
Reassessment of Reliability and Reproducibility for Triple-Negative Breast Cancer Subtyping



Abstract
:Simple Summary
Abstract
1. Introduction
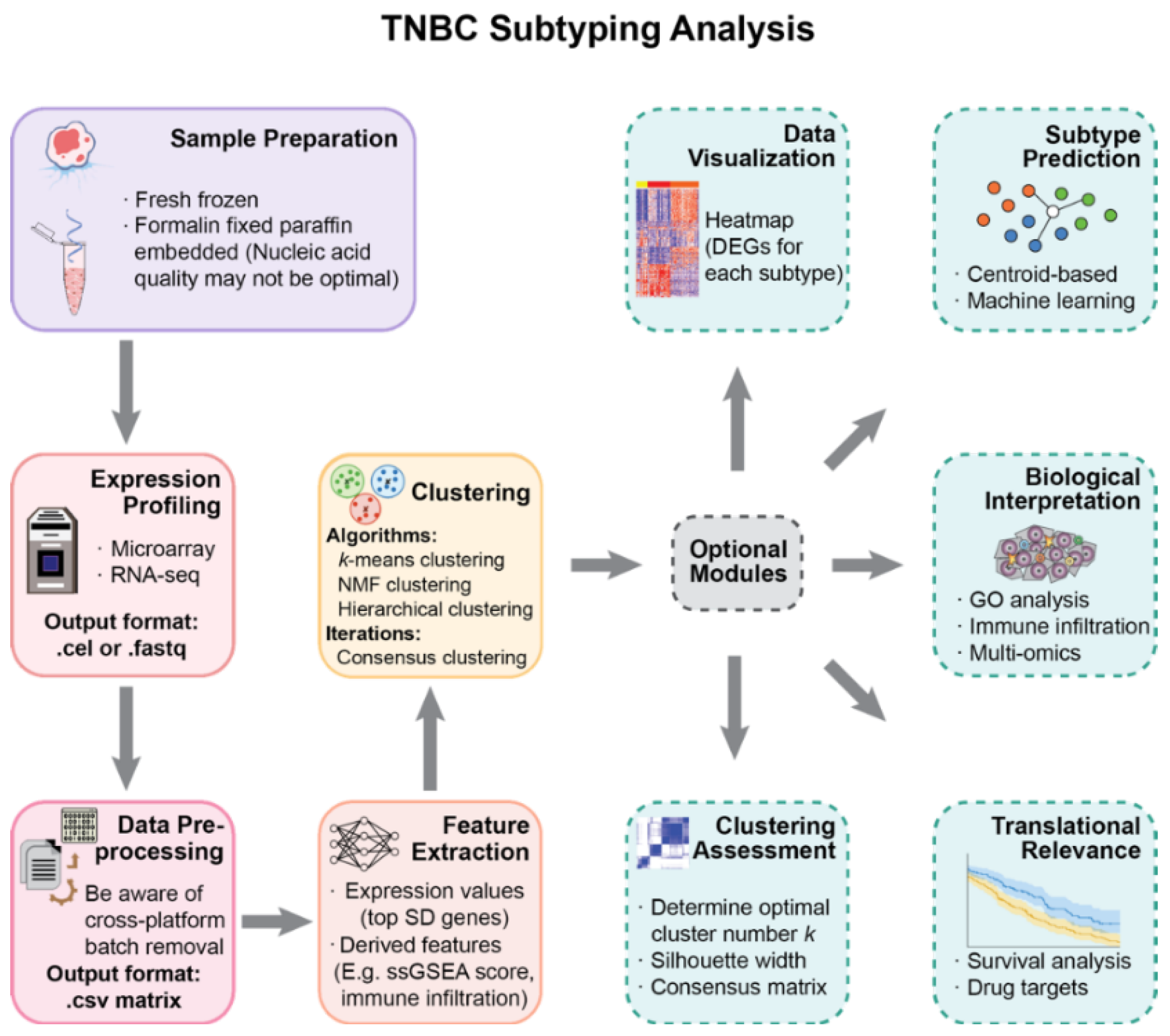
2. Materials and Methods
2.1. Data Collection
2.2. Microarray Data Preprocessing
2.3. Gene Selection and Subtype Discovery
2.4. Differential Expression and Pathway Enrichment Analysis
2.5. Comparison with Established Subtypes
2.6. Survival Analysis
2.7. Microenvironment Cell Abundance Calculation
3. Results
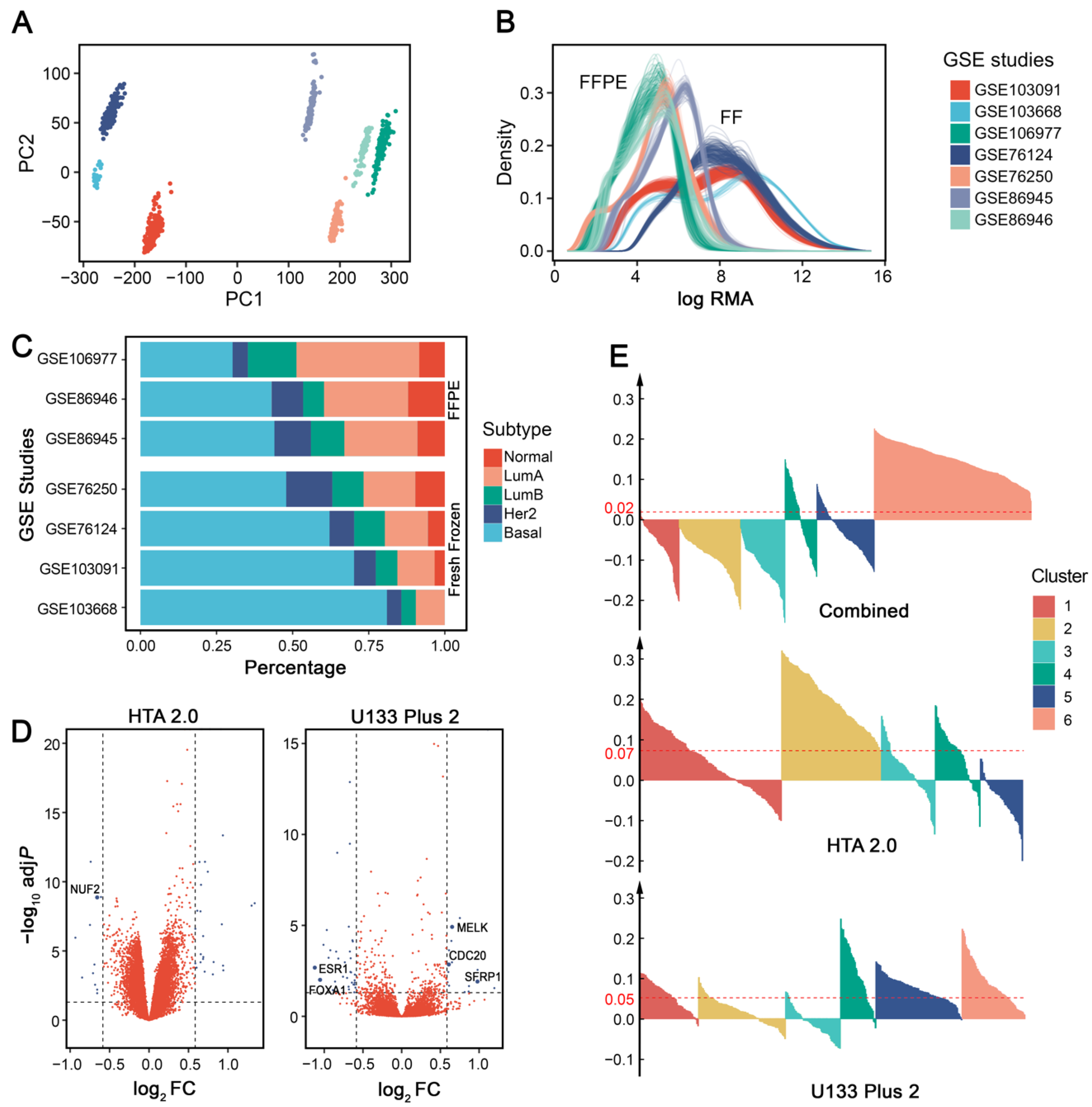
3.1. Intrinsic Subtype Distributions Are Impacted by Sample Preservation Methods
3.2. Cross-Platform Batch Effect Removal Results in Less Stable Clusters
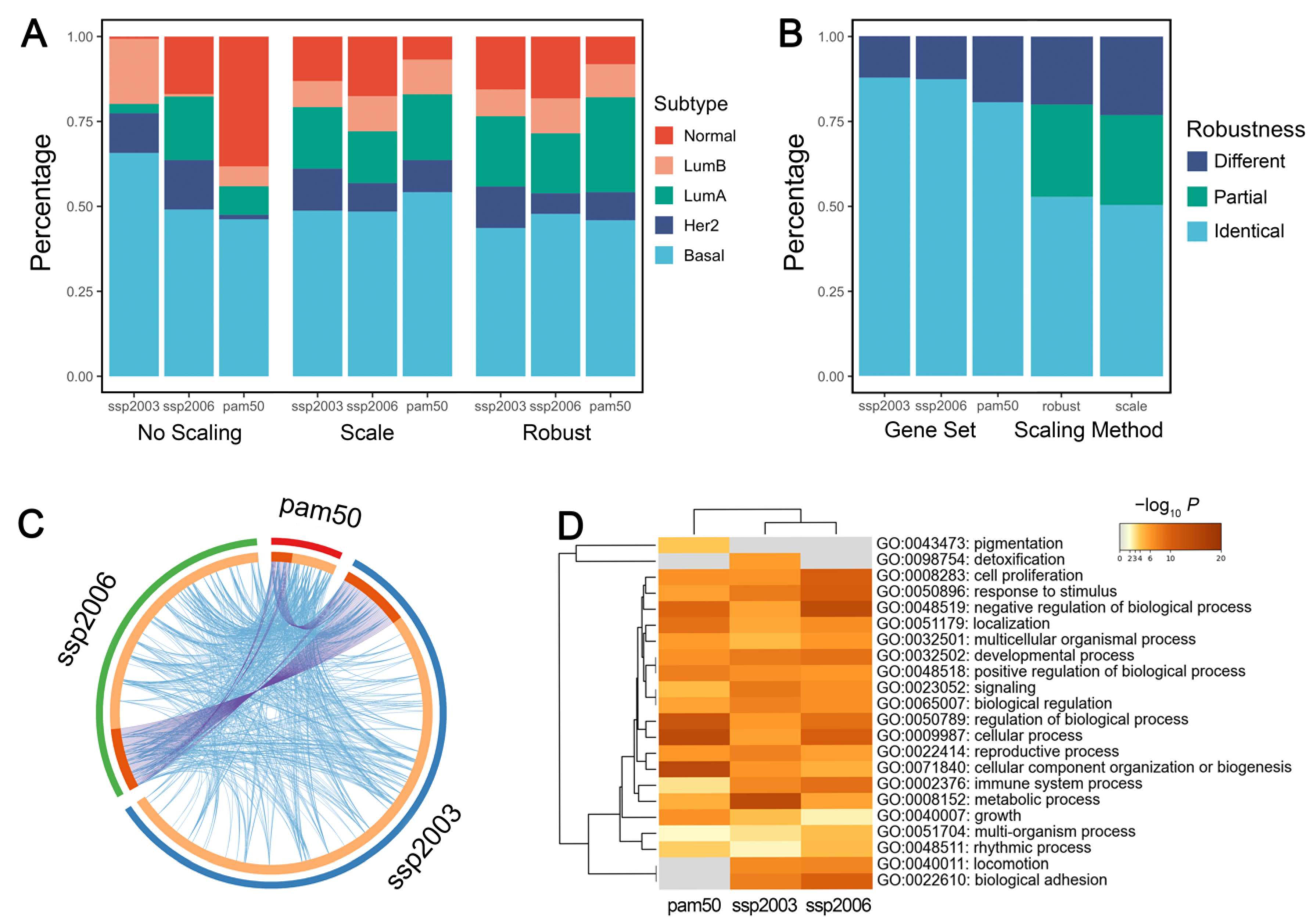
3.3. Proper Clustering Features and Algorithms Should Be Chosen for Subtype Discovery
3.4. Existing Algorithms Could Be More Suitable for TNBC Subtype Prediction than the Simple Nearest Centroid Classifier
3.5. Intrinsic Subtypes Could Not Be Reliably Assigned Due to Large Discrepancy of Different Gene Sets
3.6. mRNA-Based ER-Positive Filters Cause Unwanted Exclusion of IHC-Confirmed TNBC Samples
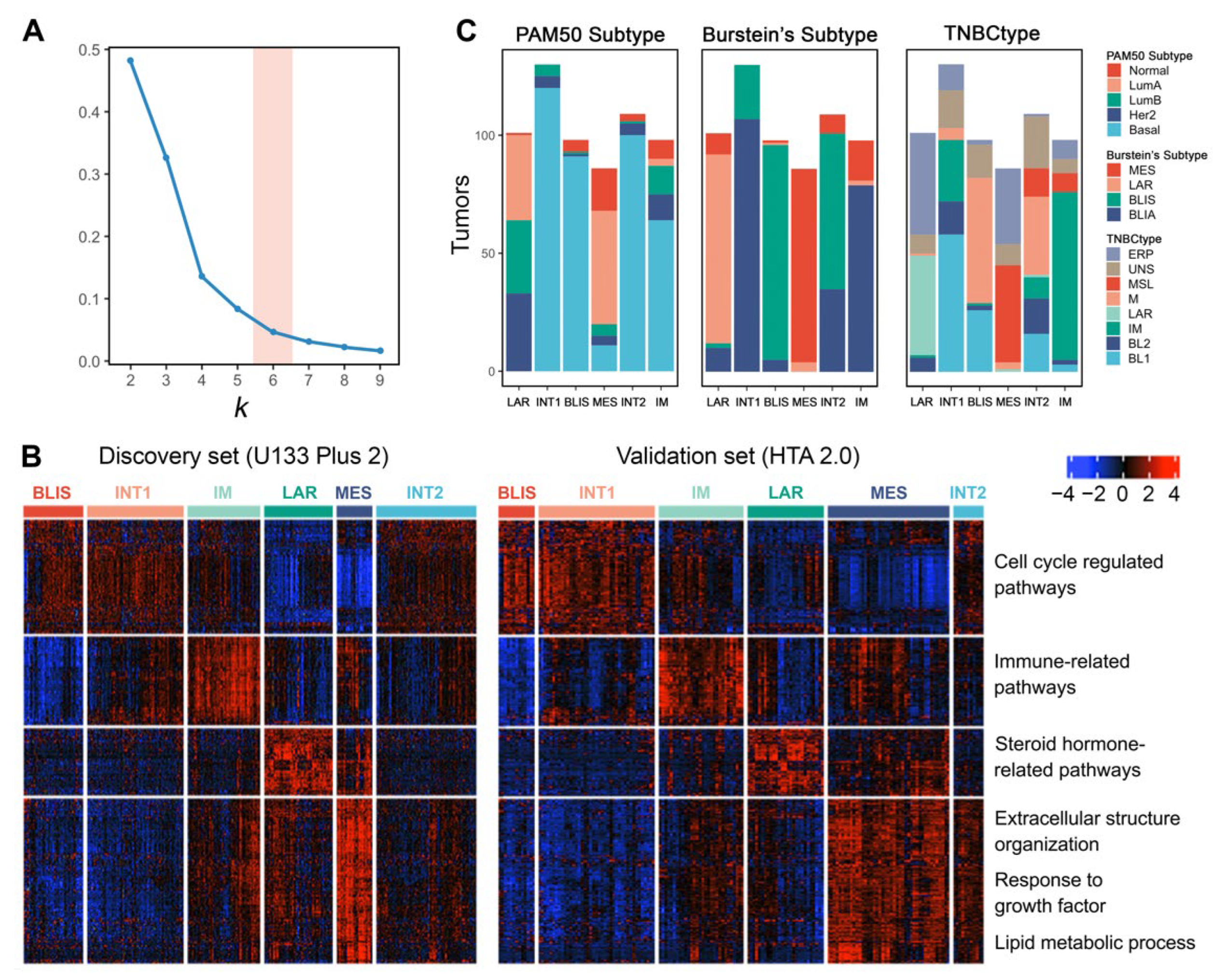
3.7. Unsupervised Clustering Reveals Six Molecular Subtypes of TNBC
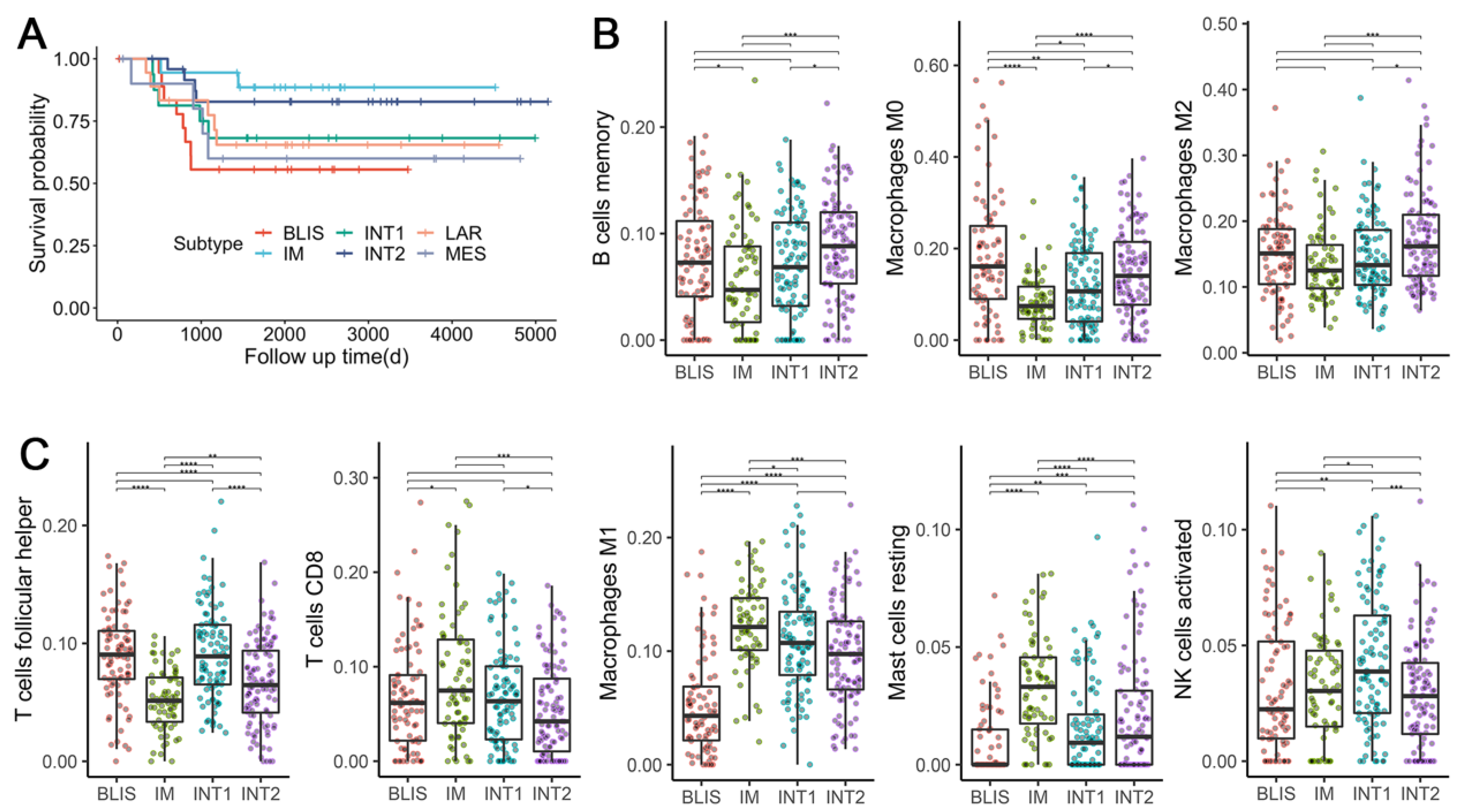
3.8. TNBC Subtypes Stratify Patients’ Survival
3.9. TNBC Subtypes Differ in Microenvironment Phenotypes
4. Conclusions and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, P.; Aggarwal, R. An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Irshad, S.; Ellis, P.; Tutt, A. Molecular heterogeneity of triple-negative breast cancer and its clinical implications. Curr. Opin. Oncol. 2011, 23, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-R.; Jiang, Y.-Z.; Xu, X.-E.; Yu, K.-D.; Jin, X.; Hu, X.; Zuo, W.-J.; Hao, S.; Wu, J.; Liu, G.-Y.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18, 33. [Google Scholar] [CrossRef] [Green Version]
