1. Introduction
How a single cytokine can induce a wide variety of downstream responses in the same cell or in different cell types is a longstanding question in the signaling field [
1,
2]. Transforming growth factor β (TGF-β) is a prototypical cytokine in this respect, as its biological responses are highly dependent on cellular context, such as cell type and its differentiation state and/or the presence of other extracellular cues [
3,
4]. TGF-β is part of a larger family of cytokines that also include activins and bone morphogenetic proteins (BMPs), which signal via selective transmembrane type I and type II serine/threonine kinase receptors and intracellular SMAD transcription factors. Examples of contextual-dependent biological roles include the action of TGF-β family ligands as morphogens during development, in which the SMAD activation duration and intensity are critical determinants. TGF-β/activin can elicit opposing tumor promoting and suppressing effects in cancer [
4], which is attributed, in part, to changes in SMAD-interacting (transcription) factors or rerouting through non-canonical signaling pathways [
5,
6,
7,
8]. Thus, TGF-β signaling spatio-temporal dynamics are determined by cell intrinsic and extrinsic factors, negative feedback loops, and self-enabling responses; however, how these contribute to different biological outputs is not well understood.
Given the multifunctional properties and wide variety of responses of TGF-β signaling, it is likely that the dynamic control of signaling plays a role [
5,
9]. Indeed, TGF-β signaling was shown to have a graded early response, and a switch-like sustained response [
10]. Others have shown that the response to a TGF-β concentration is transient and adaptive, resulting in an instructive signal for patterning [
11]. However, many other questions remain, such as whether dynamic TGF-β signaling responses are heterogeneous, oscillating, and/or coupled to cellular behavior such as cell migration.
Signal encoded states are not merely on-or-off but are highly dynamic [
2]. This is likely to be of key importance during multistep biological responses, such as cell invasion, endothelial sprouting, etc. As such, signaling can be encoded by dynamic properties, such as delay, duration, fold-change, or frequency, resulting in extra layers to modulate signaling output [
1,
2,
9]. To detect these dynamic signals, fluorescent-based signaling reporters can be used to visualize and decode these changes in signaling pathways over time [
1].
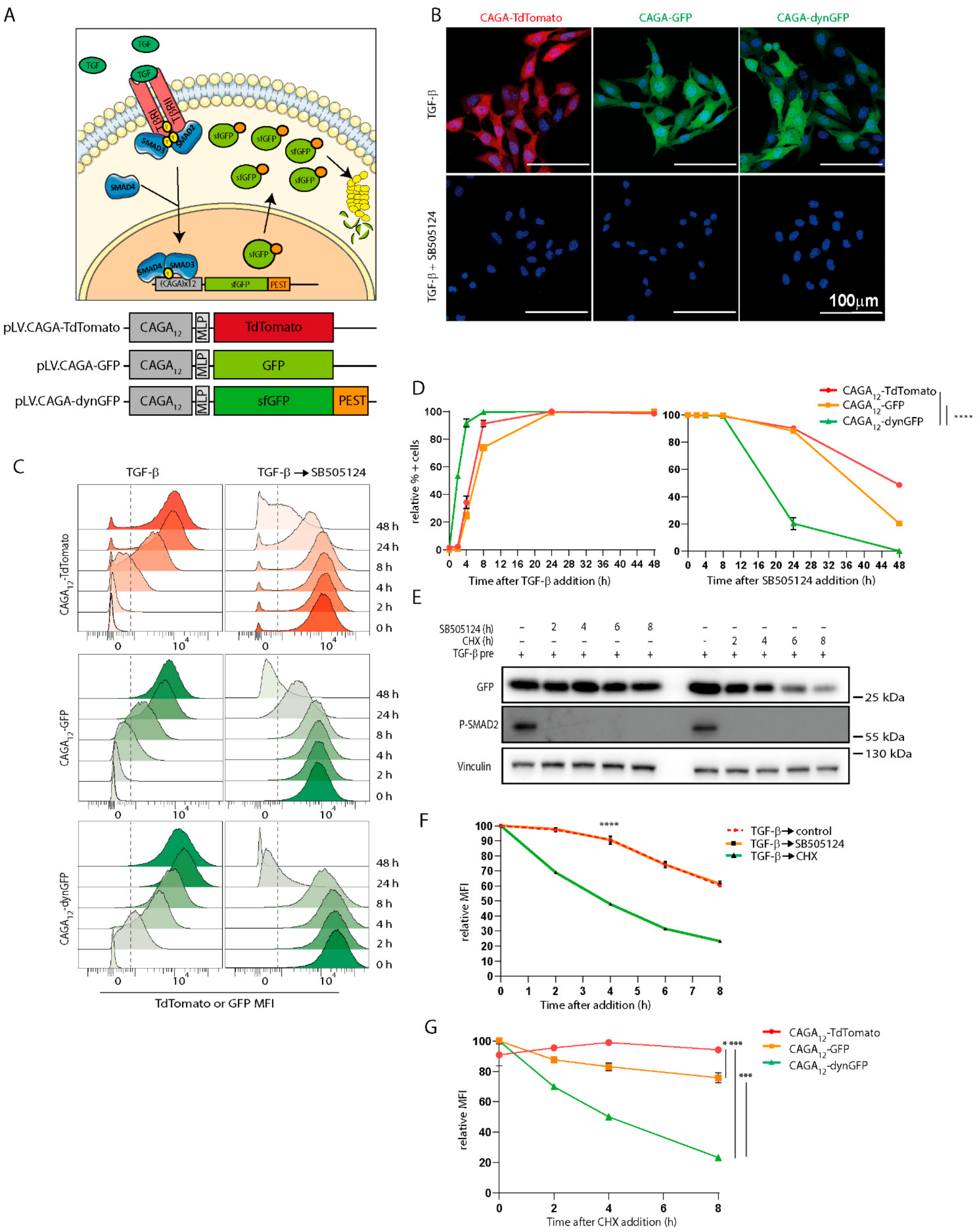
As mentioned above, TGF-β family members signal via intracellular SMAD transcription factors. Whereas activated TGF-β and activin type I receptors mediate the activation of receptor-regulated (R) mother against decapentaplegic homolog 2 (R-SMAD2) and R-SMAD3 by their carboxy (C) terminal phosphorylation on two serine residues, BMP receptors induce the C-terminal phosphorylation of R-SMAD 1, -5, and -8 proteins. Activated R-SMADs form heterodimeric and trimeric complexes with SMAD4 that translocate to the nucleus. The nuclear SMAD complexes bind to specific sequences in promoters of selected target genes, thereby regulating their transcription. The affinity of SMADs for DNA is low, and SMADs require other DNA-binding transcription factors to efficiently bind to target promoters [
4]. Target genes for TGF-β/SMAD3 signaling are, among others,
SERPINE1 (Serine proteinase inhibitor, Clade 1, Member 1, which encodes for plasminogen activator inhibitor 1 (PAI-1) protein),
SMAD7, and connective tissue growth factor (
CTGF). Within the promoter region of
SERPINE1, three 5′-CAGA-3′ boxes were identified as direct binding sites for SMAD3 and 4 [
12]. By multimerizing this SMAD Binding Element, a 5′-CAGA-3′ a transcriptional reporter construct was created: 12-times 5′-CAGA-3′ box repeat and a minimal adenovirus major late promoter (MLP) were cloned upfront of a reporter cDNA encoding luciferase [
12]. This is a widely used reporter system to interrogate TGF-β/SMAD3 signaling response in cells by preparing cell lysates of in vitro cultures or in vivo by bioluminescence but lacks good temporal or single cell resolution [
13,
14].
