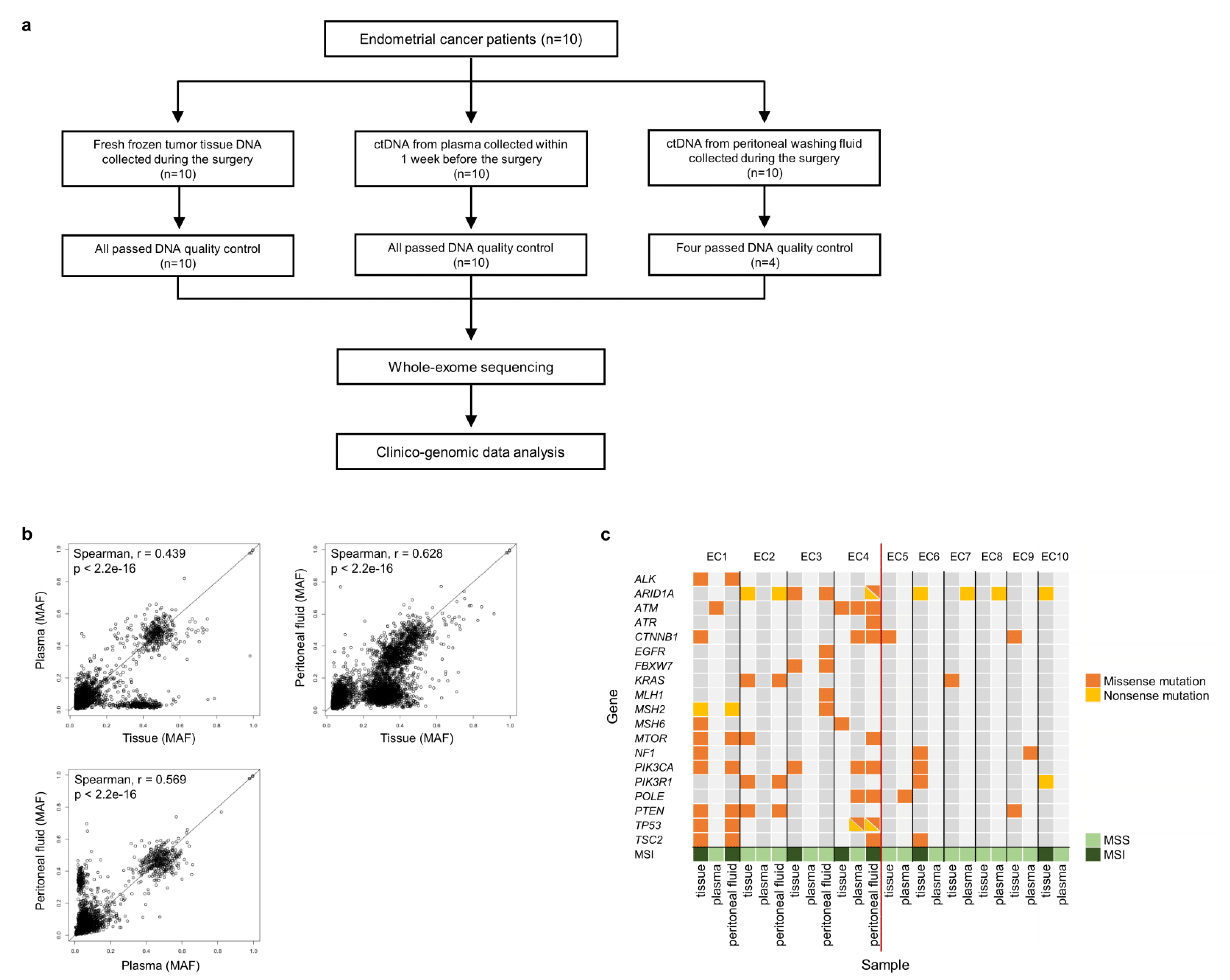
2.1. Samples
2.2. Preparation of ctDNA and gDNA
2.3. WES and Somatic Mutations
2.4. Mutational Signature Analysis
2.5. Copy Number Alteration
2.6. Microsatellite Instability
2.7. Immunohistochemistry
3. Results
3.1. Patients, Samples, and Clinical Data
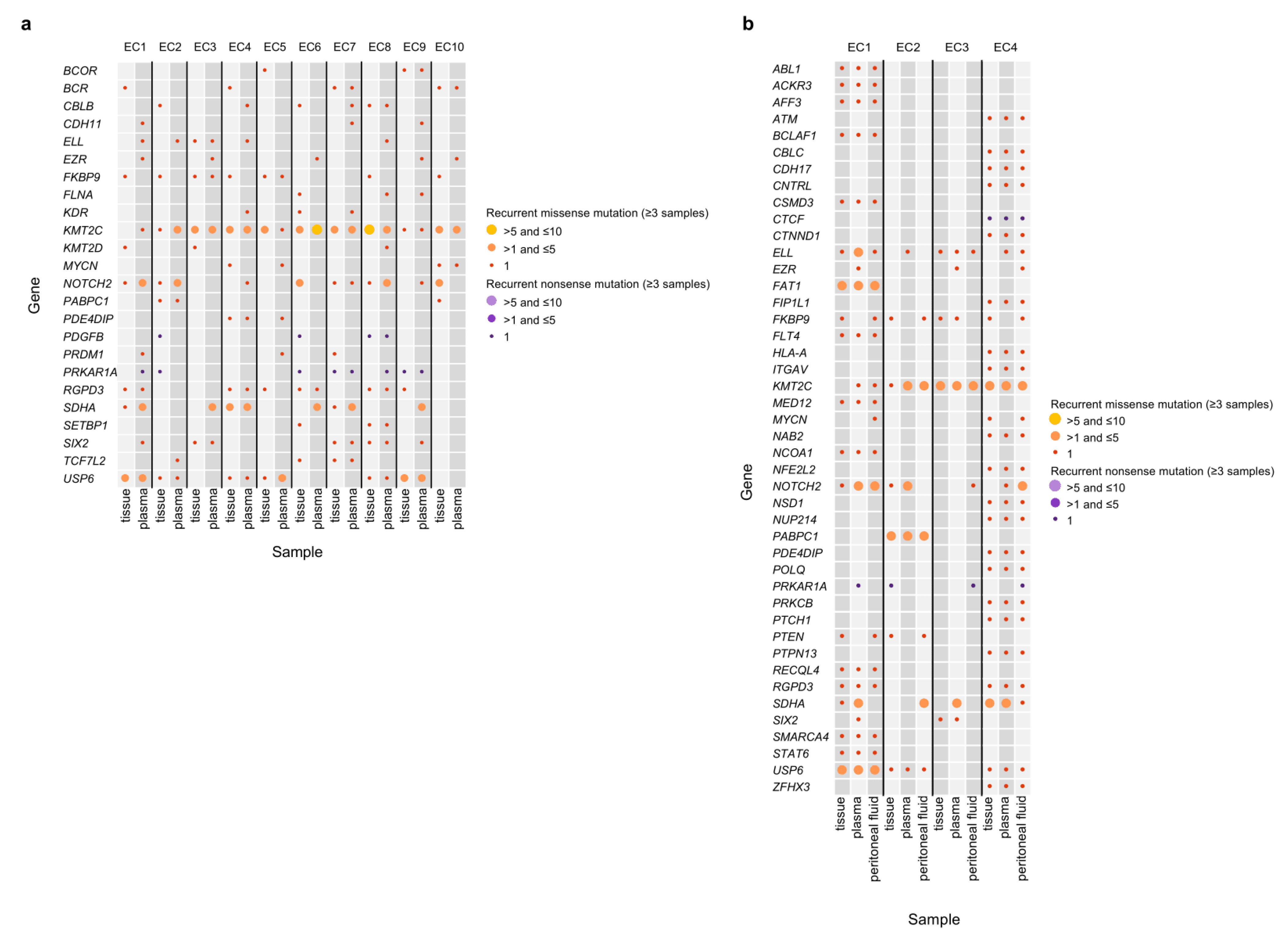
3.2. Mutation Profiles of the Peritoneal Fluid ctDNA, Plasma ctDNA, and Tumor gDNA Derived from EC Patients
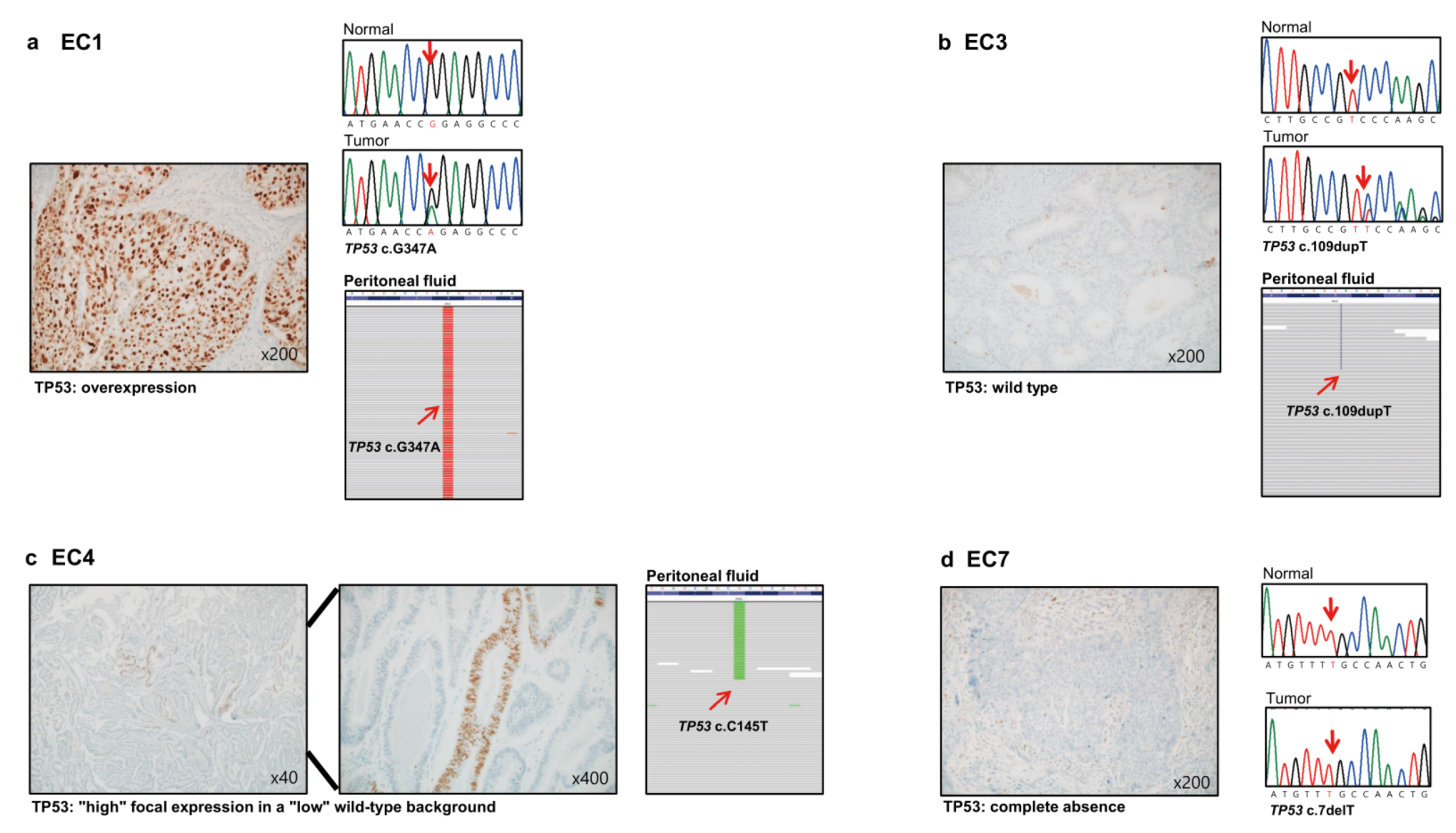
3.3. Immunohistochemistry
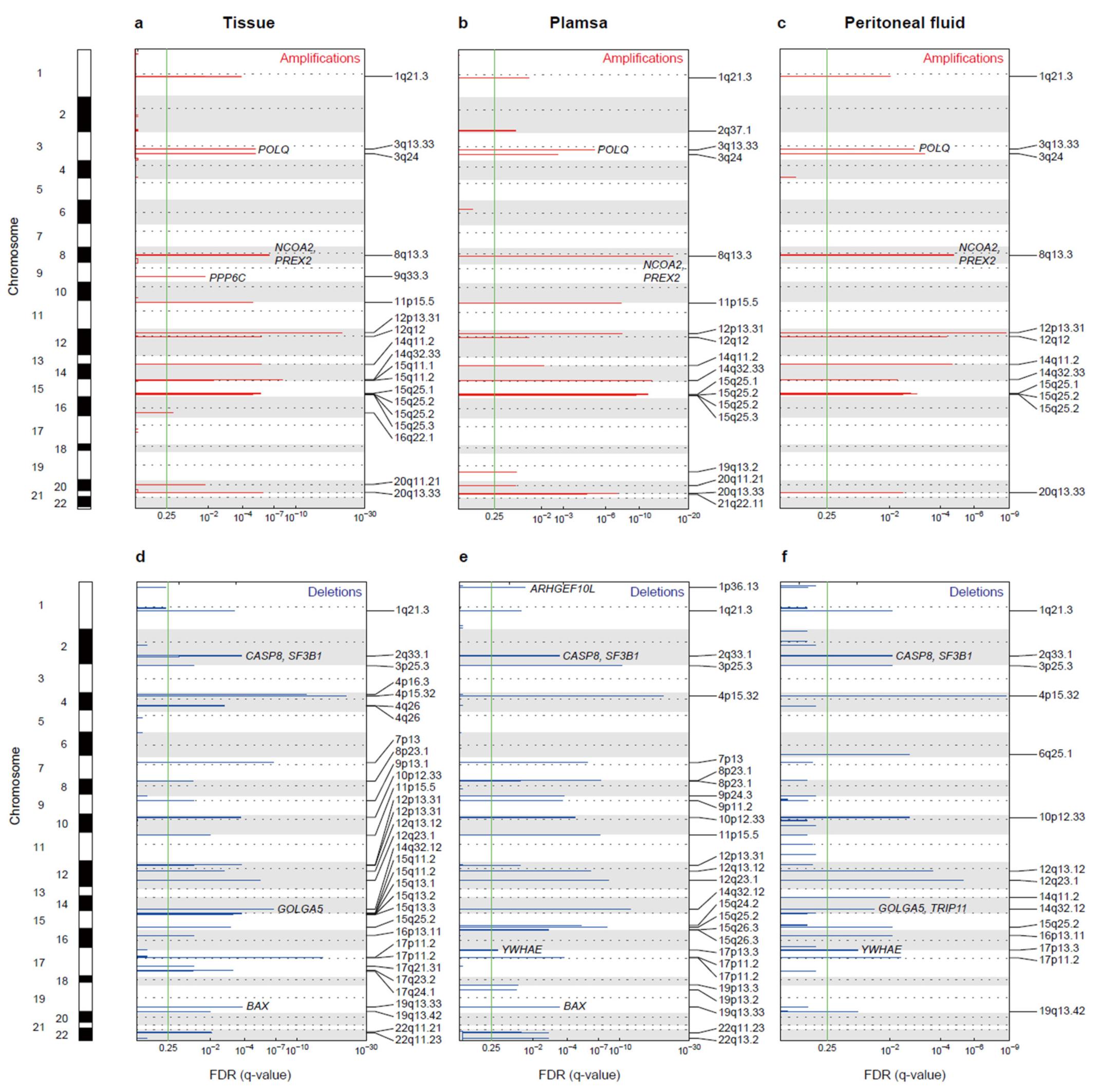
3.4. Copy Number Alterations
3.5. Microsatellite Instability
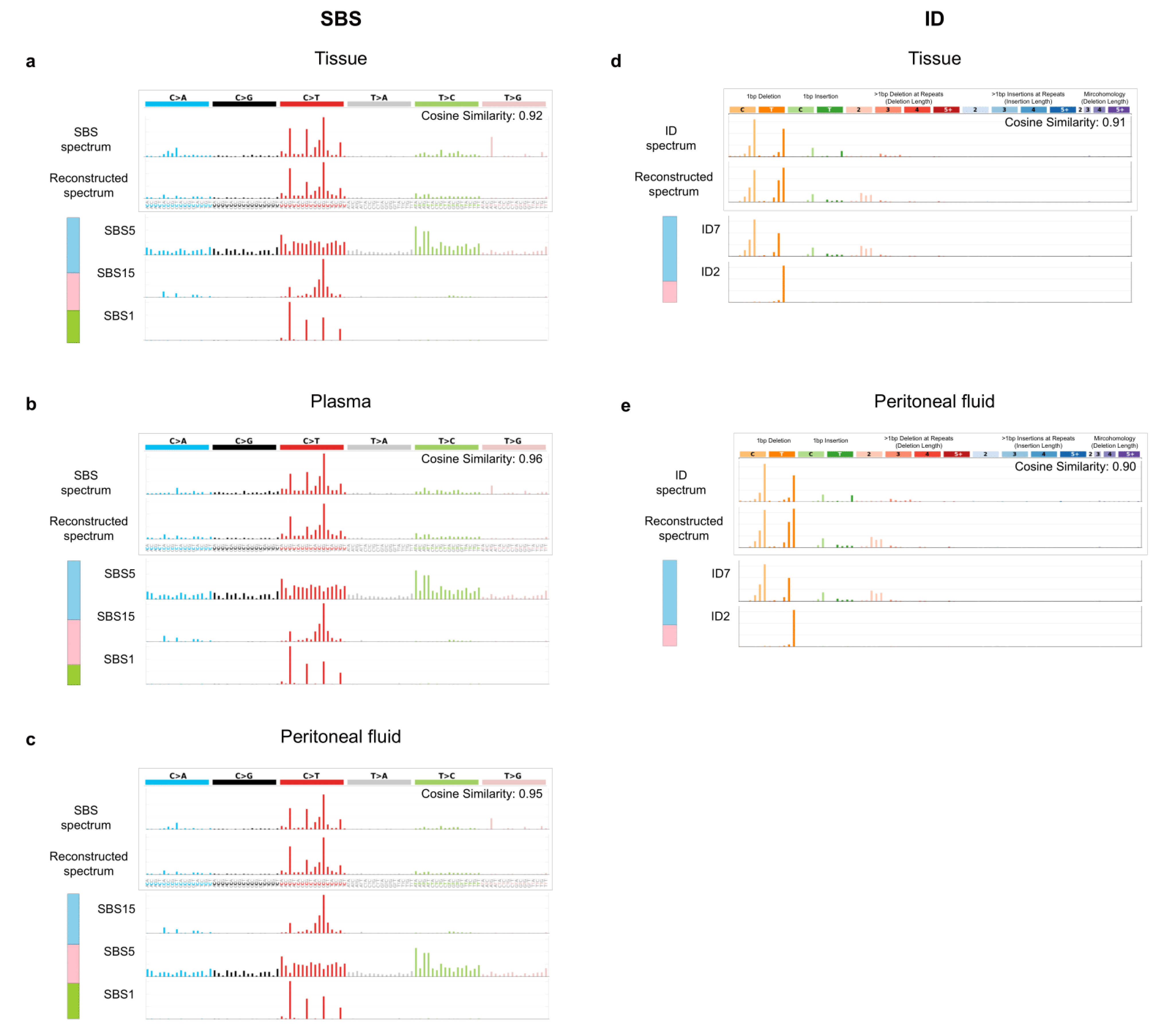
3.6. Mutational Signatures
3.7. Relation of Positive Peritoneal Fluid and Its Genetic Characteristics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levine, D.A. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- León-Castillo, A.; de Boer, S.M.; Powell, M.E.; Mileshkin, L.R.; Mackay, H.J.; Leary, A.; Nijman, H.W.; Singh, N.; Pollock, P.M.; Bessette, P.; et al. Molecular Classification of the PORTEC-3 Trial for High-Risk Endometrial Cancer: Impact on Prognosis and Benefit From Adjuvant Therapy. J. Clin. Oncol. 2020, 38, 3388–3397. [Google Scholar] [CrossRef] [PubMed]
- Talhouk, A.; McConechy, M.K.; Leung, S.; Yang, W.; Lum, A.; Senz, J.; Boyd, N.; Pike, J.; Anglesio, M.; Kwon, J.S.; et al. Confirmation of ProMisE: A simple, genomics-based clinical classifier for endometrial cancer. Cancer 2017, 123, 802–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelloo, E.; Nout, R.A.; Osse, E.M.; Jürgenliemk-Schulz, I.J.; Jobsen, J.J.; Lutgens, L.C.; van der Steen-Banasik, E.M.; Nijman, H.W.; Putter, H.; Bosse, T.; et al. Improved Risk Assessment by Integrating Molecular and Clinicopathological Factors in Early-stage Endometrial Cancer-Combined Analysis of the PORTEC Cohorts. Clin. Cancer Res. 2016, 22, 4215–4224. [Google Scholar] [CrossRef] [Green Version]
- Stelloo, E.; Bosse, T.; Nout, R.A.; MacKay, H.J.; Church, D.N.; Nijman, H.W.; Leary, A.; Edmondson, R.J.; Powell, M.E.; Crosbie, E.J.; et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Mod. Pathol. 2015, 28, 836–844. [Google Scholar] [CrossRef] [Green Version]