- Jézéquel, P.; Loussouarn, D.; Guérin-Charbonnel, C.; Campion, L.; Vanier, A.; Gouraud, W.; Lasla, H.; Guette, C.; Valo, I.; Verrièle, V.; et al. Gene-expression molecular subtyping of triple-negative breast cancer tumours: Importance of immune response. Breast Cancer Res. 2015, 17, 43. [Google Scholar] [CrossRef] [Green Version]
- Jézéquel, P.; Kerdraon, O.; Hondermarck, H.; Guérin-Charbonnel, C.; Lasla, H.; Gouraud, W.; Canon, J.-L.; Gombos, A.; Dalenc, F.; Delaloge, S.; et al. Identification of three subtypes of triple-negative breast cancer with potential therapeutic implications. Breast Cancer Res. 2019, 21, 65. [Google Scholar] [CrossRef]
- He, Y.; Jiang, Z.; Chen, C.; Wang, X. Classification of triple-negative breast cancers based on Immunogenomic profiling. J. Exp. Clin. Cancer Res. 2018, 37, 327. [Google Scholar] [CrossRef]
- Romero-Cordoba, S.; Meneghini, E.; Sant, M.; Iorio, M.V.; Sfondrini, L.; Paolini, B.; Agresti, R.; Tagliabue, E.; Bianchi, F. Decoding Immune Heterogeneity of Triple Negative Breast Cancer and Its Association with Systemic Inflammation. Cancers 2019, 11, 911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Ma, D.; Zhao, S.; Suo, C.; Shi, J.; Xue, M.-Z.; Ruan, M.; Wang, H.; Zhao, J.; Li, Q.; et al. Multi-Omics Profiling Reveals Distinct Microenvironment Characterization and Suggests Immune Escape Mechanisms of Triple-Negative Breast Cancer. Clin. Cancer Res. 2019, 25, 5002–5014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palermo, M.; Driscoll, H.; Tighe, S.; Dragon, J.; Bond, J.; Shukla, A.; Vangala, M.; Vincent, J.; Hunter, T. Expression Profiling Smackdown: Human Transcriptome Array HTA 2.0 vs. RNA-Seq. J. Biomol. Tech. 2014, 25, S20–S21. [Google Scholar]