Most signaling studies have been performed using population average methods. The advent of live cell imaging methods and fluorescent tagging of proteins or reporters driving the expression of fluorescent proteins have allowed to monitor signaling dynamics in single cells in real time. TGF-β signaling dynamics have been measured by translocation of fluorescently-tagged (phosphorylated) SMAD2, 3, and 4 from cytoplasm to nucleus, one of the early events of the TGF-β signaling pathway [
9,
11,
15,
16,
17,
18]. These studies and others have provided great insights in direct SMAD shuttling and signaling dynamics. Nuclear translocation of SMAD4, rather than SMAD2, can be correlated to a transcriptional response [
15,
17]. Transcriptional output can also be determined by other factors than the nuclear translocation of SMAD proteins [
19,
20].
Fluorescent-based transcriptional reporters allow for single cell visualization over time, and report for transcriptional output of signaling. The latter is more downstream of measuring SMAD2/3 phosphorylation or R-SMAD- or SMAD4 nuclear accumulation, and therefore, more subject to crosstalk with other signaling pathways. For example, SMAD transcriptional activity is determined by the partner transcription factors of which the activation state is dependent on activation by specific stimuli [
4]. Luwor et al. coupled a TdTomato fluorophore to a 12x 5′-CAGA-3′ box to monitor effects of TGF-β on breast cancer cell migration in vitro [
21]. This TGF-β/SMAD3 fluorescent reporter can give insights in transcription at a single time point and allows for cellular tracking at a single cell level. However, the stability of fluorescent proteins may hinder dynamic visualization over time, and therefore, this (and similar) reporters are not suited for visualizing transcription in a dynamic and temporal manner at a single cell level. Taken together, there is a clear need for a dynamic transcriptional TGF-β reporter to assess TGF-β signaling in vitro, as well as in living animals, at a single cell resolution. Measurement of the TGF-β/SMAD3 transcriptional temporal behavior in living cells will provide new insights into how dynamical features, such as duration, intensity, and frequency of temporal signals, lead to specific cellular responses.
2. Materials and Methods
2.1. Reagents
TGF-β family ligands, dissolved in 4 mM HCl/0.1% recombinant bovine serum albumin (BSA): TGF-β1 (1 ng/mL unless indicated otherwise, 7666, R&D Systems, Minneapolis, MN, USA), TGF-β2 (1 ng/mL unless indicated otherwise; kind gift from Joachim Nickel, University of Würzburg, Würzburg, Germany), TGF-β3 (1 ng/mL unless indicated otherwise; kind gift from A. Hinck, University of Pittsburgh; this ligand is generally used, unless stated otherwise), Activin A (50 ng/mL, R&D Systems), BMP2 (50 ng/mL; kind gift from Joachim Nickel, University of Würzburg, Würzburg, Germany), BMP6 (50 ng/mL; kind gift from Prof. Slobodan Vukicevic, University of Zagreb, Zagreb, Croatia), BMP7 (50 ng/mL; kind gift from Prof. Slobodan Vukicevic, University of Zagreb, Zagreb, Croatia), BMP9 (50 ng/mL, R&D Systems). Selective small molecule TβRI kinase inhibitor SB505124 (1 mM, DMSO, #3263, Tocris, Bristol, UK) and cycloheximide (50 ng/mL, C1988, Sigma, Darmstadt, Germany) were used.
2.2. Generation of Lentiviral TGF-β/SMAD3 Reporter Constructs
First, the pGLS3-CAGA
12-LUC plasmid [
12,
22] was used to generate the pGLS3-CAGA
12-TdTomato construct by replacing the luciferase gene for the TdTomato gene using
BamHI/
HindIII and
XbaI. Next,
ClaI and
EcoRV/
SmaI restriction sites were used to clone the CAGA
12-TdTomato fragment from the pGLS3-CAGA
12 into a pLV- phosphoglycerate kinase 1 (PGK)-puromycin vector to create the pLV-CAGA
12-TdTomato reporter, expressing TdTomato under transcriptional control of 12 5′-CAGA-3′ SMAD3 response elements.
For subcloning, the CAGA12-MLP element was amplified through polymerase chain reaction (PCR) using the CAGA12-MLP FW and REV (containing Age1 restriction site) primers. The purified CAGA12-MLP PCR product was digested using Cla1 and AgeI, whereas the pLV vector was digested using Cla1 and EcoR1. The digested pLV vector was treated with shrimp alkaline phosphatase (SAP) to prevent self-ligation. The isolated pLV vector and CAGA12-MLP promoter were used for subsequent cloning.
eGFP was amplified from CMV-d2eGFP-empty (gift from Phil Sharp (Addgene plasmid # 26164;
http://n2t.net/addgene:26164 (accessed on 3 September 2021); RRID:Addgene_26164)) [
23] through PCR using T7 FW GFP rev (including
EcoR1 restriction site) primers (
Table S1). The PCR product was digested using
Age1 and
EcoRI, and was thereafter subcloned with the pLV vector and CAGA
12-MLP promoter using a 3-way ligation (1:1:1) through incubation overnight at 4 °C with T4 ligase to generate pLV-CAGA
12-eGFP expressing eGFP under transcriptional control of 12 5′-CAGA-3′ SMAD3 response elements.
