
Drug Resistance in Colorectal Cancer: From Mechanism to Clinic


Abstract
:Simple Summary
Abstract
1. Introduction
2. Drug Metabolism
2.1. Reduced Drug Activation

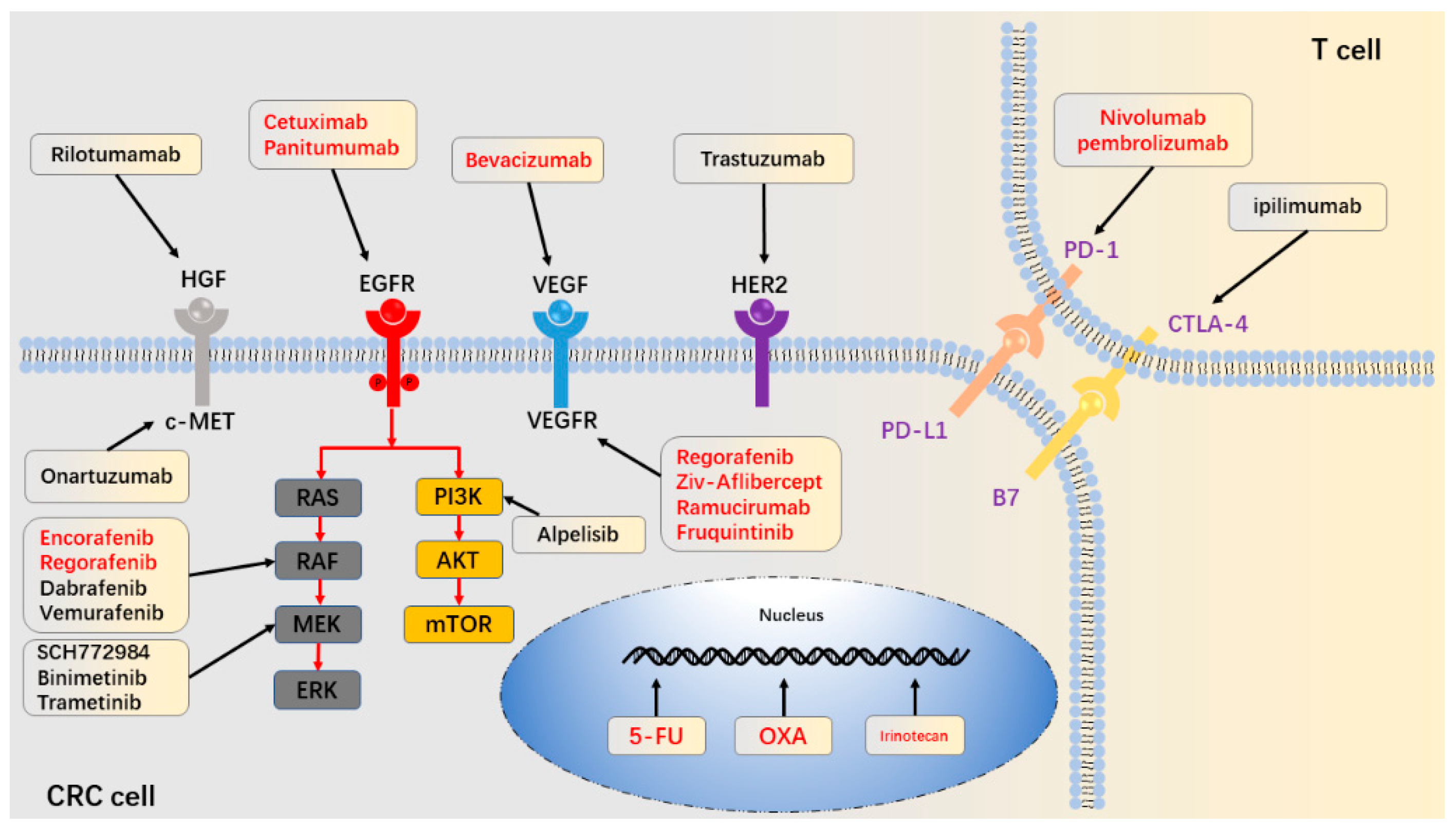{kind=link}
{kind=link}
{kind=link}
| Therapy | Treatment Regimen | Target | Subpopulation | Model System | Reference |
|---|---|---|---|---|---|
| ABC inhibitors | Cryptotanshinone/dihydrotanshinone/cinobufagin/8-oxocoptisine | P-gp | / | Cell | [12,13,14] |
| Liposomal antisense oligonucleotides | P-gp, MRP1, MRP2, Bcl-2, Bcl-XL | / | Cell | [15,16] | |
| Y-box binding protein 1 (YB-1) | TOP-1 | / | Cell | [17]. | |
| New anti-EGFR mAbs | Sym004 | EGFR ECD | EGFR G465E mutant | Cell/mice | [18] |
| MM-151 | EGFR ECD | EGFR G465E mutant | PDX | [19] | |
| Necitumumab | EGFR ECD | EGFR S468R mutant | Cell | [20] | |
| MEK inhibitors | BAY86-9766 + cetuximab | MEK | KRAS mutant | PDX | [21] |
| Pimasertib + cetuximab | MEK | KRAS, NRAS or BRAF mutant | Cell/mice/PDX | [22] | |
| Trametinib + Olaparib + αPD-L1 | MEK | KRAS mutant | Mice | [23] | |
| G-38963 + αPD-1 | MEK | KRAS mutant | Mice | [24] | |
| New anti-MEK mAbs | SCH772984 | MEK | KRAS or BRAF mutant | PDX | [25] |
| IGF-IR inhibitors | NVP-AEW541(IGF-IR i) + aEGFR | IGF-IR | / | CRC primary cell line | [26] |
| HER2 mABs | Pertuzumab + cetuximab/lapatinib | HER2 | HER2 amplification | PDX | [27] |
| Trastuzumab + cetuximab | HER2 | / | Cell | [28] | |
| 4D5 + cetuximab | HER2 | / | PDX | [29] | |
| HER3 mABs | U3-1402 | HER3 | High HER3 expression; | CDX | [30] |
| β-catenin/CBP inhibitors | TrpC5 | β-catenin/CBP | / | Cell | [31] |
| AQP5 | β-catenin/CBP | / | Cell | [32] | |
| ICG-001 | β-catenin/CBP | / | Cell/mice | [32] | |
| TLR9 agonist | SD-101 + PD-1 | TLR-9 | / | Mice | [33] |
| IMO + cetuximab/bevacizumab | TLR-9 | / | Mice | [34] | |
| IMO + cetuximab + irinotecan | TLR-9 | / | Cell and mice | [35] | |
| IMO + cetuximab | TLR-9 | / | Cell and mice | [36] | |
| TGF-β inhibitors | LY2157299 + Gefitinib | TGF-βIR | / | Cell | [37] |
| PF-03446962 + Bevacizumab | ALK-1 | / | Mice and PDX | [38] | |
| Galunisertib +αPD-L1 | TGFBR1 | low mutational burden, T-cell exclusion and TGFβ-activated stroma | Mice | [39] | |
| Apoptotic inhibitors | ABT-263 + selumetinib | Bax, Bak | BRAF(V600E) or RAS mutant | Cell | [40] |
| AEG35156 + taxanes | XIAP | / | Mice | [41] | |
| Birinapant + oxaliplatin/5-FU | XIAP | / | Cell | [42] | |
| Autophagy inhibitors | Chloroquine/3-MA + 5-FU | Autophagy | / | Cell | [43,44] |
| Chloroquine/3-MA + cetuximab | Autophagy | / | Cell | [45] | |
| 3-MA + αEGFR | Autophagy | / | Cell | [46] | |
| SB02024/SAR405 + αPD-1/PD-L1 | Vps34 | / | Mice | [47] | |
| Ferroptosis agonist | β-elemene + cetuximab | / | KRAS mutant | Cell and mice | [48] |
| RSL3 + cisplatin | / | / | Cell and mice | [49] | |
| CAF | BGJ398 + 5-FU + oxaliplatin | FGFR4 | / | Cell | [50] |
| BLU9931 + cetuximab | FGFR4 | / | Cell and mice | [51] | |
| Regorafenib + cetuximab | FGFR, VEGF, PDGFR-β | / | Cell and mice | [52] | |
| GKT137831 | NOX4 | / | Mice | [53] | |
| MDSC | KTN0158 + αPD-1 + αCTLA-4 | KIT | / | Mice | [54] |
| TAM | DFMO + 5-FU | ODC | / | Mice | [55] |
| RG7155 | CSF1R | / | Mice | [56] | |
| MDSC | R848 + oxaliplatin | TLR 7/8 agonist | / | Mice | [57] |
| MDSC and TAM | TP-16 + αPD-1 | EP4 | / | Mice | [58] |
| VEGF inhibitors | αVEGF(A) + αPD-1 | VEGF-A | / | Mice | [59,60,61] |
| Vargatef + afatinib | VEGF, EGFR | / | Mice | [62] | |
| Regorafenib + cetuximab | FGFR, VEGF, PDGFR-β | KRAS or BRAF mutant | Cell and mice | [52] |
| Therapy | Treatment Regimen | Target | Subpopulation | Species | Setting | Efficiency | Reference |
|---|---|---|---|---|---|---|---|
| New anti-EGFR mAbs | Sym004 | EGFR ECD | / | anti-EGFR-refractory/unselected mCRC | Phase I trial studies | ORCD: (67%) | NCT01117428 [18] |
| Sym004 | EGFR ECD | KRAS WT | anti-EGFR-refractory mCRC | Phase II trial studies | mOS: (10.3 m:9.6 m) | 2013-003829-29 [63] | |
| MM-151 + irinotecan vs. MM-151 | EGFR ECD | KRAS WT | Refractory advanced CRC | Phase I trial studies | MM-151-SD: 31% MM-151+ irinotecan- PR: 1/3 | NCT01520389 [64] | |
| Necitumumab | EGFR ECD | / | first-line treatment for locally advanced or metastatic CRC | Phase II trial studies | ORR: (63.6%) OS: (22.5 m) | [65] | |
| BRAF inhibitor | Vemurafenib + panitumumab | BRAF V600E | BRAF-mutant | Refractory mCRC | Pilot trial | ORR: (16.7%) | [66] |
| Vemurafenib + cetuximab vs. vemurafenib | BRAF V600E | BRAF V600E mutant | CRC | Phase II trial studies | ORR: (0%:4%) | NCT01524978 [66] | |
| Vemurafenib + irinotecan + cetuximab | BRAF V600E | BRAF V600E mutant | mCRC | Phase Ib trial studies | ORR: (35%) | NCT01787500 [67]. | |
| PI3K inhibitor | cetuximab+PX-866 vs. cetuximab | PI3K | KRAS WT | irinotecan- and oxaliplatin-refractory mCRC | Phase II trial studies | mOS: (266d:333d) | [68] |
| Alpelisib + cetuximab + encorafenib + vs. cetuximab + encorafenib | PI3K | BRAF-Mutant | mCRC | Phase Ib trial studies | mOS: (4.2 m:3.7 m) | NCT0171938 [69] | |
| IGF-IR inhibitor | IMC-A12 + cetuximab vs. IMC-A12 | IGF-IR | / | anti-EGFR-refractory mCRC | Phase II trial studies | ORR: (0%:5%) | NCT00503685 [70] |
| Dalotuzumab + Cetuximab + Irinotecan vs. Cetuximab + Irinotecan | IGF-IR | KRAS WT | chemo-refractory mCRC | Phase II/III trial studies | mOS: (10.8 m:14 m) | NCT00614393 [71] | |
| HGF/IGF-IR inhibitor | Rilotumumab + panitumumab vs. ganitumab + panitumumab vs. panitumumab | HGF/IGF-IR | KRAS WT | previously treated mCRC | Phase Ib/II trial studies | mOS: (13.8 m:10.6 m:11.6 m) | NCT00788957 [72] |
| HER2 inhibitor | Neratinib + Cetuximab | HER2 | Quadruple-WT(KRAS, NRAS, BRAF, PIK3CA) | anti-EGFR-refractory mCRC | Phase Ib trial studies | There were no objective responses | NCT01960023 [73] |
| Trastuzumab + lapatinib | HER2 | KRAS WT, HER2-positive | anti-EGFR-refractory mCRC | Phase II trial studies | ORR: (30%) | [74] | |
| Trastuzumab + Pertuzumab | HER2 | HER2 amplification | treatment-refractory mCRC | Phase IIa trial studies | ORR: (32%) | NCT02091141 [75] | |
| HER3 inhibitor | Lumretuzumab | HER3 | HER3-positive | treatment-refractory mCRC | Phase I trial studies | SD: (21.3%) | NCT01482377 [76] |
| MEHD7945A | HER3/EGFR | / | treatment-refractory mCRC | Phase I trial studies | SD: (28.6%) | NCT01207323 [77] | |
| MET inhibitor | Tivantinib + Cetuximab | MET | KRAS WT | anti-EGFR-refractory mCRC | Phase II trial studies | ORR: (9.8%) | NCT01892527 [78] |
| Tivantinib + irinotecan + cetuximab vs. irinotecan + cetuximab | MET | KRAS WT | locally advanced or metastatic CRC | Phase I/II trial studies | mPFS: (8.3 m:7.3 m) mOS: (19.8 m:16.9 m) ORR: 45%:33% | NCT01075048 [79] | |
| Capmatinib + cetuximab | MET | K/NRAS WT, MET-positive | anti-EGFR-refractory mCRC | Phase Ib trial studies | SD: (46.2%) | NCT02205398 [80] | |
| VEGF/ALK-1 inhibitor | Regorafenib + PF-03446962 | VEGF/ALK-1 | / | treatment-refractory mCRC | Phase Ib trial studies | SD: (18.2%) | [81] |
| MEK inhibitor | Cobimetinib + atezolizumab | MEK | / | mCRC | Phase I/Ib trial studies | ORR: (8%) | NCT01988896 [82] |
| Atezolizumab + cobimetinib vs. Atezolizumab vs. regorafenib | MEK | / | previously treated mCRC | Phase III trial studies | mPFS: (1.91 m:1.94 m:2.0 m) mOS: (8.87 m:7.10 m:8.51 m) | NCT02788279 [83] | |
| Cobimetinib + Atezolizumab + Hydroxychloroquine | MEK | KRAS-Mutant | Advanced Malignancies | Phase I/II trial studies | None | NCT04214418 | |
| CSF1R inhibitor | Pexidartinib + Durvalumab | CSF1R | / | Metastatic/Advanced CRC | Phase I trial studies | None | NCT02777710 |
| ARRY-382 + Pembrolizumab | CSF1R | / | Advanced Solid Tumors | Phase I/II trial studies | None | NCT02880371 | |
| anti-VEGF mAbs | Bevacizumab + Atezolizumab | VEGF-A | MSI-H | mCRC | Phase Ib trial studies | ORR: (30%) SD: (90%) | NCT01633970 [84] |
| bevacizumab + capecitabine + atezolizumab vs. bevacizumab + capecitabine | VEGF-A | / | refractory mCRC | Phase II trial studies | mPFS: (3.3 m:4.4 m) ORR: (4.35%:8.45%) 12 m OS: (43%:52%) | NCT01633970 [85] | |
| nivolumab + standard of care chemotherapy + bevacizumab | VEGF-A | / | mCRC | Phase II/III trial studies | None | NCT03414983 | |