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Gendoo, D.; Ratanasirigulchai, N.; Schröder, M.; Pare, L.; Parker, J.S.; Prat, A.; Haibe-Kains, B. Genefu: An R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics 2016, 32, 1097–1099. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Chen, X.; Li, J.; Gray, W.H.; Lehmann, B.D.; Bauer, J.A.; Shyr, Y.; Pietenpol, J.A. TNBCtype: A Subtyping Tool for Triple-Negative Breast Cancer. Cancer Inform. 2012, 11, 147–156. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar]
- Fedorowicz, G.; Guerrero, S.; Wu, T.D.; Modrusan, Z. Microarray analysis of RNA extracted from formalin-fixed, paraffin-embedded and matched fresh-frozen ovarian adenocarcinomas. BMC Med. Genom. 2009, 2, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar, C.; Meganck, S.; Taminau, J.; Steenhoff, D.; Coletta, A.; Molter, C.; Weiss-Solís, D.Y.; Duque, R.; Bersini, H.; Nowé, A. Batch effect removal methods for microarray gene expression data integration: A survey. Brief. Bioinform. 2013, 14, 469–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, V.; Rødland, E.A.; Hovig, E. Methods that remove batch effects while retaining group differences may lead to exaggerated confidence in downstream analyses. Biostatistics 2016, 17, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Gatza, M.L.; Lucas, J.E.; Barry, W.T.; Kim, J.W.; Wang, Q.; Crawford, M.D.; Datto, M.B.; Kelley, M.; Mathey-Prevot, B.; Potti, A.; et al. A pathway-based classification of human breast cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 6994–6999. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kon, M.; DeLisi, C. Pathway-based classification of cancer subtypes. Biol. Direct 2012, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Gu, Y.; Hu, Z.; Sun, X. Sample-specific perturbation of gene interactions identifies breast cancer subtypes. Brief. Bioinform. 2021, 22, bbaa268. [Google Scholar] [CrossRef]