To enhance eGFP folding speed and, thus, the appearance of fluorescence signal, we created a third construct with a superfolding GFP (sfGFP) harboring the super folding mutations (S30R, Y39N, N105, Y145F, I171V, A206V) in the eGFP. sfGFP-C1 (gift from Michael Davidson and Geoffrey Waldo (Addgene plasmid # 54579;
http://n2t.net/addgene:54579 (accessed on 3 September 2021); RRID:Addgene_54579)) [
24] was isolated using
EcoR1 and
Bsrg1. Additionally, we engineered a PEST domain at the C-terminus of the construct to promote degradation by the proteasome once TGF-β signaling ceases. CMV-d2eGFP-empty (gift from Phil Sharp (Addgene plasmid # 26164;
http://n2t.net/addgene:26164 (accessed on 3 September 2021); RRID:Addgene_26164)) [
23] was used to amplify the destabilizing domain through PCR using D2 domain FW and REV primers (
Table S1). The purified product was digested using
EcoRI and
BsrgI. The purified destabilizing domain and sfGFP-C1 products were ligated by incubating at 4 °C with T4 ligase. The resulting dynGFP-C1 was digested with
Age1 and
EcoRI to create the dynGFP insertion into the final vector. The pLV vector, CAGA
12-MLP promoter, and dynGFP were ligated using a 3-way ligation using incubation overnight at 4 °C with T4 ligase. This generated CAGA
12-dynGFP, expressing superfolder GFP with destabilizing domain under transcriptional control of 12 5′-CAGA-3′ SMAD3 response elements. Plasmid sequences were verified using sanger sequencing using the eGFP FW, eGFP rev, and PGK rev primers (
Supplementary Table S1).
2.3. Cells and Culture Methods
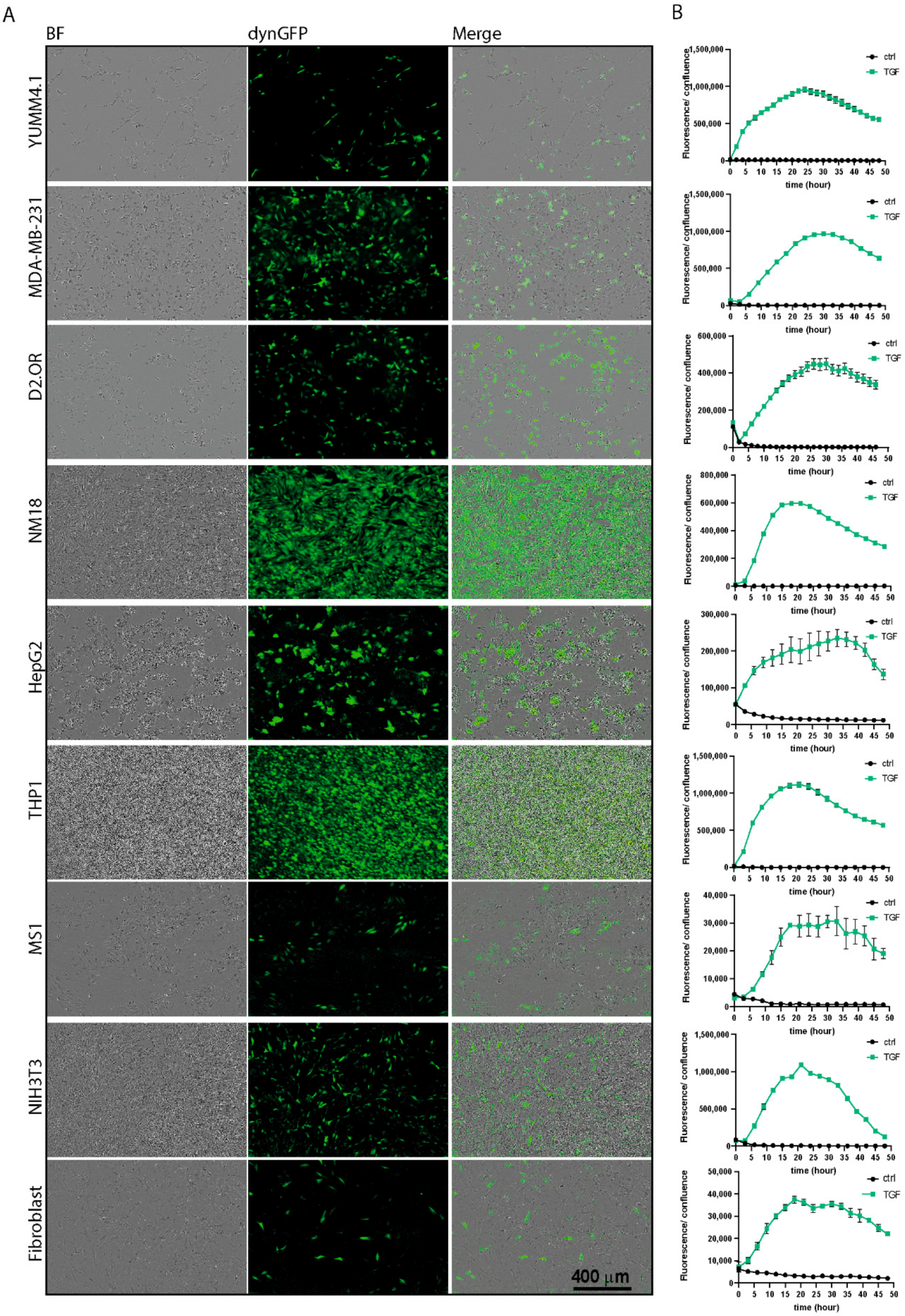
The following cell lines were used: human embryonic kidney cells HEK293T (CRL3216™, ATCC, Manassas, VA, USA), mouse melanoma cells B16F10 (CRL-6475™, ATCC) and YUMM4.1 (CRL-3366™, ATCC), human breast cancer cells MDA-MB-231 (HTB-26™, ATCC), mouse breast cancer D2.OR cells (kind gift from F.R. Miller [
25]), normal mammary gland NMuMG derivative NM18 [
26], human liver cancer cells (HepG2) (HB-8065™, ATCC), human acute monocytic leukemia cells (THP1) (TIB-202™, ATCC), mouse endothelial cells (MS1) (CRL-2279™, ATCC), pancreatic stellate cells (RLT-PSC) (kind gift from prof M. Löhr and dr. R. Heuchel [
27], Stockholm, Sweden), and primary mouse fibroblasts. HEK293T, B16F10, MDA-MB-231, D.2OR, NM18, NIH3T3, HEPG2, and primary mouse fibroblasts were maintained in DMEM containing 10% FBS and 100 U/mL penicillin-streptomycin (15140122; Thermo Fisher Scientific, Waltham, MA, USA). THP1 cells were maintained in RPMI-1640 medium containing 10% FBS and 100 U/mL penicillin-streptomycin (15140122; Thermo Fisher Scientific, Waltham, MA, USA). Mouse melanoma cells (YUMM4.1) and RLT-PSC cells were maintained in Dulbecco’s Modified Eagle Medium DMEM/F12 containing 10% fetal bovine serum (FBS) and 100 U/mL penicillin-streptomycin. MS1 cells were maintained in MEMa containing 10% fetal bovine serum (FBS) and 100 U/mL penicillin-streptomycin. All cell lines were cultured in 37 °C, 5% CO
2 incubators and routinely tested for the absence of mycoplasma infections. All human cell lines were authenticated using short tandem repeat (STR) profiling.