| anti-VEGFR mAbs | Regorafenib + cetuximab/Panitumumab | VEGFR | / | Unresectable, Locally Advanced, or Metastatic Colorectal Cancer | Phase I trial studies | Clinical Benefit: (53%) | NCT02095054 [86] |
| Phase II trial studies | None | NCT04117945 |
2.2. Increased Drug Inactivation
3. Drug Transport
4. Changes of Drug Targets
4.1. Thymidylate Synthase (TS)
4.2. Topoisomerase 1 (TOP-1)
4.3. Epidermal Growth Factor Receptor and Its Ligand
5. Aberration in Downstream Signaling Pathways
5.1. RAS/RAF/MEK/ERK
5.2. PI3K/AKT/mTOR
6. Aberrant Activations of Alternative Receptors
6.1. IGF1 and IGF2
6.2. HER2 and HER3
6.3. HGF/MET
7. Persistent Activation of Oncogenic/Bypass Signaling
7.1. Wnt/β-Catenin
7.2. JAK/STAT
7.3. TGF-β
7.4. EGFR
7.4.1. PI3K/AKT/mTOR
7.4.2. RAS/RAF/MEK/ERK
8. Pathway of Cell Death
8.1. Apoptosis
8.2. Autophagy
8.3. Ferroptosis
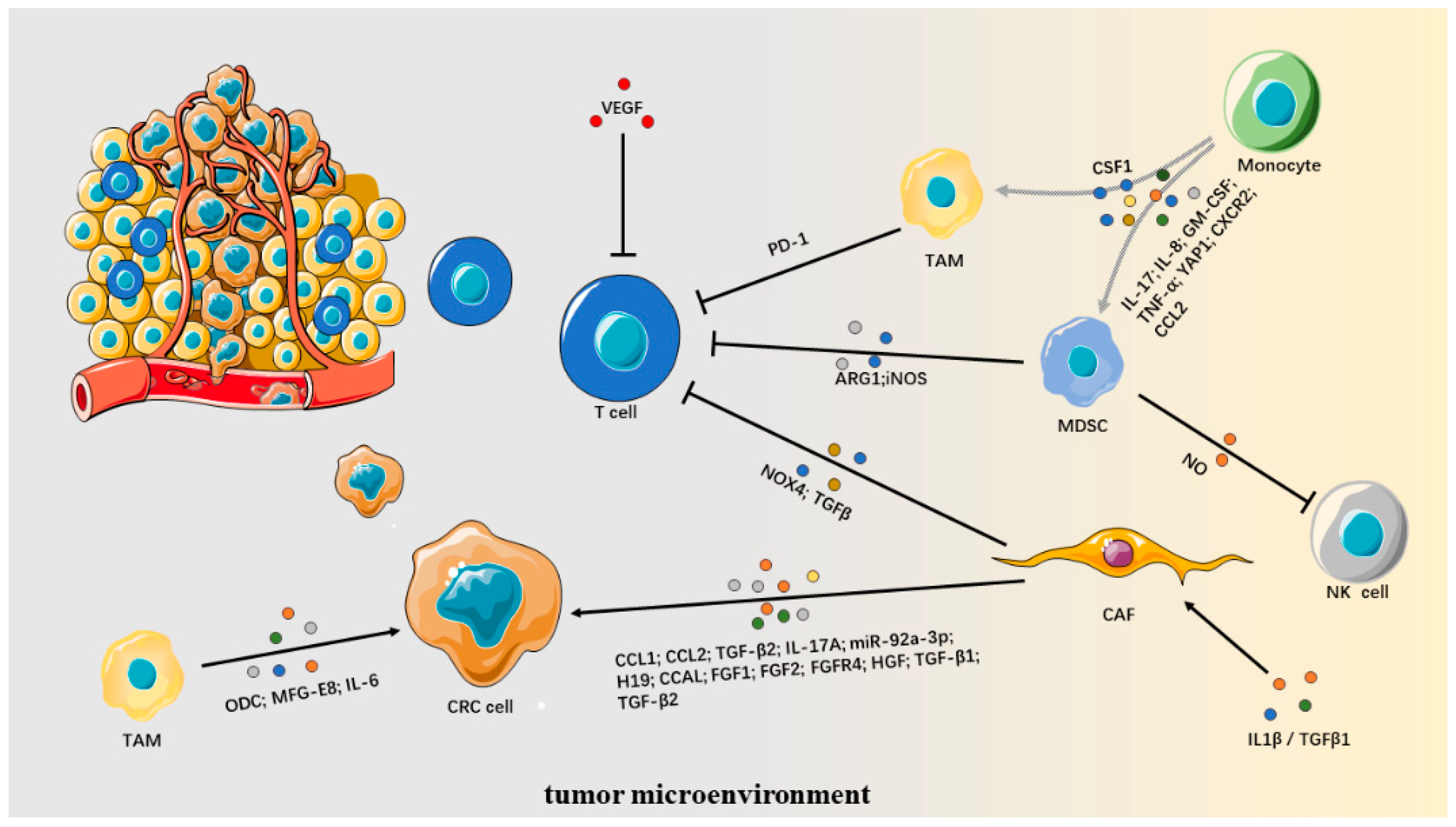
9. Tumor Microenvironment
9.1. Cancer-Associated Fibroblasts
9.2. Myeloid-Derived Suppressor Cells
9.3. Tumor-Associated Macrophages
9.4. Angiogenesis
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Malet-Martino, M.; Martino, R. Clinical studies of three oral prodrugs of 5-fluorouracil (capecitabine, UFT, S-1): A review. Oncologist 2002, 7, 288–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosuri, K.V.; Wu, X.; Wang, L.; Villalona-Calero, M.A.; Otterson, G.A. An epigenetic mechanism for capecitabine resistance in mesothelioma. Biochem. Biophys. Res. Commun. 2010, 391, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Blondy, S.; David, V.; Verdier, M.; Mathonnet, M.; Perraud, A.; Christou, N. 5-Fluorouracil resistance mechanisms in colorectal cancer: From classical pathways to promising processes. Cancer Sci. 2020, 111, 3142–3154. [Google Scholar] [CrossRef]
- Fujii, R.; Seshimo, A.; Kameoka, S. Relationships between the expression of thymidylate synthase, dihydropyrimidine dehydrogenase, and orotate phosphoribosyltransferase and cell proliferative activity and 5-fluorouracil sensitivity in colorectal carcinoma. Int. J. Clin. Oncol. 2003, 8, 72–78. [Google Scholar] [CrossRef]
- Humeniuk, R.; Menon, L.G.; Mishra, P.J.; Gorlick, R.; Sowers, R.; Rode, W.; Pizzorno, G.; Cheng, Y.-C.; Kemeny, N.; Bertino, J.R.; et al. Decreased levels of UMP kinase as a mechanism of fluoropyrimidine resistance. Mol. Cancer Ther. 2009, 8, 1037–1044. [Google Scholar] [CrossRef] [Green Version]
- Griffith, M.; Mwenifumbo, J.C.; Cheung, P.Y.; Paul, J.E.; Pugh, T.J.; Tang, M.J.; Chittaranjan, S.; Morin, R.D.; Asano, J.K.; Ally, A.A.; et al. Novel mRNA isoforms and mutations of uridine monophosphate synthetase and 5-fluorouracil resistance in colorectal cancer. Pharm. J. 2013, 13, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Salonga, D.; Danenberg, K.D.; Johnson, M.; Metzger, R.; Groshen, S.; Tsao-Wei, D.D.; Lenz, H.J.; Leichman, C.G.; Leichman, L.; Diasio, R.B.; et al. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin. Cancer Res. 2000, 6, 1322–1327. [Google Scholar]
- Soong, R.; Shah, N.; Salto-Tellez, M.; Tai, B.C.; Soo, R.A.; Han, H.C.; Ng, S.S.; Tan, W.L.; Zeps, N.; Joseph, D.; et al. Prognostic significance of thymidylate synthase, dihydropyrimidine dehydrogenase and thymidine phosphorylase protein expression in colorectal cancer patients treated with or without 5-fluorouracil-based chemotherapy. Ann. Oncol. 2008, 19, 915–919. [Google Scholar] [CrossRef]
- Lindskog, E.B.; Derwinger, K.; Gustavsson, B.; Falk, P.; Wettergren, Y. Thymidine phosphorylase expression is associated with time to progression in patients with metastatic colorectal cancer. BMC Clin. Pathol. 2014, 14, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; To, K.K.W.; Wang, L.; Zhang, L.; Lu, L.; Shen, J.; Chan, R.L.Y.; Li, M.; Yeung, J.H.K.; Cho, C.H. Reversal of P-glycoprotein (P-gp) mediated multidrug resistance in colon cancer cells by cryptotanshinone and dihydrotanshinone of Salvia miltiorrhiza. Phytomedicine 2014, 21, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Shi, X.; Qiu, Y.; Jia, T.; Yuan, X.; Zou, Y.; Liu, C.; Yu, H.; Yuan, Y.; He, X.; et al. Reversal of P-gp-mediated multidrug resistance in colon cancer by cinobufagin. Oncol. Rep. 2017, 37, 1815–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, Y.D.; Yang, M.C.; Lee, K.H.; Kim, K.R.; Choi, S.U.; Lee, K.R. Protoberberine alkaloids and their reversal activity of P-gp expressed multidrug resistance (MDR) from the rhizome of Coptis japonica Makino. Arch. Pharm. Res. 2006, 29, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.-L.; Liu, Y. Reversing multidrug resistance in Caco-2 by silencing MDR1, MRP1, MRP2, and BCL-2/BCL-xL using liposomal antisense oligonucleotides. PLoS ONE 2014, 9, e90180. [Google Scholar] [CrossRef]
- Lo, Y.-L.; Liu, Y.; Tsai, J.-C. Overcoming multidrug resistance using liposomal epirubicin and antisense oligonucleotides targeting pump and nonpump resistances in vitro and in vivo. Biomed. Pharmacother. 2013, 67, 261–267. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, K.-y.; Li, Z.; Liu, Y.-p.; Izumi, H.; Uramoto, H.; Nakayama, Y.; Ito, K.-i.; Kohno, K. Y-box binding protein 1 enhances DNA topoisomerase 1 activity and sensitivity to camptothecin via direct interaction. J. Exp. Clin. Cancer Res. 2014, 33, 112. [Google Scholar] [CrossRef] [Green Version]
- Dienstmann, R.; Patnaik, A.; Garcia-Carbonero, R.; Cervantes, A.; Benavent, M.; Roselló, S.; Tops, B.B.J.; van der Post, R.S.; Argilés, G.; Skartved, N.J.Ø.; et al. Safety and Activity of the First-in-Class Sym004 Anti-EGFR Antibody Mixture in Patients with Refractory Colorectal Cancer. Cancer Discov. 2015, 5, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Arena, S.; Siravegna, G.; Mussolin, B.; Kearns, J.D.; Wolf, B.B.; Misale, S.; Lazzari, L.; Bertotti, A.; Trusolino, L.; Adjei, A.A.; et al. MM-151 overcomes acquired resistance to cetuximab and panitumumab in colorectal cancers harboring EGFR extracellular domain mutations. Sci. Transl. Med. 2016, 8, 324ra14. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, A.; Haidar, J.N.; Eastman, S.W.; Vieth, M.; Topper, M.; Iacolina, M.D.; Walker, J.M.; Forest, A.; Shen, Y.; Novosiadly, R.D.; et al. Molecular Basis for Necitumumab Inhibition of EGFR Variants Associated with Acquired Cetuximab Resistance. Mol. Cancer Ther. 2018, 17, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Troiani, T.; Napolitano, S.; Martini, G.; Martinelli, E.; Cardone, C.; Normanno, N.; Vitagliano, D.; Morgillo, F.; Fenizia, F.; Lambiase, M.; et al. Maintenance Treatment with Cetuximab and BAY86-9766 Increases Antitumor Efficacy of Irinotecan plus Cetuximab in Human Colorectal Cancer Xenograft Models. Clin. Cancer Res. 2015, 21, 4153–4164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misale, S.; Arena, S.; Lamba, S.; Siravegna, G.; Lallo, A.; Hobor, S.; Russo, M.; Buscarino, M.; Lazzari, L.; Sartore-Bianchi, A.; et al. Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Sci. Transl. Med. 2014, 6, 224ra26. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Li, X.; Fu, Y.; Guo, E.; Ye, Y.; Li, F.; Liu, S.; Xiao, R.; Liu, C.; Lu, F.; et al. MEK Inhibition Remodels the Immune Landscape of Mutant Tumors to Overcome Resistance to PARP and Immune Checkpoint Inhibitors. Cancer Res. 2021, 81, 2714–2729. [Google Scholar] [CrossRef]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, E.J.; Jha, S.; Restaino, C.R.; Dayananth, P.; Zhu, H.; Cooper, A.; Carr, D.; Deng, Y.; Jin, W.; Black, S.; et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013, 3, 742–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, M.P.; Thomas, H.; Marks, C.; Green, M.; Fan, Z.; Modjtahedi, H. Co-targeting the EGFR and IGF-IR with anti-EGFR monoclonal antibody ICR62 and the IGF-IR tyrosine kinase inhibitor NVP-AEW541 in colorectal cancer cells. Int. J. Oncol. 2008, 33, 1107–1113. [Google Scholar]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Corà, D.; Di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef] [Green Version]