- de Souto, M.C.P.; Costa, I.G.; de Araujo, D.S.A.; Ludermir, T.B.; Schliep, A. Clustering cancer gene expression data: A comparative study. BMC Bioinform. 2008, 9, 497. [Google Scholar] [CrossRef] [Green Version]
- Brunet, J.-P.; Tamayo, P.; Golub, T.R.; Mesirov, J.P. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. USA 2004, 101, 4164–4169. [Google Scholar] [CrossRef] [Green Version]
- Gaujoux, R.; Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 2010, 11, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruspini, E.H. A new approach to clustering. Inf. Control 1969, 15, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Ankerst, M.; Breunig, M.M.; Kriegel, H.-P.; Sander, J. OPTICS: Ordering points to identify the clustering structure. ACM Sigmod Rec. 1999, 28, 49–60. [Google Scholar] [CrossRef]
- Rodriguez, A.; Laio, A. Machine learning. Clustering by fast search and find of density peaks. Science 2014, 344, 1492–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Siskind, J.M. Image segmentation with ratio cut. IEEE Trans. Pattern Anal. Mach. Intell. 2003, 25, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Jianbo, S.; Malik, J. Normalized cuts and image segmentation. IEEE Trans. Pattern Anal. Mach. Intell. 2000, 22, 888–905. [Google Scholar] [CrossRef] [Green Version]
- Tamayo, P.; Slonim, D.; Mesirov, J.; Zhu, Q.; Kitareewan, S.; Dmitrovsky, E.; Lander, E.S.; Golub, T.R. Interpreting patterns of gene expression with self-organizing maps: Methods and application to hematopoietic differentiation. Proc. Natl. Acad. Sci. USA 1999, 96, 2907–2912. [Google Scholar] [CrossRef] [Green Version]
- Monti, S.; Tamayo, P.; Mesirov, J.P.; Golub, T.R. Consensus Clustering: A Resampling-Based Method for Class Discovery and Visualization of Gene Expression Microarray Data. Mach. Learn. 2003, 52, 91–118. [Google Scholar] [CrossRef]