2.4. Generation of CAGA12-TdTomato, CAGA12-GFP and CAGA12-dynGFP Cell Lines
Lentiviruses were generated by HEK293T transfection with packaging constructs and the lentiviral constructs using standard protocols [
28]. Cells were exposed to lentivirus containing pLV.CAGA
12-TdTomato, pLV.CAGA
12-GFP or pLV.CAGA
12-dynGFP, and polybrene (8 mg/mL) for 24 h, and were allowed to recover for 1–3 days. Transduced cells were then selected using 1 mg/mL puromycin for >1 week.
B16F10 cells and RLT-PSC cells containing the CAGA12-reporter were sorted using the BD FACS Aria III 4L (BD Biosciences, San Jose, CA, USA) at the Flow Cytometry Core Facility (FCF) of Leiden University Medical Center (LUMC) in Leiden, Netherlands, through sorting the top 20% fluorescence-expressing cells after 24 h (B16F10 and RLT-PSC pLV.CAGA12-dynGFP) or 48 h (B16F10 pLV.CAGA12-TdTomato and B16F10 pLV. CAGA12-GFP) of TGF-β3 (1 ng/mL) stimulation. To generate clonal cell lines of B16F10 pLV.CAGA12-dynGFP, cells were transduced with lentiviruses containing pLV.CMV-RFP-NLS. Transduced cells were stimulated with TGF-β3 (1 ng/mL) for 24 h prior to FACS sorting of GFP+/RFP+ single cells into a 96-well plate using the BD FACS Aria III 4L (BD Biosciences, San Jose, CA, USA) at the Flow Cytometry Core Facility (FCF) of Leiden University Medical Center (LUMC) in Leiden, Netherlands. Single cells were seeded in one 96-well and expanded, and 3 clonal lines (clone 7, clone 8, clone 9) were selected for further experiments. FACS-sorted cells were used within 1 month of sorting.
2.5. Colorectal Cancer Organoid Culture
The colorectal cancer patient-derived organoid (PDO) cultures used in this study were previously established and characterized [
29]. The culture conditions used were previously described [
29]. Briefly, organoids were cultured in 75% Matrigel
® domes (growth factor reduced basement membrane matrix, phenol red-free, Corning
® (Corning, NY, USA). The culture medium conditions consist of Advanced DMEM/F12 (Thermo Fisher Scientific, Waltham, MA, USA) with 1% Penicillin/Streptomycin (Thermo Fisher Scientific, Waltham, MA, USA), 10 mM HEPES, 1% Glutamax (Thermo Fisher Scientific, Waltham, MA, USA), 100 ng/mL Noggin conditioned medium, 1X B27, 1.25 mM N-acetyl-L-cysteine, 0.5 mM A83-01 (Tocris), 10 mM SB202190. In general, the organoid medium was refreshed every 2–3 days. After around 1–2 weeks of culture, organoids were harvested and incubated with TrypLE Express (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C for 1–5 min to obtain a single cell suspension. Residual TrypLE was washed out using Advanced DMEM/F12. Subsequently, the single cells were resuspended in 75% Matrigel/25% Advanced DMEM/F12 domes, and plated in 6-, 12-, or 24- well culture plates.