- Luca, T.; Barresi, V.; Privitera, G.; Musso, N.; Caruso, M.; Condorelli, D.F.; Castorina, S. In vitro combined treatment with cetuximab and trastuzumab inhibits growth of colon cancer cells. Cell Prolif. 2014, 47, 435–447. [Google Scholar] [CrossRef]
- Kuwada, S.K.; Scaife, C.L.; Kuang, J.; Li, X.; Wong, R.F.; Florell, S.R.; Coffey, R.J.; Gray, P.D. Effects of trastuzumab on epidermal growth factor receptor-dependent and -independent human colon cancer cells. Int. J. Cancer 2004, 109, 291–301. [Google Scholar] [CrossRef]
- Koganemaru, S.; Kuboki, Y.; Koga, Y.; Kojima, T.; Yamauchi, M.; Maeda, N.; Kagari, T.; Hirotani, K.; Yasunaga, M.; Matsumura, Y.; et al. U3-1402, a Novel HER3-Targeting Antibody-Drug Conjugate, for the Treatment of Colorectal Cancer. Mol. Cancer Ther. 2019, 18, 2043–2050. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Chen, Z.; Zhu, Y.; Pan, Q.; Liu, Y.; Qi, X.; Jin, L.; Jin, J.; Ma, X.; Hua, D. Inhibition of transient receptor potential channel 5 reverses 5-Fluorouracil resistance in human colorectal cancer cells. J. Biol. Chem. 2015, 290, 448–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Yang, T.; Li, D.; Ding, F.; Bai, G.; Wang, W.; Sun, H. Knockdown of aquaporin-5 sensitizes colorectal cancer cells to 5-fluorouracil via inhibition of the Wnt-β-catenin signaling pathway. Biochem. Cell Biol. 2018, 96, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Torrejon, D.Y.; Abril-Rodriguez, G.; Champhekar, A.S.; Tsoi, J.; Campbell, K.M.; Kalbasi, A.; Parisi, G.; Zaretsky, J.M.; Garcia-Diaz, A.; Puig-Saus, C.; et al. Overcoming Genetically Based Resistance Mechanisms to PD-1 Blockade. Cancer Discov. 2020, 10, 1140–1157. [Google Scholar] [CrossRef] [PubMed]
- Damiano, V.; Caputo, R.; Garofalo, S.; Bianco, R.; Rosa, R.; Merola, G.; Gelardi, T.; Racioppi, L.; Fontanini, G.; De Placido, S.; et al. TLR9 agonist acts by different mechanisms synergizing with bevacizumab in sensitive and cetuximab-resistant colon cancer xenografts. Proc. Natl. Acad. Sci. USA 2007, 104, 12468–12473. [Google Scholar] [CrossRef] [Green Version]
- Damiano, V.; Caputo, R.; Bianco, R.; D’Armiento, F.P.; Leonardi, A.; De Placido, S.; Bianco, A.R.; Agrawal, S.; Ciardiello, F.; Tortora, G. Novel toll-like receptor 9 agonist induces epidermal growth factor receptor (EGFR) inhibition and synergistic antitumor activity with EGFR inhibitors. Clin. Cancer Res. 2006, 12, 577–583. [Google Scholar] [CrossRef] [Green Version]
- Rosa, R.; Melisi, D.; Damiano, V.; Bianco, R.; Garofalo, S.; Gelardi, T.; Agrawal, S.; Di Nicolantonio, F.; Scarpa, A.; Bardelli, A.; et al. Toll-like receptor 9 agonist IMO cooperates with cetuximab in K-ras mutant colorectal and pancreatic cancers. Clin. Cancer Res. 2011, 17, 6531–6541. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Hölzel, M.; Knijnenburg, T.; Schlicker, A.; Roepman, P.; McDermott, U.; Garnett, M.; Grernrum, W.; Sun, C.; Prahallad, A.; et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-β receptor signaling. Cell 2012, 151, 937–950. [Google Scholar] [CrossRef] [Green Version]
- Hu-Lowe, D.D.; Chen, E.; Zhang, L.; Watson, K.D.; Mancuso, P.; Lappin, P.; Wickman, G.; Chen, J.H.; Wang, J.; Jiang, X.; et al. Targeting activin receptor-like kinase 1 inhibits angiogenesis and tumorigenesis through a mechanism of action complementary to anti-VEGF therapies. Cancer Res. 2011, 71, 1362–1373. [Google Scholar] [CrossRef] [Green Version]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Sale, M.J.; Cook, S.J. The BH3 mimetic ABT-263 synergizes with the MEK1/2 inhibitor selumetinib/AZD6244 to promote BIM-dependent tumour cell death and inhibit acquired resistance. Biochem. J. 2013, 450, 285–294. [Google Scholar] [CrossRef]
- LaCasse, E.C.; Cherton-Horvat, G.G.; Hewitt, K.E.; Jerome, L.J.; Morris, S.J.; Kandimalla, E.R.; Yu, D.; Wang, H.; Wang, W.; Zhang, R.; et al. Preclinical characterization of AEG35156/GEM 640, a second-generation antisense oligonucleotide targeting X-linked inhibitor of apoptosis. Clin. Cancer Res. 2006, 12, 5231–5241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fichtner, M.; Bozkurt, E.; Salvucci, M.; McCann, C.; McAllister, K.A.; Halang, L.; Düssmann, H.; Kinsella, S.; Crawford, N.; Sessler, T.; et al. Molecular subtype-specific responses of colon cancer cells to the SMAC mimetic Birinapant. Cell Death Dis. 2020, 11, 1020. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Tsuno, N.H.; Sunami, E.; Tsurita, G.; Kawai, K.; Okaji, Y.; Nishikawa, T.; Shuno, Y.; Hongo, K.; Hiyoshi, M.; et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010, 10, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Takeuchi, T.; Kuwano, H. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann. Surg. Oncol. 2009, 16, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1alpha and Bcl-2 and activating the beclin 1/hVps34 complex. Cancer Res. 2010, 70, 5942–5952. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Gao, S.; Wang, D.; Song, D.; Feng, Y. Colorectal cancer cells are resistant to anti-EGFR monoclonal antibody through adapted autophagy. Am. J. Transl. Res. 2016, 8, 1190–1196. [Google Scholar]
- Noman, M.Z.; Parpal, S.; Van Moer, K.; Xiao, M.; Yu, Y.; Viklund, J.; De Milito, A.; Hasmim, M.; Andersson, M.; Amaravadi, R.K.; et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci. Adv. 2020, 6, eaax7881. [Google Scholar] [CrossRef]
- Chen, P.; Li, X.; Zhang, R.; Liu, S.; Xiang, Y.; Zhang, M.; Chen, X.; Pan, T.; Yan, L.; Feng, J.; et al. Combinative treatment of β-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics 2020, 10, 5107–5119. [Google Scholar] [CrossRef]
- Zhang, X.; Sui, S.; Wang, L.; Li, H.; Zhang, L.; Xu, S.; Zheng, X. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J. Cell Physiol. 2020, 235, 3425–3437. [Google Scholar] [CrossRef]
- Turkington, R.C.; Longley, D.B.; Allen, W.L.; Stevenson, L.; McLaughlin, K.; Dunne, P.D.; Blayney, J.K.; Salto-Tellez, M.; Van Schaeybroeck, S.; Johnston, P.G. Fibroblast growth factor receptor 4 (FGFR4): A targetable regulator of drug resistance in colorectal cancer. Cell Death Dis. 2014, 5, e1046. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.-S.; Sun, E.-G.; Choi, J.-N.; Kim, D.-H.; Kim, J.-H.; Ryu, K.-H.; Shim, H.-J.; Hwang, J.-E.; Bae, W.-K.; Kim, H.-R.; et al. Fibroblast growth factor receptor 4 increases epidermal growth factor receptor (EGFR) signaling by inducing amphiregulin expression and attenuates response to EGFR inhibitors in colon cancer. Cancer Sci. 2020, 111, 3268–3278. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, S.; Martini, G.; Rinaldi, B.; Martinelli, E.; Donniacuo, M.; Berrino, L.; Vitagliano, D.; Morgillo, F.; Barra, G.; De Palma, R.; et al. Primary and Acquired Resistance of Colorectal Cancer to Anti-EGFR Monoclonal Antibody Can Be Overcome by Combined Treatment of Regorafenib with Cetuximab. Clin. Cancer Res. 2015, 21, 2975–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanley, C.J.; Mellone, M.; Ford, K.; Thirdborough, S.M.; Mellows, T.; Frampton, S.J.; Smith, D.M.; Harden, E.; Szyndralewiez, C.; Bullock, M.; et al. Targeting the Myofibroblastic Cancer-Associated Fibroblast Phenotype Through Inhibition of NOX4. J. Natl. Cancer Inst. 2018, 110, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Garton, A.J.; Seibel, S.; Lopresti-Morrow, L.; Crew, L.; Janson, N.; Mandiyan, S.; Trombetta, E.S.; Pankratz, S.; LaVallee, T.M.; Gedrich, R. Anti-KIT Monoclonal Antibody Treatment Enhances the Antitumor Activity of Immune Checkpoint Inhibitors by Reversing Tumor-Induced Immunosuppression. Mol. Cancer Ther. 2017, 16, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, Y.; Hao, L.; Hou, A.; Chen, X.; Li, Y.; Wang, R.; Luo, P.; Ruan, Z.; Ou, J.; et al. Macrophages induce resistance to 5-fluorouracil chemotherapy in colorectal cancer through the release of putrescine. Cancer Lett. 2016, 381, 305–313. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Xie, Y.; Xiong, Y.; Liu, S.; Qiu, C.; Zhu, Z.; Mao, H.; Yu, M.; Wang, X. TLR 7/8 agonist reverses oxaliplatin resistance in colorectal cancer via directing the myeloid-derived suppressor cells to tumoricidal M1-macrophages. Cancer Lett. 2020, 469, 173–185. [Google Scholar] [CrossRef]