- Concin, N.; Matias-Guiu, X.; Vergote, I.; Cibula, D.; Mirza, M.R.; Marnitz, S.; Ledermann, J.; Bosse, T.; Chargari, C.; Fagotti, A.; et al. ESGO/ESTRO/ESP guidelines for the management of patients with endometrial carcinoma. Int. J. Gynecol. Cancer 2021, 31, 12–39. [Google Scholar] [CrossRef]
- Mandel, P. Les acides nucleiques du plasma sanguin chez 1 homme. C R Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Perakis, S.; Speicher, M.R. Emerging concepts in liquid biopsies. BMC Med. 2017, 15, 75. [Google Scholar] [CrossRef] [Green Version]
- Husain, H.; Nykin, D.; Bui, N.; Quan, D.; Gomez, G.; Woodward, B.; Venkatapathy, S.; Duttagupta, R.; Fung, E.; Lippman, S.M.; et al. Cell-Free DNA from Ascites and Pleural Effusions: Molecular Insights into Genomic Aberrations and Disease Biology. Mol. Cancer Ther. 2017, 16, 948–955. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Xu, B.; Qi, L.; Zhu, D.; Liu, B.; Wei, J. Next-generation sequencing reveals mutational accordance between cell-free DNA from plasma, malignant pleural effusion and ascites and directs targeted therapy in a gastric cancer patient. Cancer Biol. Ther. 2019, 20, 15–20. [Google Scholar] [CrossRef]
- Han, M.-R.; Lee, S.H.; Park, J.Y.; Hong, H.; Ho, J.Y.; Hur, S.Y.; Choi, Y.J. Clinical Implications of Circulating Tumor DNA from Ascites and Serial Plasma in Ovarian Cancer. Cancer Res. Treat. 2020, 52, 779–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayo-de-Las-Casas, C.; Velasco, A.; Sanchez, D.; Martínez-Bueno, A.; Garzón-Ibáñez, M.; Gatius, S.; Ruiz-Miró, M.; Gonzalez-Tallada, X.; Llordella, I.; Tresserra, F.; et al. Detection of somatic mutations in peritoneal lavages and plasma of endometrial cancer patients: A proof-of-concept study. Int. J. Cancer 2020, 147, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Bolivar, A.M.; Luthra, R.; Mehrotra, M.; Chen, W.; Barkoh, B.A.; Hu, P.; Zhang, W.; Broaddus, R.R. Targeted next-generation sequencing of endometrial cancer and matched circulating tumor DNA: Identification of plasma-based, tumor-associated mutations in early stage patients. Mod. Pathol. 