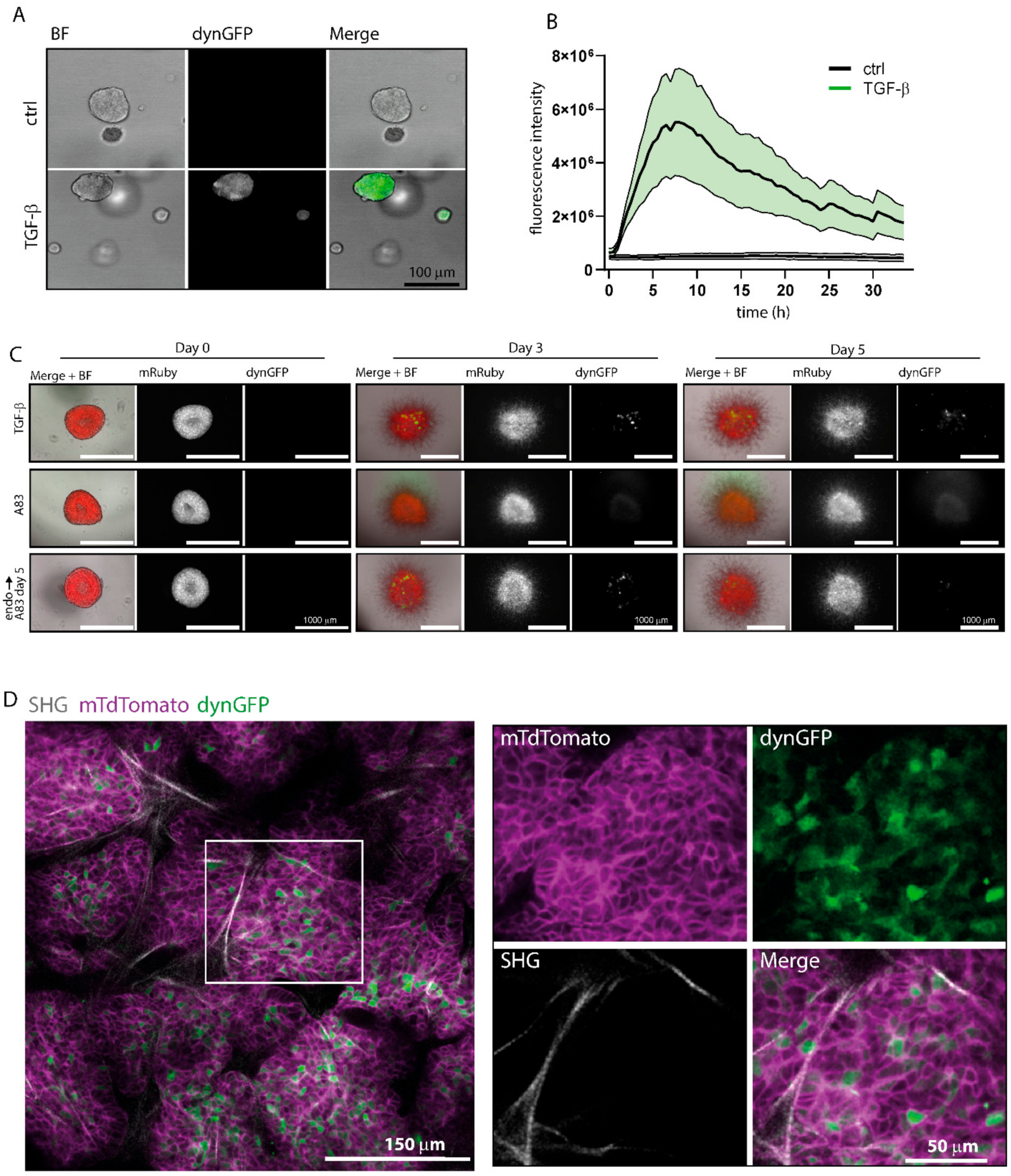
The PDOs were stably transduced with the pLV.CAGA12-dynGFP reporter, and selected with puromycin and subsequent FACS-sorting of 24 h stimulated TGF-β1 (5 ng/mL) dynGFP-positive cells. PDOs were dissociated into single cells, and subsequently plated in a 384-well glass bottom plate (4581, Corning). All TGF-β1 stimulation experiments were conducted in organoid medium without noggin, A83-01, and N-acetyl-L-cysteine. PDOs were allowed to grow for 7–10 days, after which, the experimental medium (5 ng/mL carrier-free recombinant human TGF-β1, R&D 240-B) was added. Organoids were scanned for 72 h with 30 min time intervals using a 40X water objective lens on a Leica SP8X confocal microscope equipped with a culture chamber for adequate conditions (37 °C and 5.0% CO2 overflow).
2.6. Pancreatic Organoid Co-Culture
Resection-derived pancreatic tumor organoid PDO1 (HUB-08-B2-029B), was obtained from the Hubrecht Organoid Technology Biobank (HUB) [
30]. Organoids were cultured in Matrigel
® (growth factor reduced basement membrane matrix, phenol red-free, Corning
®) domes overlaid with advanced DMEM/F12 supplemented with Glutamine/Glutamax (200 nM 100×, Invitrogen, Waltham, MA, USA), 100 UI/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific, Waltham, MA, USA), B27 (1x, Invitrogen), recombinant Noggin, 100 ng/mL (Peprotech, London, UK), Nicotinamide 10 mM (Sigma, Darmstadt, Germany), N-Acetyl Cysteine 1.25 mM, (Sigma, Darmstadt, Germany), Primocin 50 μg (Invivogen, San Diego, CA, USA), Gastrin 10 nM (Sigma, Darmstadt, Germany), recombinant R-Spondin 3 200 ng/mL (Peprotech), recombinant FGF10A 100 ng/mL (Peprotech), recombinant EGF (5 ng/mL), A83-01 500 nM (Selleckhem, Munich, Germany), WNT-surrogate–Fc fusion protein 0.5 nM (Utrecht Protein Express, Utrecht, The Netherlands). The pancreatic stellate cell line RLT-PSC was a kind gift from Professor M. Löhr and Doctor R. Heuchel (Stockholm, Sweden) [
27]. RLT-PSC cells were transduced with pLentiPGK Hygro DEST H2B-mRuby2 (pLentiPGK Hygro DEST H2B-mRuby2 was a gift from Markus Covert, Addgene plasmid #90236) and selected with 100 mg/mL Hygromycin B (Sigma, Darmstadt, Germany) for one week. Subsequently, RLT-PSC-mRuby2 was transduced with pLV.CAGA
12-dynGFP and selected with 1 mg/mL Puromycin (Invitrogen). After puromycin selection, cells were stimulated with TGF-β3 (1 ng/mL) for 24 h, and FACs-sorted for the top 20% cells expressing dynGFP.
For co-culture experiments, PDOs and RLT-PSCs were trypsinized to single cell suspensions; PDO and RLT-PSC cells were then mixed together in a 1:10 ratio in RLT-PSC medium containing 1% Matrigel. The mixtures were plated in ultra-low attachment 96-well round-bottom plates (Corning, NY, USA; n = 1000 PDO and n = 10,000 RLT-PSC cells per well), centrifuged (1200 RPM, 1 min), and incubated overnight (37 °C, 5% CO2). The next day, the formed aggregates were collected, and the supernatant was removed. Individual aggregates were taken up in 100% Matrigel and plated in flat-bottom 96-well plates (Greiner, Kremsmünster, Austria) which were pre-coated with 50 mL Matrigel. PDO medium was added after polymerization of the Matrigel. The co-culture experiment was performed without the presence of the TβRI kinase inhibitor A83-01, unless specifically indicated. The different experimental conditions containing either TGF-β1 (5 ng/mL), A83-01 (500 nM, Selleckhem), or control pancreatic organoid medium without A83-01 (endogenous condition) were added and refreshed every 2 days for a period of 5 days. At day 4, A83-01 (500 nM, Selleckhem) was added to the endogenous condition. Photos were taken daily using the Cytation 5 live-cell imaging microscope (Agilent, Santa Clara, CA, USA).