- Lu, W.; Yu, W.; He, J.; Liu, W.; Yang, J.; Lin, X.; Zhang, Y.; Wang, X.; Jiang, W.; Luo, J.; et al. Reprogramming immunosuppressive myeloid cells facilitates immunotherapy for colorectal cancer. EMBO Mol. Med. 2021, 13, e12798. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.-L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Kim, C.G.; Jang, M.; Kim, Y.; Leem, G.; Kim, K.H.; Lee, H.; Kim, T.-S.; Choi, S.J.; Kim, H.-D.; Han, J.W.; et al. VEGF-A drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci. Immunol. 2019, 4, eaay0555. [Google Scholar] [CrossRef]
- Ding, C.; Li, L.; Yang, T.; Fan, X.; Wu, G. Combined application of anti-VEGF and anti-EGFR attenuates the growth and angiogenesis of colorectal cancer mainly through suppressing AKT and ERK signaling in mice model. BMC Cancer 2016, 16, 791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poindessous, V.; Ouaret, D.; El Ouadrani, K.; Battistella, A.; Mégalophonos, V.F.; Kamsu-Kom, N.; Petitprez, A.; Escargueil, A.E.; Boudou, P.; Dumont, S.; et al. EGFR- and VEGF(R)-targeted small molecules show synergistic activity in colorectal cancer models refractory to combinations of monoclonal antibodies. Clin. Cancer Res. 2011, 17, 6522–6530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagut, C.; Argilés, G.; Ciardiello, F.; Poulsen, T.T.; Dienstmann, R.; Kragh, M.; Kopetz, S.; Lindsted, T.; Ding, C.; Vidal, J.; et al. Efficacy of Sym004 in Patients With Metastatic Colorectal Cancer with Acquired Resistance to Anti-EGFR Therapy and Molecularly Selected by Circulating Tumor DNA Analyses: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2018, 4, e175245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieu, C.H.; Beeram, M.; Harb, W.A.; Kearns, J.D.; Sloss, C.M.; Nering, R.; Adjei, A.A.; Wolf, B.B. Safety, pharmacology, and preliminary clinical activity of MM-151: An oligocolnal anti-EGFR theraputic in patients with cetuximab-resistant CRC and other refractory solid tumors. J. Clin. Oncol. 2015, 33, 647. [Google Scholar] [CrossRef]
- Elez, E.; Hendlisz, A.; Delaunoit, T.; Sastre, J.; Cervantes, A.; Varea, R.; Chao, G.; Wallin, J.; Tabernero, J. Phase II study of necitumumab plus modified FOLFOX6 as first-line treatment in patients with locally advanced or metastatic colorectal cancer. Br. J. Cancer 2016, 114, 372–380. [Google Scholar] [CrossRef] [Green Version]
- Yaeger, R.; Cercek, A.; O’Reilly, E.M.; Reidy, D.L.; Kemeny, N.; Wolinsky, T.; Capanu, M.; Gollub, M.J.; Rosen, N.; Berger, M.F.; et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin. Cancer Res. 2015, 21, 1313–1320. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Morris, V.K.; El Osta, B.; Sorokin, A.V.; Janku, F.; Fu, S.; Overman, M.J.; Piha-Paul, S.; Subbiah, V.; Kee, B.; et al. Phase IB Study of Vemurafenib in Combination with Irinotecan and Cetuximab in Patients with Metastatic Colorectal Cancer with BRAFV600E Mutation. Cancer Discov. 2016, 6, 1352–1365. [Google Scholar] [CrossRef] [Green Version]
- Bowles, D.W.; Kochenderfer, M.; Cohn, A.; Sideris, L.; Nguyen, N.; Cline-Burkhardt, V.; Schnadig, I.; Choi, M.; Nabell, L.; Chaudhry, A.; et al. A Randomized, Phase II Trial of Cetuximab With or Without PX-866, an Irreversible Oral Phosphatidylinositol 3-Kinase Inhibitor, in Patients With Metastatic Colorectal Carcinoma. Clin. Colorectal Cancer 2016, 15, 337–344.e2. [Google Scholar] [CrossRef]
- van Geel, R.M.J.M.; Tabernero, J.; Elez, E.; Bendell, J.C.; Spreafico, A.; Schuler, M.; Yoshino, T.; Delord, J.-P.; Yamada, Y.; Lolkema, M.P.; et al. A Phase Ib Dose-Escalation Study of Encorafenib and Cetuximab with or without Alpelisib in Metastatic -Mutant Colorectal Cancer. Cancer Discov. 2017, 7, 610–619. [Google Scholar] [CrossRef] [Green Version]
- Reidy, D.L.; Vakiani, E.; Fakih, M.G.; Saif, M.W.; Hecht, J.R.; Goodman-Davis, N.; Hollywood, E.; Shia, J.; Schwartz, J.; Chandrawansa, K.; et al. Randomized, phase II study of the insulin-like growth factor-1 receptor inhibitor IMC-A12, with or without cetuximab, in patients with cetuximab- or panitumumab-refractory metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 4240–4246. [Google Scholar] [CrossRef] [Green Version]
- Sclafani, F.; Kim, T.Y.; Cunningham, D.; Kim, T.W.; Tabernero, J.; Schmoll, H.J.; Roh, J.K.; Kim, S.Y.; Park, Y.S.; Guren, T.K.; et al. A Randomized Phase II/III Study of Dalotuzumab in Combination With Cetuximab and Irinotecan in Chemorefractory, KRAS Wild-Type, Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2015, 107, djv258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.; Eng, C.; Nowara, E.; Swieboda-Sadlej, A.; Tebbutt, N.C.; Mitchell, E.; Davidenko, I.; Stephenson, J.; Elez, E.; Prenen, H.; et al. Randomized phase Ib/II trial of rilotumumab or ganitumab with panitumumab versus panitumumab alone in patients with wild-type KRAS metastatic colorectal cancer. Clin. Cancer Res. 2014, 20, 4240–4250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, S.A.; Lee, J.J.; George, T.J.; Wade, J.L.; Stella, P.J.; Wang, D.; Sama, A.R.; Piette, F.; Pogue-Geile, K.L.; Kim, R.S.; et al. Neratinib-Plus-Cetuximab in Quadruple-WT () Metastatic Colorectal Cancer Resistant to Cetuximab or Panitumumab: NSABP FC-7, A Phase Ib Study. Clin. Cancer Res. 2021, 27, 1612–1622. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; VanderWalde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C.; et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): An updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019, 20, 518–530. [Google Scholar] [CrossRef]
- Meulendijks, D.; Jacob, W.; Martinez-Garcia, M.; Taus, A.; Lolkema, M.P.; Voest, E.E.; Langenberg, M.H.G.; Fleitas Kanonnikoff, T.; Cervantes, A.; De Jonge, M.J.; et al. First-in-Human Phase I Study of Lumretuzumab, a Glycoengineered Humanized Anti-HER3 Monoclonal Antibody, in Patients with Metastatic or Advanced HER3-Positive Solid Tumors. Clin. Cancer Res. 2016, 22, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Juric, D.; Dienstmann, R.; Cervantes, A.; Hidalgo, M.; Messersmith, W.; Blumenschein, G.R.; Tabernero, J.; Roda, D.; Calles, A.; Jimeno, A.; et al. Safety and Pharmacokinetics/Pharmacodynamics of the First-in-Class Dual Action HER3/EGFR Antibody MEHD7945A in Locally Advanced or Metastatic Epithelial Tumors. Clin. Cancer Res. 2015, 21, 2462–2470. [Google Scholar] [CrossRef] [Green Version]
- Rimassa, L.; Bozzarelli, S.; Pietrantonio, F.; Cordio, S.; Lonardi, S.; Toppo, L.; Zaniboni, A.; Bordonaro, R.; Di Bartolomeo, M.; Tomasello, G.; et al. Phase II Study of Tivantinib and Cetuximab in Patients With KRAS Wild-type Metastatic Colorectal Cancer With Acquired Resistance to EGFR Inhibitors and Emergence of MET Overexpression: Lesson Learned for Future Trials With EGFR/MET Dual Inhibition. Clin. Colorectal Cancer 2019, 18, 125–132.e2. [Google Scholar] [CrossRef]
- Eng, C.; Bessudo, A.; Hart, L.L.; Severtsev, A.; Gladkov, O.; Müller, L.; Kopp, M.V.; Vladimirov, V.; Langdon, R.; Kotiv, B.; et al. A randomized, placebo-controlled, phase 1/2 study of tivantinib (ARQ 197) in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with wild-type KRAS who have received first-line systemic therapy. Int. J. Cancer 2016, 139, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Delord, J.-P.; Argilés, G.; Fayette, J.; Wirth, L.; Kasper, S.; Siena, S.; Mesia, R.; Berardi, R.; Cervantes, A.; Dekervel, J.; et al. A phase 1b study of the MET inhibitor capmatinib combined with cetuximab in patients with MET-positive colorectal cancer who had progressed following anti-EGFR monoclonal antibody treatment. Investig. New Drugs 2020, 38, 1774–1783. [Google Scholar] [CrossRef]
- Clarke, J.M.; Blobe, G.C.; Strickler, J.H.; Uronis, H.E.; Zafar, S.Y.; Morse, M.; Dropkin, E.; Howard, L.; O’Neill, M.; Rushing, C.N.; et al. A phase Ib study of the combination regorafenib with PF-03446962 in patients with refractory metastatic colorectal cancer (REGAL-1 trial). Cancer Chemother. Pharmacol. 2019, 84, 909–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellmann, M.D.; Kim, T.W.; Lee, C.B.; Goh, B.C.; Miller, W.H.; Oh, D.Y.; Jamal, R.; Chee, C.E.; Chow, L.Q.M.; Gainor, J.F.; et al. Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann. Oncol. 2019, 30, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.; Kim, T.W.; Bendell, J.; Argilés, G.; Tebbutt, N.C.; Di Bartolomeo, M.; Falcone, A.; Fakih, M.; Kozloff, M.; Segal, N.H.; et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): A multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2019, 20, 849–861. [Google Scholar] [CrossRef]