2019, 32, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Moss, E.L.; Gorsia, D.N.; Collins, A.; Sandhu, P.; Foreman, N.; Gore, A.; Wood, J.; Kent, C.; Silcock, L.; Guttery, D.S. Utility of Circulating Tumor DNA for Detection and Monitoring of Endometrial Cancer Recurrence and Progression. Cancers 2020, 12, 2231. [Google Scholar] [CrossRef]
- Robinson, K.M.; Hawkins, A.S.; Santana-Cruz, I.; Adkins, R.S.; Shetty, A.C.; Nagaraj, S.; Sadzewicz, L.; Tallon, L.J.; Rasko, D.A.; Fraser, C.M.; et al. Aligner optimization increases accuracy and decreases compute times in multi-species sequence data. Microb. Genom. 2017, 3, e000122. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Seo, J.; Park, J.; Nam, J.-Y.; Choi, A.; Ignatius, J.S.; Bjornson, R.D.; Chae, J.-H.; Jang, I.-J.; Lee, S.; et al. Korean Variant Archive (KOVA): A reference database of genetic variations in the Korean population. Sci. Rep. 2017, 7, 4287. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, E.N.; Huang, M.N.; Mahto, U.; Barnes, M.; Stratton, M.R.; Rozen, S.G.; Alexandrov, L.B. SigProfilerMatrixGenerator: A tool for visualizing and exploring patterns of small mutational events. BMC Genom. 2019, 20, 685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering Signatures of Mutational Processes Operative in Human Cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Han, M.-R.; Shin, S.; Park, H.-C.; Kim, M.S.; Lee, S.H.; Jung, S.H.; Song, S.Y.; Lee, S.H.; Chung, Y.-J. Mutational signatures and chromosome alteration profiles of squamous cell carcinomas of the vulva. Exp. Mol. Med. 2018, 50, e442. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef] [Green Version]
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef]
- Hemming, M.L.; Klega, K.S.; Rhoades, J.; Ha, G.; Acker, K.E.; Andersen, J.L.; Thai, E.; Nag, A.; Thorner, A.R.; Raut, C.P.; et al. Detection of Circulating Tumor DNA in Patients With Leiomyosarcoma With Progressive Disease. JCO Precis. Oncol. 