2.7. Flow Cytometry Analysis
Cells were treated with TGF-β3 (1 ng/mL), SB505124 (1 mM), cycloheximide (50 mg/mL), or vehicle solvent control for the indicated time points. Samples treated with TGF-β type I receptor kinase inhibitor SB505124 were pre-stimulated with TGF-β 24 h (CAGA12-dynGFP) or 48 h (CAGA12-TdTomato/CAGA12-GFP) prior to SB505124 treatment. To collect cells, cells were trypsinized and resuspended in DMEM containing 10% FBS and pen-strep. Sample were washed and resuspended in PBS containing 0.2% BSA. For cell cycle analysis, cells were treated with Hoechst33342 (20 mg/mL) (#62249, Thermo Fisher) for 45 min at 37 °C prior to trypsinization of cells, and were treated with TGF-β (1 ng/mL) for 2 h prior to trypsinization of cells. Fluorescence was directly measured by BD LSRII (BD Biosciences, Franklin Lakes NJ, USA). Flow cytometry data were analyzed using FlowJoTM (v10, BD Biosciences, Franklin Lakes, NJ, USA).
2.8. rt-qPCR
B16F10 CAGA
12-TdTomato/CAGA
12-GFP/CAGA
12-dynGFP cells were treated with TGF-β3 (1 ng/mL) and/or SB505124 (1 mM). Samples treated with SB505124 were pre-stimulated with TGF-β 24 h (CAGA
12-dynGFP) or 48 h (CAGA
12-TdTomato/CAGA
12-GFP) prior to SB505124 treatment. Total RNA extraction was performed using the NucleoSpin RNA II kit (Macherey-Nagel, Dűren, Germany). An amount of 1 mg of RNA was used for cDNA synthesis using the Revert Aid First Strand cDNA synthesis kit (Thermo Fisher). Quantitative PCR was performed using SYBBR GoTaq qPCR master mix (Promega, Madison, WI, USA) and 0.5 mM of primers targeting (dyn)GFP, SMAD7,
Serpine1,
Ctgf,
Gapdh, and
Hprt (
Table S2). RT-qPCR was performed on the CFX Connect Real-Time PCR detection system (Bio-Rad, Hercules, CA, USA). Measurements were performed in technical duplicate and independent biological triplicate; target gene expression was normalized for
Gapdh and
Hprt expression.
2.9. Western Blotting
Protein lysates were harvested in Laemmli buffer (0.12 M Tris-HCl pH 6.8, 4% SDS, 20% glycerol, 35 mM β-mercaptoethanol, and bromophenol blue) and boiled for 5 min. Western blotting was performed using standard procedures. Membranes were blocked in 5% non-fat dry milk for 1 h at room temperature and incubated with primary antibody in 2.5% non-fat dry milk overnight at 4 °C. The primary antibodies used were GFP (sc-8334, Santa Cruz), PAI-1 (ab222754, Abcam, Cambridge, UK), phospho-SMAD1 and 2 [
31], GAPDH, and Vinculin (V9131, Sigma, Darmstadt, Germany). As the C-termini of SMAD3, and BMP R-SMAD1 and -5 are identical, p-SMAD2 was used to assess TβRI activity. Experiments were performed in independent biological triplicate; representative images are shown.
2.10. CAGA12-Luciferase Assay
B16F10 cells were seeded in a 24-well plate and transfected with plasmids containing the CAGA12-luc transcriptional reporter and SV40 promoter-controlled β-Gal cDNA expression vector expressing β-galactosidase using polyethylenimine (PEI). Cells were washed and stimulated with TGF-β3 (1 ng/mL) and/or SB505124 (1 mM) overnight. Cells were lysed, and luciferase and β-galactosidase signal were measured with a PerkinElmer plate reader. Luciferase signals were corrected for signals in untransfected control samples and normalized for b-galactosidase expression. Experiments were performed by integrating results of three independent wells.
2.11. Live Cell Imaging Using Incucyte®
CAGA12-GFP/CAGA12-dynGFP reporter B16F10 cells were plated in a 96-well plate (#3595, Corning) and were stimulated with TGF-β3 (1 ng/mL or otherwise indicated), SB505124 (1 mM), cycloheximide (50 mg/mL), or vehicle solvent control for the indicated time points. Measurements were taken every 2 h using the Incucyte
® S3 live-cell analysis system using brightfield and a GFP laser with 300 msec exposure. Fluorescence intensity and confluency were measured using Incucyte
® S3 Software. Experiments were performed by integrating results of three independent wells. Signal over background (s/b) scores were calculated by dividing the average fluorescence intensity upon TGF-β stimulation by unstimulated controls at the indicated timepoints. Z′ scores were calculated using the following formula:
2.12. Confocal Microscopy
B16F10 CAGA12-TdTomato/CAGA12-GFP/CAGA12-dynGFP were plated in a chambered coverslip (80826 Ibidi, Gräfelfing Germany). Cells were treated with TGF-β3 (1 ng/mL) and/or SB505124 (1 mM) for 24 h (CAGA12-dynGFP) or 48 h (CAGA12-TdTomato/CAGA12-GFP). Live cells were stained with Hoechst 33342 (10 mg/mL) (#62249, Thermo Fisher) for 15 min, prior to imaging. Confocal images were taken on a Leica TCS SP5 confocal laser scanning microscope.
2.13. Intravital Imaging Experiments
Experimental Model and Subject Details
All mice were housed under a 12 h light/dark cycle and under specific pathogen-free laboratory conditions, receiving food and water ad libitum. All experiments were approved and performed according to the guidelines of the Animal Welfare Committees of the Netherlands Cancer Institute, protocol code 9.1.957, The Netherlands.
MMTV-PyMT,
R26-mTmG, and E-cad-mCFP transgenic mice were crossed to generate donor organoids, i.e., MMTV-
PyMT and
R26-mTmG mice [
32,
33] (mice were purchased from Jackson Laboratory, Sacramento, CA, USA); Ecad-mCFP mice [
34] were a gift from Hans Clevers. The process of isolation and culturing of the donor mouse organoids are described in [
35]. As acceptors for orthotopic transplantation, 8-to-16 weeks old female Friend Virus B NIH Jackson mice (referred in the text to as FVB) were used. For transplantation, 250,000 single cells were plated 3 days prior to transplantation. At the day of transplantation, BME was dissolved by mechanical disruption, and cells were dissolved in 100 μL Basal Membrane Extract (BME) type 2 (RGF BME type 2 PathClear):PBS, and injected into the fat pad of the 4th mammary gland of acceptor mice.