- Hochster, H.S.; Bendell, J.C.; Cleary, J.M.; Foster, P.; Zhang, W.; He, X.; Hernandez, G.; Iizuka, K.; Eckhardt, S.G. Efficacy and safety of atezolizumab (atezo) and bevacizumab (bev) in a phase Ib study of microsatellite instability (MSI)-high metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2017, 35, 673. [Google Scholar] [CrossRef]
- Mettu, N.B.; Twohy, E.; Ou, F.S.; Halfdanarson, T.R.; Lenz, H.J.; Breakstone, R.; Boland, P.M.; Crysler, O.; Wu, C.; Grothey, A.; et al. 533PD-BACCI: A phase II randomized, double-blind, multicenter, placebo-controlled study of capecitabine (C) bevacizumab (B) plus atezolizumab (A) or placebo (P) in refractory metastatic colorectal cancer (mCRC): An ACCRU network study. Ann. Oncol. 2019, 30, v203. [Google Scholar] [CrossRef]
- Subbiah, V.; Khawaja, M.R.; Hong, D.S.; Amini, B.; Yungfang, J.; Liu, H.; Johnson, A.; Schrock, A.B.; Ali, S.M.; Sun, J.X.; et al. First-in-human trial of multikinase VEGF inhibitor regorafenib and anti-EGFR antibody cetuximab in advanced cancer patients. JCI Insight 2017, 2, e90380. [Google Scholar] [CrossRef]
- Kciuk, M.; Marciniak, B.; Kontek, R. Irinotecan-Still an Important Player in Cancer Chemotherapy: A Comprehensive Overview. Int. J. Mol. Sci. 2020, 21, 4919. [Google Scholar] [CrossRef]
- Xu, Y.; Villalona-Calero, M.A. Irinotecan: Mechanisms of tumor resistance and novel strategies for modulating its activity. Ann. Oncol. 2002, 13, 1841–1851. [Google Scholar] [CrossRef]
- Shaojun, C.; Li, H.; Haixin, H.; Guisheng, L. Expression of Topoisomerase 1 and carboxylesterase 2 correlates with irinotecan treatment response in metastatic colorectal cancer. Cancer Biol. Ther. 2018, 19, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Zhang, W.; Ma, M.K.; McLeod, H.L. Human carboxylesterase 2 is commonly expressed in tumor tissue and is correlated with activation of irinotecan. Clin. Cancer Res. 2002, 8, 2605–2611. [Google Scholar]
- Uchino, J.; Takayama, K.; Harada, A.; Sone, T.; Harada, T.; Curiel, D.T.; Kuroki, M.; Nakanishi, Y. Tumor targeting carboxylesterase fused with anti-CEA scFv improve the anticancer effect with a less toxic dose of irinotecan. Cancer Gene Ther. 2008, 15, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oosterhoff, D.; Overmeer, R.M.; de Graaf, M.; van der Meulen, I.H.; Giaccone, G.; van Beusechem, V.W.; Haisma, H.J.; Pinedo, H.M.; Gerritsen, W.R. Adenoviral vector-mediated expression of a gene encoding secreted, EpCAM-targeted carboxylesterase-2 sensitises colon cancer spheroids to CPT-11. Br. J. Cancer 2005, 92, 882–887. [Google Scholar] [CrossRef] [Green Version]
- Wierdl, M.; Morton, C.L.; Weeks, J.K.; Danks, M.K.; Harris, L.C.; Potter, P.M. Sensitization of human tumor cells to CPT-11 via adenoviral-mediated delivery of a rabbit liver carboxylesterase. Cancer Res. 2001, 61, 5078–5082. [Google Scholar] [PubMed]
- Gutova, M.; Najbauer, J.; Chen, M.Y.; Potter, P.M.; Kim, S.U.; Aboody, K.S. Therapeutic targeting of melanoma cells using neural stem cells expressing carboxylesterase, a CPT-11 activating enzyme. Curr. Stem Cell Res. Ther. 2010, 5, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Yi, B.-R.; Kim, S.U.; Choi, K.-C. Co-treatment with therapeutic neural stem cells expressing carboxyl esterase and CPT-11 inhibit growth of primary and metastatic lung cancers in mice. Oncotarget 2014, 5, 12835–12848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.S.; Yoon, K.; Choi, S.-A.; Yoon, S.-B.; Kim, S.U.; Lee, H.J. Tumor-specific gene therapy for pancreatic cancer using human neural stem cells encoding carboxylesterase. Oncotarget 2016, 7, 75319–75327. [Google Scholar] [CrossRef] [Green Version]
- Saif, M.W.; Syrigos, K.N.; Katirtzoglou, N.A. S-1: A promising new oral fluoropyrimidine derivative. Expert Opin. Investig. Drugs 2009, 18, 335–348. [Google Scholar] [CrossRef]
- Zhang, X.; Soong, R.; Wang, K.; Li, L.; Davie, J.R.; Guarcello, V.; Diasio, R.B. Suppression of DPYD expression in RKO cells via DNA methylation in the regulatory region of the DPYD promoter: A potentially important epigenetic mechanism regulating DPYD expression. Biochem. Cell Biol. 2007, 85, 337–346. [Google Scholar] [CrossRef]
- Ezzeldin, H.H.; Lee, A.M.; Mattison, L.K.; Diasio, R.B. Methylation of the DPYD promoter: An alternative mechanism for dihydropyrimidine dehydrogenase deficiency in cancer patients. Clin. Cancer Res. 2005, 11, 8699–8705. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.J.; Chestnut, W.G.; Merrill, B.M.; Spector, T. Mechanism-based inactivation of dihydropyrimidine dehydrogenase by 5-ethynyluracil. J. Biol. Chem. 1992, 267, 5236–5242. [Google Scholar] [CrossRef]
- Spector, T.; Cao, S.; Rustum, Y.M.; Harrington, J.A.; Porter, D.J. Attenuation of the antitumor activity of 5-fluorouracil by (R)-5-fluoro-5,6-dihydrouracil. Cancer Res. 1995, 55, 1239–1241. [Google Scholar] [PubMed]
- Takechi, T.; Fujioka, A.; Matsushima, E.; Fukushima, M. Enhancement of the antitumour activity of 5-fluorouracil (5-FU) by inhibiting dihydropyrimidine dehydrogenase activity (DPD) using 5-chloro-2,4-dihydroxypyridine (CDHP) in human tumour cells. Eur. J. Cancer 2002, 38, 1271–1277. [Google Scholar] [CrossRef]
- Johnson, M.R.; Hageboutros, A.; Wang, K.; High, L.; Smith, J.B.; Diasio, R.B. Life-threatening toxicity in a dihydropyrimidine dehydrogenase-deficient patient after treatment with topical 5-fluorouracil. Clin Cancer Res. 1999, 5, 2006–2011. [Google Scholar]
- De Man, F.M.; Goey, A.K.L.; van Schaik, R.H.N.; Mathijssen, R.H.J.; Bins, S. Individualization of Irinotecan Treatment: A Review of Pharmacokinetics, Pharmacodynamics, and Pharmacogenetics. Clin. Pharmacokinet. 2018, 57, 1229–1254. [Google Scholar] [CrossRef] [Green Version]
- Van der Bol, J.M.; Mathijssen, R.H.J.; Loos, W.J.; Friberg, L.E.; van Schaik, R.H.N.; de Jonge, M.J.A.; Planting, A.S.T.; Verweij, J.; Sparreboom, A.; de Jong, F.A. Cigarette smoking and irinotecan treatment: Pharmacokinetic interaction and effects on neutropenia. J. Clin. Oncol. 2007, 25, 2719–2726. [Google Scholar] [CrossRef] [PubMed]
- Mathijssen, R.H.J.; Verweij, J.; de Bruijn, P.; Loos, W.J.; Sparreboom, A. Effects of St. John’s wort on irinotecan metabolism. J. Natl. Cancer Inst. 2002, 94, 1247–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innocenti, F.; Undevia, S.D.; Ramírez, J.; Mani, S.; Schilsky, R.L.; Vogelzang, N.J.; Prado, M.; Ratain, M.J. A phase I trial of pharmacologic modulation of irinotecan with cyclosporine and phenobarbital. Clin. Pharmacol. Ther. 2004, 76, 490–502. [Google Scholar] [CrossRef]
- van der Bol, J.M.; Visser, T.J.; Loos, W.J.; de Jong, F.A.; Wiemer, E.A.C.; van Aken, M.O.; Planting, A.S.; Schellens, J.H.; Verweij, J.; Mathijssen, R.H.J. Effects of methimazole on the elimination of irinotecan. Cancer Chemother. Pharmacol. 2011, 67, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Kehrer, D.F.; Sparreboom, A.; Verweij, J.; de Bruijn, P.; Nierop, C.A.; van de Schraaf, J.; Ruijgrok, E.J.; de Jonge, M.J. Modulation of irinotecan-induced diarrhea by cotreatment with neomycin in cancer patients. Clin. Cancer Res. 2001, 7, 1136–1141. [Google Scholar]