2019, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Westin, S.N.; Broaddus, R.R. Personalized therapy in endometrial cancer: Challenges and opportunities. Cancer Biol. Ther. 2012, 13, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.A.; Howitt, B.E.; Wu, C.J.; Konstantinopoulos, P.A. Predicted neoantigen load in non-hypermutated endometrial cancers: Correlation with outcome and tumor-specific genomic alterations. Gynecol. Oncol. Rep. 2016, 19, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Nair, N.; Camacho-Vanegas, O.; Rykunov, D.; Dashkoff, M.; Camacho, S.C.; Schumacher, C.A.; Irish, J.C.; Harkins, T.T.; Freeman, E.; Garcia, I.; et al. Genomic Analysis of Uterine Lavage Fluid Detects Early Endometrial Cancers and Reveals a Prevalent Landscape of Driver Mutations in Women without Histopathologic Evidence of Cancer: A Prospective Cross-Sectional Study. PLoS Med. 2016, 13, e1002206. [Google Scholar] [CrossRef]
- Kautto, E.A.; Bonneville, R.; Miya, J.; Yu, L.; Krook, M.A.; Reeser, J.W.; Roychowdhury, S. Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget 2017, 8, 7452–7463. [Google Scholar] [CrossRef] [Green Version]
- Kommoss, S.; McConechy, M.K.; Kommoss, F.; Leung, S.; Bunz, A.; Magrill, J.; Britton, H.; Kommoss, F.; Grevenkamp, F.; Karnezis, A.; et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann. Oncol. 2018, 29, 1180–1188. [Google Scholar] [CrossRef]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.-Y.; Andre, F.; et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef] [Green Version]
- Middha, S.; Zhang, L.; Nafa, K.; Jayakumaran, G.; Wong, D.; Kim, H.R.; Sadowska, J.; Berger, M.F.; Delair, D.F.; Shia, J.; et al. Reliable Pan-Cancer Microsatellite Instability Assessment by Using Targeted Next-Generation Sequencing Data. JCO Precis. Oncol. 2017, 1, 1–17. [Google Scholar] [CrossRef]
- Köbel, M.; Ronnett, B.M.; Singh, N.; Soslow, R.A.; Gilks, C.B.; McCluggage, W.G. Interpretation of P53 Immunohistochemistry in Endometrial Carcinomas: Toward Increased Reproducibility. Int. J. Gynecol. Pathol. 2019, 38 (Suppl. 1), S123–S131. [Google Scholar] [CrossRef]
- Colombo, N.; Creutzberg, C.; Amant, F.; Bosse, T.; González-Martín, A.; Ledermann, J.; Marth, C.; Nout, R.; Querleu, D.; Mirza, M.R.; et al. ESMO-ESGO-ESTRO Consensus Conference on Endometrial Cancer: Diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, 16–41. [Google Scholar] [CrossRef]