2.14. PYMT Organoid Generation and Viral Transductions
MMTV-PyMT, R26-mTmG, and E-cad-mCFP transgenic mice spontaneously developed mammary tumors at the age of 8–14 weeks. Upon tumor formation, mice were sacrificed, and mammary tumor organoids were isolated from one donor. Mammary tumors were minced and enzymatically digested: gently shaken for 30 min at 37 °C in digestion mix (0.2% trypsin (from bovine pancreas, Sigma, Darmstadt, Germany) and 0.2% collagenase A (Roche, Basel, Swtizerland). The digested tumors were spun down, and cell fragments were embedded in Basal Membrane Extract (BME) type 2 (RGF BME type 2 PathClear). The mammary tumor organoid medium contained DMEM/F12 Glutamax (Thermo Fisher Scientific, Waltham, MA, USA, 2% B27 (Invitrogen), and 10 ng/mL FGF.
For lentiviral production, HEK293T cells (ATCC® CRL-1573™), were plated at 7 × 106 cells per 15 cm dish. After 24 h, cells were transfected with 32 mg of the pLV. CAGA12-dynGFP lentiviral plasmid, and the lentiviral packaging and envelope expression plasmids (pMDL, REV, VSV-G) were diluted in 1 mL of Optimem (Gibco) and 150 mL PEI. After mixing and 5 min incubation at RT, the mixture was added dropwise to the dish. Two days after transfection, the supernatant of transfected cells was harvested, filtered through 0.45 mm filters, and concentrated using Amnico Ultra centrifugal filter units (Merck, Darmstadt, Germany). For lentiviral organoid transduction, organoids were dissociated into single cells by 10 min digestion at 37 °C in TrypLE (Thermo Fisher Scientific, Waltham, MA, USA), followed by mechanical disruption. Single cells were resuspended in concentrated virus supplemented with Y-27632 (1/1000, #M1817, Abmole Bioscience, Houston, TX, USA) and polybrene, and spin-incubated at RT for 60 min at 600 rpm in a 48-well plate. After incubation for 6 h at 37 °C, the cell suspension was collected, and single cells were embedded in BME. The mammary tumor organoid medium was supplemented with Y-27632 (1/1000, #M1817, Abmole) until organoids recovered.
After recovery and expansion, organoids were treated for 48 h with 5 ng/mL TGF-β1. Organoids were collected, washed, and dissociated into single cells by enzymatic digestion for 10 min at 37 °C in TrypLE and mechanical disruption. Cells were resuspended in FACS buffer (PBS + 5 mM EDTA, 2% FBS), and the mTdTom+/GFP+ cell population was sorted on a FACS ARIA Fusion (BD biosciences) to >90% purity.
2.15. Single Cell Live Cell Imaging
Before conducting experiments, 100K cells B16F10 CAGA
12-dynGFP containing a pLV.CMV-RFP-NLS were seeded on fibronectin (0.1 mg/mL)-coated 14 mm-glass bottom dishes in DMEM medium without phenol red. Experiments were performed 16–24 h after plating on the glass-bottom dishes. The cells were simultaneously imaged using 460 nm (dynGFP; 10 mW/cm
2) and 532 nm (RFP-NLS10; mW/cm
2) excitation for 1 day at 1 frame per 3 min with the 1x objective (numerical aperture = 0.5) in the Ultrawide field-of-view optical microscope [
36]. The field of view size (~2.5 × 3.5 mm) was chosen for imaging containing a fraction of the cells. For the Clone 7 data, 2575 cells were detected by the end of day 1; for the Clone 9 data, 10748 cells were detected by the end of day 1. We have successfully tracked 83.1% (=2139/2575) of the Clone 7 population, and 96.9% (=10,414/10,748) of the Clone 9 population.
The movement of CAGA
12-dynGFP B16F10 cells expressing red fluorescent protein (RFP) containing a nuclear localization signal (NLS) was tracked by RFP signal and analyzed using the mTGMM (modified Tracking using Gaussian Mixture Model) cell-tracking algorithm [
36]. In brief, the intensity profile of individual nuclei/cell was modelled as a 2D Gaussian distribution. Nuclei tracking was performed by forwarding every Gaussian from time point
t to
t + 1 using Bayesian inference, with a priori knowledge that the position, shape, and overall intensity of nuclei could not change dramatically between two consecutive time points. After nuclei/cell detection and tracking, nuclei masks and positions over time were recorded. The RFP signal of individual cells was then extracted from the GFP channel over time using the nuclei/cell masks.
After segmentation and tracking of the cells, the GFP signal of individual cells from the GFP channel over time was extracted. A heatmap was generated to visualize the dynamical process over all cells of this dataset. The dataset could be further classified into groups via Euclidean distance-based hierarchical clustering analysis. After the analysis, two distinct groups were clearly observed: the first group had the GFP intensity trace going up at around 300 min, and the second group had the GFP intensity trace going up at around 700 min. We, therefore, called these two groups the early and late responders, respectively.
2.16. Statistical Analysis
Experiments were performed in independent biological triplicates, unless otherwise indicated. Error bars represent ± standard error of the mean (SEM), unless otherwise indicated. Statistics were calculated using GraphPad Prism 7 software.
4. Discussion
TGF-β is a highly context dependent cytokine that elicits its multifunctional effects via intracellular SMAD transcriptional effectors, of which the activity is determined by the integration of diverse extracellular and internal cues. Visualization of the dynamic aspects of TGF-β/SMAD-induced transcriptional responses will allow for refining and provide novel insights into the role of temporal behavior of SMAD signaling in the physio-pathological processes and consequences of pharmacological/(epi)genetic manipulation. However, the folding and degradation time of fluorescent proteins limits the ability of fluorescent transcriptional reporters to reflect dynamics in signaling. By using a quickly folded and highly unstable fluorescent GFP, we were able to create a TGF-β/SMAD3 reporter that allows the monitoring of SMAD3-driven transcriptional temporal responses in single living cells. Using this reporter in a clonal B16F10 melanoma cell line, we observed heterogeneity in TGF-β/SMAD3-induced transcriptional response, distinguishing slow, fast, and non-responders. We further highlight the use of this reporter in different 2D cell cultures and 3D organoid (co-) cultures, as well is in a biologically relevant context in vivo using intravital imaging. These observations will promote studies in which the activity/expression or localization of signaling molecules requires (single cell) time-lapse imaging of thousands of living cells, and studies on (endogenous) TGF-β signaling dynamics in vitro and in vivo.