- Kong, R.; Liu, T.; Zhu, X.; Ahmad, S.; Williams, A.L.; Phan, A.T.; Zhao, H.; Scott, J.E.; Yeh, L.-A.; Wong, S.T.C. Old drug new use—Amoxapine and its metabolites as potent bacterial β-glucuronidase inhibitors for alleviating cancer drug toxicity. Clin. Cancer Res. 2014, 20, 3521–3530. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, J.-F.; Bernard, O.; Villeneuve, L.; Têtu, B.; Guillemette, C. Irinotecan inactivation is modulated by epigenetic silencing of UGT1A1 in colon cancer. Clin. Cancer Res. 2006, 12, 1850–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bélanger, A.-S.; Tojcic, J.; Harvey, M.; Guillemette, C. Regulation of UGT1A1 and HNF1 transcription factor gene expression by DNA methylation in colon cancer cells. BMC Mol. Biol. 2010, 11, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.-H.; Chen, Z.-S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updates 2016, 27, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Huang, S.-M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M.; Ling, V. The molecular basis of multidrug resistance in cancer: The early years of P-glycoprotein research. FEBS Lett. 2006, 580, 998–1009. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Lin, C.; Zhang, Y.; Zhang, X.; Zhang, C.; Zhang, P.; Xie, X.; Ren, Z. miR-506 enhances the sensitivity of human colorectal cancer cells to oxaliplatin by suppressing MDR1/P-gp expression. Cell Prolif. 2017, 50, e12341. [Google Scholar] [CrossRef]
- Du, J.; He, Y.; Li, P.; Wu, W.; Chen, Y.; Ruan, H. IL-8 regulates the doxorubicin resistance of colorectal cancer cells via modulation of multidrug resistance 1 (MDR1). Cancer Chemother. Pharmacol. 2018, 81, 1111–1119. [Google Scholar] [CrossRef]
- Robey, R.W.; Shukla, S.; Steadman, K.; Obrzut, T.; Finley, E.M.; Ambudkar, S.V.; Bates, S.E. Inhibition of ABCG2-mediated transport by protein kinase inhibitors with a bisindolylmaleimide or indolocarbazole structure. Mol. Cancer Ther. 2007, 6, 1877–1885. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Qiu, M.; Tang, Q.-L.; Liu, M.; Lang, N.; Bi, F. Establishment and biological characteristics of oxaliplatin-resistant human colon cancer cell lines. Chin. J. Cancer 2010, 29, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Herraez, E.; Gonzalez-Sanchez, E.; Vaquero, J.; Romero, M.R.; Serrano, M.A.; Marin, J.J.G.; Briz, O. Cisplatin-induced chemoresistance in colon cancer cells involves FXR-dependent and FXR-independent up-regulation of ABC proteins. Mol. Pharm. 2012, 9, 2565–2576. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-P.; Ohnuma, S.; Ambudkar, S.V. Discovering natural product modulators to overcome multidrug resistance in cancer chemotherapy. Curr. Pharm. Biotechnol. 2011, 12, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Lu, F.-Y.; Shi, R.-H.; Feng, Y.-D.; Zhao, X.-D.; Lu, Z.-P.; Xiao, L.; Zhou, G.-Q.; Qiu, J.-M.; Cheng, C.-E. MiR-26b regulates 5-FU-resistance in human colorectal cancer via down-regulation of Pgp. Am. J. Cancer Res. 2018, 8, 2518–2527. [Google Scholar] [PubMed]
- Gao, R.; Fang, C.; Xu, J.; Tan, H.; Li, P.; Ma, L. LncRNA CACS15 contributes to oxaliplatin resistance in colorectal cancer by positively regulating ABCC1 through sponging miR-145. Arch. Biochem. Biophys. 2019, 663, 183–191. [Google Scholar] [CrossRef]
- Fan, H.; Zhu, J.-H.; Yao, X.-Q. Knockdown of long non-coding RNA PVT1 reverses multidrug resistance in colorectal cancer cells. Mol. Med. Rep. 2018, 17, 8309–8315. [Google Scholar] [CrossRef] [Green Version]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Grogan, L.; Sotos, G.A.; Allegra, C.J. Leucovorin modulation of fluorouracil. Oncology 1993, 7, 63–72. [Google Scholar]
- Tan, K.B.; Mattern, M.R.; Eng, W.K.; McCabe, F.L.; Johnson, R.K. Nonproductive rearrangement of DNA topoisomerase I and II genes: Correlation with resistance to topoisomerase inhibitors. J. Natl. Cancer Inst. 1989, 81, 1732–1735. [Google Scholar] [CrossRef]
- Li, X.G.; Haluska, P.; Hsiang, Y.H.; Bharti, A.; Kufe, D.W.; Rubin, E.H. Identification of topoisomerase I mutations affecting both DNA cleavage and interaction with camptothecin. Ann. N. Y. Acad. Sci. 1996, 803, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Fornasier, G.; Francescon, S.; Baldo, P. An Update of Efficacy and Safety of Cetuximab in Metastatic Colorectal Cancer: A Narrative Review. Adv. Ther. 2018, 35, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Cremolini, C.; Fioravanti, A.; Orlandi, P.; Salvatore, L.; Masi, G.; Schirripa, M.; Di Desidero, T.; Antoniotti, C.; Canu, B.; et al. EGFR ligands as pharmacodynamic biomarkers in metastatic colorectal cancer patients treated with cetuximab and irinotecan. Target Oncol. 2014, 9, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Xian, C.J. Roles of epidermal growth factor family in the regulation of postnatal somatic growth. Endocr Rev 2007, 28, 284–296. [Google Scholar] [CrossRef] [Green Version]
- Khambata-Ford, S.; Garrett, C.R.; Meropol, N.J.; Basik, M.; Harbison, C.T.; Wu, S.; Wong, T.W.; Huang, X.; Takimoto, C.H.; Godwin, A.K.; et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J. Clin. Oncol. 2007, 25, 3230–3237. [Google Scholar] [CrossRef]
- Jacobs, B.; De Roock, W.; Piessevaux, H.; Van Oirbeek, R.; Biesmans, B.; De Schutter, J.; Fieuws, S.; Vandesompele, J.; Peeters, M.; Van Laethem, J.-L.; et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J. Clin. Oncol. 2009, 27, 5068–5074. [Google Scholar] [CrossRef]
- Seligmann, J.F.; Elliott, F.; Richman, S.D.; Jacobs, B.; Hemmings, G.; Brown, S.; Barrett, J.H.; Tejpar, S.; Quirke, P.; Seymour, M.T. Combined Epiregulin and Amphiregulin Expression Levels as a Predictive Biomarker for Panitumumab Therapy Benefit or Lack of Benefit in Patients With RAS Wild-Type Advanced Colorectal Cancer. JAMA Oncol. 2016, 2, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Fukuoka, M.; Wu, Y.-L.; Thongprasert, S.; Sunpaweravong, P.; Leong, S.-S.; Sriuranpong, V.; Chao, T.-Y.; Nakagawa, K.; Chu, D.-T.; Saijo, N.; et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J. Clin. Oncol. 2011, 29, 2866–2874. [Google Scholar] [CrossRef]
- Chung, K.Y.; Shia, J.; Kemeny, N.E.; Shah, M.; Schwartz, G.K.; Tse, A.; Hamilton, A.; Pan, D.; Schrag, D.; Schwartz, L.; et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J. Clin. Oncol. 2005, 23, 1803–1810. [Google Scholar] [CrossRef]
- Hecht, J.R.; Mitchell, E.; Neubauer, M.A.; Burris, H.A.; Swanson, P.; Lopez, T.; Buchanan, G.; Reiner, M.; Gansert, J.; Berlin, J. Lack of correlation between epidermal growth factor receptor status and response to Panitumumab monotherapy in metastatic colorectal cancer. Clin. Cancer Res. 2010, 16, 2205–2213. [Google Scholar] [CrossRef] [Green Version]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Bellosillo, B.; Siravegna, G.; Martínez, A.; Cañadas, I.; Lazzari, L.; Ferruz, N.; Russo, M.; Misale, S.; González, I.; et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin. Cancer Res. 2015, 21, 2157–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Emburgh, B.O.; Arena, S.; Siravegna, G.; Lazzari, L.; Crisafulli, G.; Corti, G.; Mussolin, B.; Baldi, F.; Buscarino, M.; Bartolini, A.; et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat. Commun. 2016, 7, 13665. [Google Scholar] [CrossRef]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.-W.; Hsu, J.-M.; Xia, W.; Wang, H.-L.; Wang, Y.-N.; Chang, W.-C.; Arold, S.T.; Chou, C.-K.; Tsou, P.-H.; Yamaguchi, H.; et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J. Clin. Investig. 2015, 125, 4529–4543. [Google Scholar] [CrossRef] [Green Version]
- Hochster, H.S.; Hart, L.L.; Ramanathan, R.K.; Childs, B.H.; Hainsworth, J.D.; Cohn, A.L.; Wong, L.; Fehrenbacher, L.; Abubakr, Y.; Saif, M.W.; et al. Safety and efficacy of oxaliplatin and fluoropyrimidine regimens with or without bevacizumab as first-line treatment of metastatic colorectal cancer: Results of the TREE Study. J. Clin. Oncol. 2008, 26, 3523–3529. [Google Scholar] [CrossRef]
- Sánchez-Martín, F.J.; Bellosillo, B.; Gelabert-Baldrich, M.; Dalmases, A.; Cañadas, I.; Vidal, J.; Martinez, A.; Argilés, G.; Siravegna, G.; Arena, S.; et al. The First-in-class Anti-EGFR Antibody Mixture Sym004 Overcomes Cetuximab Resistance Mediated by EGFR Extracellular Domain Mutations in Colorectal Cancer. Clin. Cancer Res. 2016, 22, 3260–3267. [Google Scholar] [CrossRef] [Green Version]