- Billingsley, C.C.; Cohn, D.E.; Mutch, D.G.; Stephens, J.A.; Suarez, A.A.; Goodfellow, P.J. Polymerase ɛ (POLE) mutations in endometrial cancer: Clinical outcomes and implications for Lynch syndrome testing. Cancer 2015, 121, 386–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Shao, H.; Ban, X.; Zhang, H.; You, Y.; Zhou, N.; Mao, X.; Zhao, H.; Chen, J.; Lu, Z. Detection of POLE Subtypes in High-Grade Endometrioid Carcinoma by BaseScope-ISH Assay. Front. Oncol. 2019, 9, 831. [Google Scholar] [CrossRef] [PubMed]
- Soumerai, T.E.; Donoghue, M.T.A.; Bandlamudi, C.; Srinivasan, P.; Chang, M.T.; Zamarin, D.; Cadoo, K.A.; Grisham, R.N.; O’Cearbhaill, R.E.; Tew, W.P.; et al. Clinical Utility of Prospective Molecular Characterization in Advanced Endometrial Cancer. Clin. Cancer Res. 2018, 24, 5939–5947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, D.I.; Sowers, L.C.; Loeb, L.A. Reverse chemical mutagenesis: Identification of the mutagenic lesions resulting from reactive oxygen species-mediated damage to DNA. Proc. Natl. Acad. Sci. USA 1994, 91, 6609–6613. [Google Scholar] [CrossRef] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [Green Version]
- Ashley, C.W.; Da Cruz Paula, A.; Kumar, R.; Mandelker, D.; Pei, X.; Riaz, N.; Reis-Filho, J.S.; Weigelt, B. Analysis of mutational signatures in primary and metastatic endometrial cancer reveals distinct patterns of DNA repair defects and shifts during tumor progression. Gynecol. Oncol. 2019, 152, 11–19. [Google Scholar] [CrossRef]
- Vermij, L.; Smit, V.; Nout, R.; Bosse, T. Incorporation of molecular characteristics into endometrial cancer management. Histopathology 2020, 76, 52–63. [Google Scholar] [CrossRef]
- León-Castillo, A.; Gilvazquez, E.; Nout, R.; Smit, V.T.; McAlpine, J.N.; McConechy, M.; Kommoss, S.; Brucker, S.Y.; Carlson, J.W.; Epstein, E.; et al. Clinicopathological and molecular characterisation of “multiple-classifier” endometrial carcinomas. J. Pathol. 2020, 250, 312–322. [Google Scholar] [CrossRef]
- Pinelli, C.; Bogani, G.; Casarin, J.; Raspagliesi, F.; Ghezzi, F. Malignant peritoneal cytology in endometrial cancer. Eur. J. Cancer 2020, 140, 147–148. [Google Scholar] [CrossRef]