- Tibshirani, R.; Hastie, T.; Narasimhan, B.; Chu, G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6567–6572. [Google Scholar] [CrossRef] [Green Version]
- Dabney, A.R. Classification of microarrays to nearest centroids. Bioinformatics 2005, 21, 4148–4154. [Google Scholar] [CrossRef] [Green Version]
- Mullins, M.; Perreard, L.; Quackenbush, J.F.; Gauthier, N.; Bayer, S.; Ellis, M.; Parker, J.; Perou, C.M.; Szabo, A.; Bernard, P.S. Agreement in breast cancer classification between microarray and quantitative reverse transcription PCR from fresh-frozen and formalin-fixed, paraffin-embedded tissues. Clin. Chem. 2007, 53, 1273–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Zhang, C.; Ogihara, M. A comparative study of feature selection and multiclass classification methods for tissue classification based on gene expression. Bioinformatics 2004, 20, 2429–2437. [Google Scholar] [CrossRef] [PubMed]
- Dudoit, S.; Fridlyand, J.; Speed, T.P. Comparison of Discrimination Methods for the Classification of Tumors Using Gene Expression Data. J. Am. Stat. Assoc. 2002, 97, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Sørlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Fan, C.; Oh, D.S.; Marron, J.S.; He, X.; Qaqish, B.F.; Livasy, C.; Carey, L.A.; Reynolds, E.; Dressler, L.; et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genom. 2006, 7, 96. [Google Scholar] [CrossRef] [Green Version]
- Weigelt, B.; Mackay, A.; A’hern, R.; Natrajan, R.; Tan, D.S.; Dowsett, M.; Ashworth, A.; Reis-Filho, J.S. Breast cancer molecular profiling with single sample predictors: A retrospective analysis. Lancet Oncol. 2010, 11, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, C.; Chen, K.; Tang, H.; Tang, J.; Song, C.; Xie, X. ERβ1 inversely correlates with PTEN/PI3K/AKT pathway and predicts a favorable prognosis in triple-negative breast cancer. Breast Cancer Res. Treat. 2015, 152, 255–269. [Google Scholar] [CrossRef]