- El-Deiry, W.S.; Goldberg, R.M.; Lenz, H.-J.; Shields, A.F.; Gibney, G.T.; Tan, A.R.; Brown, J.; Eisenberg, B.; Heath, E.I.; Phuphanich, S.; et al. The current state of molecular testing in the treatment of patients with solid tumors, 2019. CA Cancer J. Clin. 2019, 69, 305–343. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Köhne, C.-H.; Láng, I.; Folprecht, G.; Nowacki, M.P.; Cascinu, S.; Shchepotin, I.; Maurel, J.; Cunningham, D.; Tejpar, S.; et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: Updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J. Clin. Oncol. 2011, 29, 2011–2019. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chang Chien, C.-R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Peeters, M.; Price, T.J.; Cervantes, A.; Sobrero, A.F.; Ducreux, M.; Hotko, Y.; André, T.; Chan, E.; Lordick, F.; Punt, C.J.A.; et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 4706–4713. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Normanno, N.; Maiello, E.; Martinelli, E.; Troiani, T.; Pisconti, S.; Giuliani, F.; Barone, C.; Cartenì, G.; Rachiglio, A.M.; et al. Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next-generation sequencing: Findings from the CAPRI-GOIM trial. Ann. Oncol. 2014, 25, 1756–1761. [Google Scholar] [CrossRef] [PubMed]
- Bokemeyer, C.; Köhne, C.H.; Ciardiello, F.; Lenz, H.J.; Heinemann, V.; Klinkhardt, U.; Beier, F.; Duecker, K.; van Krieken, J.H.; Tejpar, S. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur. J. Cancer 2015, 51, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Blons, H.; Emile, J.F.; Le Malicot, K.; Julié, C.; Zaanan, A.; Tabernero, J.; Mini, E.; Folprecht, G.; Van Laethem, J.L.; Thaler, J.; et al. Prognostic value of KRAS mutations in stage III colon cancer: Post hoc analysis of the PETACC8 phase III trial dataset. Ann. Oncol. 2014, 25, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Liu, Y.; Wu, K.; Luo, H.; Cui, L. AMPK activation overcomes anti-EGFR antibody resistance induced by KRAS mutation in colorectal cancer. Cell Commun. Signal. 2020, 18, 115. [Google Scholar] [CrossRef]
- Dunn, E.F.; Iida, M.; Myers, R.A.; Campbell, D.A.; Hintz, K.A.; Armstrong, E.A.; Li, C.; Wheeler, D.L. Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab. Oncogene 2011, 30, 561–574. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef]
- Shinozaki, E.; Yoshino, T.; Yamazaki, K.; Muro, K.; Yamaguchi, K.; Nishina, T.; Yuki, S.; Shitara, K.; Bando, H.; Mimaki, S.; et al. Clinical significance of BRAF non-V600E mutations on the therapeutic effects of anti-EGFR monoclonal antibody treatment in patients with pretreated metastatic colorectal cancer: The Biomarker Research for anti-EGFR monoclonal Antibodies by Comprehensive Cancer genomics (BREAC) study. Br. J. Cancer 2017, 117, 1450–1458. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, H.; Bardelli, A.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.E. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 2002, 418, 934. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.-Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated V600E-Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J. Clin. Oncol. 2021, 39, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Hobor, S.; Bertotti, A.; Zecchin, D.; Huang, S.; Galimi, F.; Cottino, F.; Prahallad, A.; Grernrum, W.; Tzani, A.; et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014, 7, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.-H.; Wang, Y.-Z.; Tu, J.; Liu, C.-W.; Yuan, Y.-J.; Lin, R.; He, W.-L.; Cai, S.-R.; He, Y.-L.; Ye, J.-N. Anti-EGFR therapy in metastatic colorectal cancer: Mechanisms and potential regimens of drug resistance. Gastroenterol. Rep. 2020, 8, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Martini, M.; Molinari, F.; Veronese, S.; Nichelatti, M.; Artale, S.; Di Nicolantonio, F.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009, 69, 1851–1857. [Google Scholar] [CrossRef] [Green Version]
- Samuels, Y.; Diaz, L.A.; Schmidt-Kittler, O.; Cummins, J.M.; Delong, L.; Cheong, I.; Rago, C.; Huso, D.L.; Lengauer, C.; Kinzler, K.W.; et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005, 7, 561–573. [Google Scholar] [CrossRef] [Green Version]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Mao, C.; Yang, Z.Y.; Hu, X.F.; Chen, Q.; Tang, J.L. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: A systematic review and meta-analysis. Ann. Oncol. 2012, 23, 1518–1525. [Google Scholar] [CrossRef]
- Malek, M.; Kielkowska, A.; Chessa, T.; Anderson, K.E.; Barneda, D.; Pir, P.; Nakanishi, H.; Eguchi, S.; Koizumi, A.; Sasaki, J.; et al. PTEN Regulates PI(3,4)P Signaling Downstream of Class I PI3K. Mol. Cell 2017, 68, 566–580.e10. [Google Scholar] [CrossRef] [Green Version]
- Jhawer, M.; Goel, S.; Wilson, A.J.; Montagna, C.; Ling, Y.-H.; Byun, D.-S.; Nasser, S.; Arango, D.; Shin, J.; Klampfer, L.; et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008, 68, 1953–1961. [Google Scholar] [CrossRef] [Green Version]
- Loupakis, F.; Pollina, L.; Stasi, I.; Ruzzo, A.; Scartozzi, M.; Santini, D.; Masi, G.; Graziano, F.; Cremolini, C.; Rulli, E.; et al. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 2622–2629. [Google Scholar] [CrossRef] [PubMed]
- Scartozzi, M.; Giampieri, R.; Maccaroni, E.; Mandolesi, A.; Giustini, L.; Silva, R.; Zaniboni, A.; Biscotti, T.; Biagetti, S.; Galizia, E.; et al. Analysis of HER-3, insulin growth factor-1, nuclear factor-kB and epidermal growth factor receptor gene copy number in the prediction of clinical outcome for K-RAS wild-type colorectal cancer patients receiving irinotecan-cetuximab. Ann. Oncol. 2012, 23, 1706–1712. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, A. Insulin-Like Growth Factor 1 (IGF-1) Signaling in Glucose Metabolism in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 6434. [Google Scholar] [CrossRef]
- Scartozzi, M.; Mandolesi, A.; Giampieri, R.; Pierantoni, C.; Loupakis, F.; Zaniboni, A.; Galizia, E.; Giustini, L.; Silva, R.R.; Bisonni, R.; et al. Insulin-like growth factor 1 expression correlates with clinical outcome in K-RAS wild type colorectal cancer patients treated with cetuximab and irinotecan. Int. J. Cancer 2010, 127, 1941–1947. [Google Scholar] [CrossRef]
- Zanella, E.R.; Galimi, F.; Sassi, F.; Migliardi, G.; Cottino, F.; Leto, S.M.; Lupo, B.; Erriquez, J.; Isella, C.; Comoglio, P.M.; et al. IGF2 is an actionable target that identifies a distinct subpopulation of colorectal cancer patients with marginal response to anti-EGFR therapies. Sci. Transl. Med. 2015, 7, 272ra12. [Google Scholar] [CrossRef]
- Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [CrossRef] [Green Version]
- Yonesaka, K.; Zejnullahu, K.; Okamoto, I.; Satoh, T.; Cappuzzo, F.; Souglakos, J.; Ercan, D.; Rogers, A.; Roncalli, M.; Takeda, M.; et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci. Transl. Med. 2011, 3, 99ra86. [Google Scholar] [CrossRef] [Green Version]
- Kavuri, S.M.; Jain, N.; Galimi, F.; Cottino, F.; Leto, S.M.; Migliardi, G.; Searleman, A.C.; Shen, W.; Monsey, J.; Trusolino, L.; et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015, 5, 832–841. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, B.S.; Kljavin, N.M.; Stawiski, E.W.; Chan, E.; Parikh, C.; Durinck, S.; Chaudhuri, S.; Pujara, K.; Guillory, J.; Edgar, K.A.; et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell 2013, 23, 603–617. [Google Scholar] [CrossRef] [Green Version]
- Loree, J.M.; Bailey, A.M.; Johnson, A.M.; Yu, Y.; Wu, W.; Bristow, C.A.; Davis, J.S.; Shaw, K.R.; Broaddus, R.; Banks, K.C.; et al. Molecular Landscape of ERBB2/ERBB3 Mutated Colorectal Cancer. J. Natl. Cancer Inst. 2018, 110, 1409–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seligmann, J.F.; Hatch, A.J.; Richman, S.D.; Elliott, F.; Jacobs, B.; Brown, S.; Hurwitz, H.; Barrett, J.H.; Quirke, P.; Nixon, A.B.; et al. Association of Tumor HER3 Messenger RNA Expression With Panitumumab Efficacy in Advanced Colorectal Cancer. JAMA Oncol. 2018, 4, 564–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, M.; Stolz, D.B.; Esplen, J.E.; Dorko, K.; Michalopoulos, G.K.; Strom, S.C. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J. Biol. Chem. 2000, 275, 8806–8811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrantonio, F.; Vernieri, C.; Siravegna, G.; Mennitto, A.; Berenato, R.; Perrone, F.; Gloghini, A.; Tamborini, E.; Lonardi, S.; Morano, F.; et al. Heterogeneity of Acquired Resistance to Anti-EGFR Monoclonal Antibodies in Patients with Metastatic Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2414–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrantonio, F.; Oddo, D.; Gloghini, A.; Valtorta, E.; Berenato, R.; Barault, L.; Caporale, M.; Busico, A.; Morano, F.; Gualeni, A.V.; et al. MET-Driven Resistance to Dual EGFR and BRAF Blockade May Be Overcome by Switching from EGFR to MET Inhibition in BRAF-Mutated Colorectal Cancer. Cancer Discov. 2016, 6, 963–971. [Google Scholar] [CrossRef] [Green Version]