- García-Sanz, P.; Triviño, J.C.; Mota, A.; López, M.P.; Colás, E.; Rojo-Sebastián, A.; García, Á.; Gatius, S.; Ruiz, M.; Prat, J.; et al. Chromatin remodelling and DNA repair genes are frequently mutated in endometrioid endometrial carcinoma. Int. J. Cancer 2017, 140, 1551–1563. [Google Scholar] [CrossRef]
- Salvesen, H.B.; Carter, S.L.; Mannelqvist, M.; Dutt, A.; Getz, G.; Stefansson, I.M.; Raeder, M.B.; Sos, M.L.; Engelsen, I.B.; Trovik, J.; et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc. Natl. Acad. Sci. USA 2009, 106, 4834–4839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanekar, Y.; Sadasivam, S. In silico analysis reveals a shared immune signature in CASP8-mutated carcinomas with varying correlations to prognosis. PeerJ 2019, 7, e6402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, R.R.; Pluciennik, A.; Burdett, V.; Modrich, P.L. DNA mismatch repair: Functions and mechanisms. Chem. Rev. 2006, 106, 302–323. [Google Scholar] [CrossRef]
- Makker, V.; Rasco, D.; Vogelzang, N.J.; Brose, M.S.; Cohn, A.L.; Mier, J.; Di Simone, C.; Hyman, D.M.; Stepan, D.E.; Dutcus, C.E.; et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: An interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 711–718. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Pathologic Diagnosis | Age (yr) | FIGO Stage (2009) | Histologic Grade | Tumor Size (cm) | LVSI | Peritoneal Fluid | Recurrence/Progression | Current Status | Follow Up Period (mo) | Adjuvant Treatment | Others |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EC1 | Endometroid adenocarcinoma | 62 | IIIA | G3 | 7 | (−) | (−) | NED | 62 | Chemotherapy and radiotherapy | ||
| EC2 | Endometroid adenocarcinoma | 50 | IIIC1 | G1 | 8 | (+) | (+) | NED | 53 | Chemotherapy and radiotherapy | ||
| EC3 | Endometroid adenocarcinoma | 74 | IB | G1 | 3 | (+) | (−) | NED | 47 | Radiotherapy | ||
| EC4 | Endometroid adenocarcinoma | 55 | IIIC2 | G3 | 10 | (−) | (+) | Recur 15 mo after the surgery. | NED | 47 | Chemotherapy and radiotherapy | |