- Anestis, A.; Sarantis, P.; Theocharis, S.; Zoi, I.; Tryfonopoulos, D.; Korogiannos, A.; Koumarianou, A.; Xingi, E.; Thomaidou, D.; Kontos, M.; et al. Estrogen receptor beta increases sensitivity to enzalutamide in androgen receptor-positive triple-negative breast cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- Felder, M.; Kapur, A.; Gonzalez-Bosquet, J.; Horibata, S.; Heintz, J.; Albrecht, R.; Fass, L.; Kaur, J.; Hu, K.; Shojaei, H.; et al. MUC16 (CA125): Tumor biomarker to cancer therapy, a work in progress. Mol. Cancer 2014, 13, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollern, D.P.; Xu, N.; Thennavan, A.; Glodowski, C.; Garcia-Recio, S.; Mott, K.R.; He, X.; Garay, J.P.; Carey-Ewend, K.; Marron, D.; et al. B Cells and T Follicular Helper Cells Mediate Response to Checkpoint Inhibitors in High Mutation Burden Mouse Models of Breast Cancer. Cell 2019, 179, 1191–1206.e21. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-H.; Li, C.-X.; Liu, M.; Jiang, J.-Y. Predictive and prognostic role of tumour-infiltrating lymphocytes in breast cancer patients with different molecular subtypes: A meta-analysis. BMC Cancer 2020, 20, 1150. [Google Scholar] [CrossRef] [PubMed]
- Rønnov-Jessen, L.; Petersen, O.W.; Bissell, M.J. Cellular changes involved in conversion of normal to malignant breast: Importance of the stromal reaction. Physiol. Rev. 1996, 76, 69–125. [Google Scholar] [CrossRef] [PubMed]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Kosok, M.; Alli-Shaik, A.; Bay, B.H.; Gunaratne, J. Comprehensive Proteomic Characterization Reveals Subclass-Specific Molecular Aberrations within Triple-negative Breast Cancer. Iscience 2020, 23, 100868. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, R.T.; Perez, E.; Hernández, D.; Miller, C.P.; Haas, K.M.; Irie, H.Y.; Lee, S.