To study TGF-β/SMAD-induced transcription, luciferase-based reporters coupled with multimerized SMAD3/4 binding elements have been used extensively in the literature, both in vitro and in vivo [
11,
12,
15,
44,
45,
46,
47]. Zhu and colleagues used the CAGA
12-luciferase reporter in breast cancer cells to study TGF-β/SMAD3 activity by measuring luminescence during tumorigenesis in vivo, whereas others created a transgenic CAGA
12-luciferase mouse, studying TGF-β/SMAD3 response to injury [
45,
48]. To achieve a more dynamic visualization with the CAGA
12-luciferase reporter, Sorre and colleagues coupled a nano luciferase (NLuc) as reporter to the CAGA
12 box and used microfluidic technology to apply an unstable Nluc substrate prior to image signal readout [
11]. This setup, however, requires specialized microfluidics equipment and operator expertise, and is therefore not easily adaptable by other laboratories. Moreover, luciferase-based assays in vivo do not allow for a single-cell resolution.
Fluorescent transcriptional reporters allow for single-cell resolution. However, since the protein half-life of fluorophores can be long (e.g., >24 h for GFP), signaling dynamics cannot be appropriately studied with the current reporters. The half-life of our new dynGFP reporter is significantly reduced (~4 h), and is therefore, in combination with enhanced protein folding, better suited to study signaling dynamics. The sensitivity of our reporter will, however, be dependent on the sensitivity of the experimental readout, the sensitivity of the cell line, and the level of integration of the lentiviral reporter. The current improvements were made on the protein level. Additionally, to enhance instability of the dynGFP reporter on an mRNA level, an unstable polyA tail could be added [
49]. However, this is not suited for lentiviral vectors, as an additional polyA tail behind the gene of interest in lentiviral vectors reduces viral titers.
The CAGA12-dynGFP reporter can be used in live cell imaging systems, such as the Incucyte®, to assess the dynamic response to TGF-β in a bulk population of cells. This is a very suitable set-up for examining, e.g., the effect of drug responses on TGF-β activity. However, this system is less qualified to observe different responses to TGF-β between single cells, due to its limited resolution and capability to track single cells over time. Using microscope systems specialized for single cell imaging, CAGA12-dynGFP increases can be monitored in single cells to study TGF-β responses over time.
Some limitations of the reporter should also be discussed. We used clonal B16F10 CAGA12-dynGFP cells to investigate TGF-β responses throughout the cell cycle. We observed a higher CAGA12-dynGFP signal in later phases of the cell cycle; however, using a clonal control cell line constitutively expressing dynGFP, we observed a very similar pattern. We therefore encourage the use of a constitutive control reporter in addition to the dynGFP for cell-cycle related studies.
The dynamic TGF-β/SMAD3 reporter is highly suited for investigating dynamic responses to TGF-β in different settings. However, as heterogeneity in dynGFP intensity within a (clonal) CAGA
12-dynGFP cell line can exist for technical reasons, we encourage the use of the reporter for studying TGF-β responses over time, in contrast to comparing reporter signal at one time point. Lastly, we noted a difference in fluorescence reporter activity 48 h after a single TGF-β treatment (when comparing
Figure 1C and
Figure S2A). We hypothesize that these differences are the result of differences in confluency and well size, which resulted in a different TGF-β turnover. Proper controls should be included when interpreting these results.
Visualizing endogenous dynamic TGF-β/SMAD3-induced responses without the addition of exogenous TGF-β in biologically relevant environments in vitro or in vivo may provide novel insights. Based on our in vitro results, the reporter can pick up (exogenously stimulated) TGF-β levels as low as 0.04 ng/mL in various cell lines. This is sensitive enough to pick up endogenous TGF-β levels in tissues, as we observed dynGFP+ cells in organoid co-cultures of fibroblasts and pancreatic cancer cells, and in vivo in mouse mammary tumors. This reporter can visualize both paracrine and autocrine signaling, and it will be interesting to distinguish between the two using additional controls. In mouse mammary tumors, we could observe a heterogenous pattern of TGF-β signaling within the tumor. These heterogeneous patterns were also observed in vitro upon TGF-β stimulation, showing that the in vivo (endogenous) TGF-β activation mimics TGF-β stimulation in vitro. As the cells were not sorted to a single cell clone, this heterogeneity could be due to differences in incorporation of the reporter (
Figure 3). Alternatively, the heterogeneity in intensity is the result of heterogeneity in response, similar to what Zhu and colleagues observed when treating breast cancer cells with TGF-β. These researchers used the CAGA
12-TdTomato reporter in MDA-MB-231 breast cancer cells and observed a population showing no TdTomato signal upon TGF-β treatment, as well as populations showing low, medium, and high TdTomato signal. It would be interesting to use the dynamic CAGA
12-dynGFP reporter in this case to follow these dynamics over time [
21]. Giampieri et al. used a CAGA
12-CFP reporter and SMAD2 nuclear translocation to monitor TGF-β signaling in primary breast cancer and lymph node metastasis [
18]. They also reported a heterogeneity in CAGA
12-CFP signal in primary MTLn3E tumors, similar to the heterogeneity we observed in PYMT tumors. Additionally, Giampieri reported that, upon metastasizing to the lymph node, breast cancer cells lost their motility. Motile cells in the primary tumor were mostly CAGA
12-CFP-positive. Interestingly, once metastasized to the lymph nodes, they observed mostly non-motile, CAGA
12-CFP negative cells in late lymph node metastasis, suggesting that the TGF-β/SMAD3 transcriptional response during lymph node metastasis is transient [
18]. Taking the results together, it would be of interest to examine the contribution of TGF-β/SMAD3 signaling dynamics during lymph node metastasis of breast cancer cells containing the CAGA
12-dynGFP reporter by performing long-term intravital imaging.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}