| EC5 | Endometroid adenocarcinoma | 45 | II | G1 | 9 | (+) | (−) | NED | 47 | Radiotherapy | ||
| EC6 | Endometroid adenocarcinoma | 54 | IB | G3 | 5 | (+) | (−) | NED | 45 | Radiotherapy | ||
| EC7 | Endometroid adenocarcinoma | 67 | IIIC1 | G3 | 4 | (+) | (−) | NED | 42 | Chemotherapy and radiotherapy | ||
| EC8 | Endometroid adenocarcinoma | 74 | IIIC1 | G2 | 6 | (+) | (+) | Progression 5 mo after the surgery. | NED | 39 | Chemotherapy and radiotherapy | |
| EC9 | Endometroid adenocarcinoma | 59 | IVB | G1 | 7 | (+) | (−) | Never NED | Under treatment | 33 | Chemotherapy and radiotherapy | Incomplete surgical resection and lung metastasis |
| EC10 | Endometroid adenocarcinoma | 51 | IIIC2 | G2 | 12 | (+) | (−) | NED | 29 | Chemotherapy, radiotherapy |
| EC1 | EC2 | EC3 | EC4 | EC5 | EC6 | EC7 | EC8 | EC9 | EC10 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| P53 mut ** | Tissue | mut | wt | mut | wt | wt | wt | mut | wt | wt | wt |
| Plasma | wt | wt | wt | mut | wt | wt | wt | wt | wt | wt | |
| Peritoneal fluid * | mut | wt | mut | mut | |||||||
| TP53 immunohisto-chemistry | Tissue | Overexpression | wt | wt | wt *** | wt | wt | Complete absence | wt | wt | wt |
| POLE mut ** | Tissue | wt | wt | wt | wt | wt | wt | wt | wt | wt | wt |
| Plasma | wt | mut | wt | EDM | mut | wt | wt | wt | wt | wt | |
| Peritoneal fluid * | wt | wt | wt | EDM | |||||||
| MSI status | Tissue | MSI-H | MSS | MSI-H | MSI-H | MSS | MSI-H | MSS | MSS | MSS | MSI-H |
| Plasma | MSS | MSS | MSS | MSS | MSS | MSS | MSS | MSS | MSS | MSS | |
| Peritoneal fluid * | MSI-H | MSS | MSS | MSI-H | |||||||
| Copy number low/ high | Tissue | low | low | low | low | low | high | low | low | low | low |
| Plasma | low | low | low | low | low | low | low | low | low | low | |
| Peritoneal fluid * | low | low | low | low | |||||||
| Risk group (ESGO 2016) | Tissue | High | High | High-intermediate | High | High | High | High | High | Metastatic | High |
| Risk group (ESGO 2021), molecular classification known | Tissue | High | High | High-intermediate | High | High | High-intermediate | High | High | Metastatic | High |
| Tissue (N = 10) | Plasma (N = 10) | Peritoneal Fluid (N = 4) | |