-I.; Blau, C.A.; Villén, J. The proteomic landscape of triple-negative breast cancer. Cell Rep. 2015, 11, 630–644. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Chen, L.; Li, Y.; Qin, Y.; Pan, R.; Zhao, W.; Gu, Y.; Wang, H.; Wang, R.; Chen, X.; et al. Critical limitations of prognostic signatures based on risk scores summarized from gene expression levels: A case study for resected stage I non-small-cell lung cancer. Brief. Bioinform. 2016, 17, 233–242. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods a | Descriptions |
|---|---|
| Partitioning-based | Partitioning clustering iteratively assigns samples between clusters based on their similarity. It is relatively efficient, but sensitive to outliers and needs the number of clusters to be specified in advance. |
| k-means | |
| k-medoids b | |
| Fuzzy c-means [32] | |
| Hierarchical clustering | Hierarchical clustering creates homogeneous groups of samples by either a top-down (divisive) or a bottom-up (agglomerative) approach. The output dendrogram is easy to understand. |
| Divisive | |
| Agglomerative | |
| Density-based | Density-based clustering works by detecting densely connected regions. It does not require the number of clusters to be specified and can deal with noisy data and non-convex clusters. However, it is not suitable when there are significant density differences. |
| DBSCAN c | |
| OPTICS d [33] | |
| DPCA e [34] | |
| Spectral clustering RatioCut [35] | Spectral clustering is based on graph theory. It reduces the dimensionality of the dataset and then applies a basic clustering algorithm. |
| Ncut [36] | |
| Non-negative matrix factorization (NMF) [30] | NMF reduces the dimension of expression data, and in the meantime, places each sample into a cluster corresponding to the metagene. |