|---|---|---|---|
| Somatic mutation number | 7908.6/tumor | 8127.1/tumor | 12,241.3/tumor |
| Mutation allele frequency | 0.05/variant | 0.20/variant | 0.07/variant |
| Nonsilent mutation number | 8087.3/tumor | 7546.3/tumor | 12,526/tumor |
| Mutation allele frequency (nonsilent) | 0.07/variant | 0.19/variant | 0.09/variant |
| Silent mutation number | 861.2/tumor | 1150.4/tumor | 1517/tumor |
| Mutation allele frequency (silent) | 0.03/variant | 0.24/variant | 0.04/variant |
| Putative driver mutation number | 72.2/tumor | 99.7/tumor | 154.75/tumor |
| Representative driver mutation | FKBP9, KMT2C, USP6 | KMT2C, NOTCH2, SDHA, USP6 | KMT2C, NOTCH2, USP6 |
| No. of CNAs | 290 (median: 27.5) | 232 (median: 23.0) | 128 (median: 32.5) |
| Length of CNAs (Mb) | 2046.8 (median: 106.1) | 568.3 (median: 50.9) | 337.6 (median: 62.4) |
| Most recurrent copy gain | 12p | 8q | 12p |
| Most recurrent copy loss | 4p | 4p | 4p |
| MSI | MSI (n = 5) | MSI (n = 0) | MSI (n = 2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ju, H.-Y.; Ho, J.Y.; Kang, J.; Hur, S.Y.; Kim, S.; Choi, Y.J.; Han, M.-R. Whole-Exome Sequencing Reveals Clinical Potential of Circulating Tumor DNA from Peritoneal Fluid and Plasma in Endometrial Cancer. Cancers 2022, 14, 2506. https://doi.org/10.3390/cancers14102506
Ju H-Y, Ho JY, Kang J, Hur SY, Kim S, Choi YJ, Han M-R. Whole-Exome Sequencing Reveals Clinical Potential of Circulating Tumor DNA from Peritoneal Fluid and Plasma in Endometrial Cancer. Cancers. 2022; 14(10):2506. https://doi.org/10.3390/cancers14102506
Chicago/Turabian StyleJu, Hye-Yeon, Jung Yoon Ho, Jun Kang, Soo Young Hur, Sejin Kim, Youn Jin Choi, and Mi-Ryung Han. 2022. "Whole-Exome Sequencing Reveals Clinical Potential of Circulating Tumor DNA from Peritoneal Fluid and Plasma in Endometrial Cancer" Cancers 14, no. 10: 2506. https://doi.org/10.3390/cancers14102506
APA StyleJu, H. -Y., Ho, J. Y., Kang, J., Hur, S. Y., Kim, S., Choi, Y. J., & Han, M. -R. (2022). Whole-Exome Sequencing Reveals Clinical Potential of Circulating Tumor DNA from Peritoneal Fluid and Plasma in Endometrial Cancer. Cancers, 14(10), 2506. https://doi.org/10.3390/cancers14102506