| Model-based | Unlike traditional algorithms, model-based clustering attempts to provide soft assignment and measures the probability of a sample belonging to each cluster. |
| Gaussian mixture models | |
| Self-organizing maps [37] |
| Methods a | Descriptions |
|---|---|
| Centroid-based | Centroid-based methods assign new samples to one of the existing classes based on centroids computed from the discovery set. PAM and ClaNC are examples of modified simple nearest centroid methods. |
| Simple nearest centroid classifier (SNCC) | |
| Prediction analysis of microarrays (PAM) [39] | |
| Classification to nearest centroids (ClaNC) [40] | |
| Supervised machine learning | Supervised machine learning methods can learn rules from labeled training data. For cancer subtyping, machine learning models could be trained on the discovery set and used to predict class assignment on the new datasets. Traditional and simple methods could already achieve good performance. |
| Support vector machine (SVM) | |
| k-nearest neighbor (k-NN) | |
| Linear discriminant analysis (LDA) | |
| Decision trees |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Liu, Y.; Chen, M. Reassessment of Reliability and Reproducibility for Triple-Negative Breast Cancer Subtyping. Cancers 2022, 14, 2571. https://doi.org/10.3390/cancers14112571
Yu X, Liu Y, Chen M. Reassessment of Reliability and Reproducibility for Triple-Negative Breast Cancer Subtyping. Cancers. 2022; 14(11):2571. https://doi.org/10.3390/cancers14112571
Chicago/Turabian StyleYu, Xinjian, Yongjing Liu, and Ming Chen. 2022. "Reassessment of Reliability and Reproducibility for Triple-Negative Breast Cancer Subtyping" Cancers 14, no. 11: 2571. https://doi.org/10.3390/cancers14112571
APA StyleYu, X., Liu, Y., & Chen, M. (2022). Reassessment of Reliability and Reproducibility for Triple-Negative Breast Cancer Subtyping. Cancers, 14(11), 2571. https://doi.org/10.3390/cancers14112571