- Troiani, T.; Martinelli, E.; Napolitano, S.; Vitagliano, D.; Ciuffreda, L.P.; Costantino, S.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A.; et al. Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin. Cancer Res. 2013, 19, 6751–6765. [Google Scholar] [CrossRef] [Green Version]
- Adjei, A.A.; Schwartz, B.; Garmey, E. Early clinical development of ARQ 197, a selective, non-ATP-competitive inhibitor targeting MET tyrosine kinase for the treatment of advanced cancers. Oncologist 2011, 16, 788–799. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Bugter, J.M.; Fenderico, N.; Maurice, M.M. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat. Rev. Cancer 2021, 21, 5–21. [Google Scholar] [CrossRef]
- Yuan, S.; Tao, F.; Zhang, X.; Zhang, Y.; Sun, X.; Wu, D. Role of Wnt/-Catenin Signaling in the Chemoresistance Modulation of Colorectal Cancer. BioMed Res. Int. 2020, 2020, 9390878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Li, C. Convergence between Wnt-β-catenin and EGFR signaling in cancer. Mol. Cancer 2010, 9, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grainger, S.; Nguyen, N.; Richter, J.; Setayesh, J.; Lonquich, B.; Oon, C.H.; Wozniak, J.M.; Barahona, R.; Kamei, C.N.; Houston, J.; et al. EGFR is required for Wnt9a-Fzd9b signalling specificity in haematopoietic stem cells. Nat. Cell Biol. 2019, 21, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhao, X.; Liu, Q.; Li, C.; Graves-Deal, R.; Cao, Z.; Singh, B.; Franklin, J.L.; Wang, J.; Hu, H.; et al. lncRNA MIR100HG-derived miR-100 and miR-125b mediate cetuximab resistance via Wnt/β-catenin signaling. Nat. Med. 2017, 23, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Siddiqui, B.A.; Anandhan, S.; Yadav, S.S.; Subudhi, S.K.; Gao, J.; Goswami, S.; Allison, J.P. The Next Decade of Immune Checkpoint Therapy. Cancer Discov. 2021, 11, 838–857. [Google Scholar] [CrossRef]
- Grasso, C.S.; Giannakis, M.; Wells, D.K.; Hamada, T.; Mu, X.J.; Quist, M.; Nowak, J.A.; Nishihara, R.; Qian, Z.R.; Inamura, K.; et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov. 2018, 8, 730–749. [Google Scholar] [CrossRef] [Green Version]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008052. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Wu, J.; Wang, W.-J.; Chen, S.; Zheng, Y.; Yu, X.; Meeth, K.; Sahraei, M.; Bothwell, A.L.M.; Chen, L.; et al. DKK2 imparts tumor immunity evasion through β-catenin-independent suppression of cytotoxic immune-cell activation. Nat. Med. 2018, 24, 262–270. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- van Loosdregt, J.; Fleskens, V.; Tiemessen, M.M.; Mokry, M.; van Boxtel, R.; Meerding, J.; Pals, C.E.G.M.; Kurek, D.; Baert, M.R.M.; Delemarre, E.M.; et al. Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity 2013, 39, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 2020, 80, 106210. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-T.; Wang, S.; Ye, Y.-J.; Du, R.-Y.; Cui, Z.-R.; Somsouk, M. Constitutive activation of Stat3 signaling pathway in human colorectal carcinoma. World J. Gastroenterol. 2004, 10, 1569–1573. [Google Scholar] [CrossRef] [PubMed]
- Spitzner, M.; Roesler, B.; Bielfeld, C.; Emons, G.; Gaedcke, J.; Wolff, H.A.; Rave-Fränk, M.; Kramer, F.; Beissbarth, T.; Kitz, J.; et al. STAT3 inhibition sensitizes colorectal cancer to chemoradiotherapy in vitro and in vivo. Int. J. Cancer 2014, 134, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, D.; Chen, X.; He, L.; Li, T.; Xu, X.; Li, M. Nuclear PKM2 contributes to gefitinib resistance via upregulation of STAT3 activation in colorectal cancer. Sci. Rep. 2015, 5, 16082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, J.D.; Young, H.A. IFN-γ: A cytokine at the right time, is in the right place. Semin. Immunol. 2019, 43, 101280. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int. Rev. Immunol. 2009, 28, 239–260. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Kursunel, M.A.; Esendagli, G. The untold story of IFN-γ in cancer biology. Cytokine Growth Factor Rev. 2016, 31, 73–81. [Google Scholar] [CrossRef]
- Ikeda, H.; Old, L.J.; Schreiber, R.D. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002, 13, 95–109. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Murthy, A.; Gerber, S.A.; Koch, C.J.; Lord, E.M. Intratumoral Hypoxia Reduces IFN-γ-Mediated Immunity and MHC Class I Induction in a Preclinical Tumor Model. Immunohorizons 2019, 3, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriegsman, B.A.; Vangala, P.; Chen, B.J.; Meraner, P.; Brass, A.L.; Garber, M.; Rock, K.L. Frequent Loss of IRF2 in Cancers Leads to Immune Evasion through Decreased MHC Class I Antigen Presentation and Increased PD-L1 Expression. J. Immunol. 2019, 203, 1999–2010. [Google Scholar] [CrossRef] [PubMed]
- Fend, L.; Yamazaki, T.; Remy, C.; Fahrner, C.; Gantzer, M.; Nourtier, V.; Préville, X.; Quéméneur, E.; Kepp, O.; Adam, J.; et al. Immune Checkpoint Blockade, Immunogenic Chemotherapy or IFN-α Blockade Boost the Local and Abscopal Effects of Oncolytic Virotherapy. Cancer Res. 2017, 77, 4146–4157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef]
- Romano, G.; Santi, L.; Bianco, M.R.; Giuffrè, M.R.; Pettinato, M.; Bugarin, C.; Garanzini, C.; Savarese, L.; Leoni, S.; Cerrito, M.G.; et al. The TGF-β pathway is activated by 5-fluorouracil treatment in drug resistant colorectal carcinoma cells. Oncotarget 2016, 7, 22077–22091. [Google Scholar] [CrossRef] [Green Version]
- Quan, Q.; Zhong, F.; Wang, X.; Chen, K.; Guo, L. PAR2 Inhibition Enhanced the Sensitivity of Colorectal Cancer Cells to 5-FU and Reduced EMT Signaling. Oncol. Res. 2019, 27, 779–788. [Google Scholar] [CrossRef]
- Brunen, D.; Willems, S.M.; Kellner, U.; Midgley, R.; Simon, I.; Bernards, R. TGF-β: An emerging player in drug resistance. Cell Cycle 2013, 12, 2960–2968. [Google Scholar] [CrossRef] [Green Version]
- Cunha, S.I.; Pietras, K. ALK1 as an emerging target for antiangiogenic therapy of cancer. Blood 2011, 117, 6999–7006. [Google Scholar] [CrossRef] [Green Version]
- Mazzocca, A.; Fransvea, E.; Lavezzari, G.; Antonaci, S.; Giannelli, G. Inhibition of transforming growth factor beta receptor I kinase blocks hepatocellular carcinoma growth through neo-angiogenesis regulation. Hepatology 2009, 50, 1140–1151. [Google Scholar] [CrossRef]
- Zhang, M.; Kleber, S.; Röhrich, M.; Timke, C.; Han, N.; Tuettenberg, J.; Martin-Villalba, A.; Debus, J.; Peschke, P.; Wirkner, U.; et al. Blockade of TGF-β signaling by the TGFβR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011, 71, 7155–7167. [Google Scholar] [CrossRef] [Green Version]
- Akbari, A.; Amanpour, S.; Muhammadnejad, S.; Ghahremani, M.H.; Ghaffari, S.H.; Dehpour, A.R.; Mobini, G.R.; Shidfar, F.; Abastabar, M.; Khoshzaban, A.; et al. Evaluation of antitumor activity of a TGF-beta receptor I inhibitor (SD-208) on human colon adenocarcinoma. DARU J. Pharm. Sci. 2014, 22, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Duran, A.; L’Hermitte, A.; Shelton, P.M.; Nakanishi, N.; Reina-Campos, M.; Huang, J.; Soldevila, F.; Baaten, B.J.G.; Tauriello, D.V.F.; et al. Simultaneous Loss of Both Atypical Protein Kinase C Genes in the Intestinal Epithelium Drives Serrated Intestinal Cancer by Impairing Immunosurveillance. Immunity 2018, 49, 1132–1147.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef]
- Bachman, K.E.; Argani, P.; Samuels, Y.; Silliman, N.; Ptak, J.; Szabo, S.; Konishi, H.; Karakas, B.; Blair, B.G.; Lin, C.; et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol. Ther. 2004, 3, 772–775. [Google Scholar] [CrossRef] [Green Version]
- Lastwika, K.J.; Wilson, W.; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Sharma, A.; Lin, Y.; Wu, Y.; He, Q.; Gu, Y.; Xu, Z.P.; Monteiro, M.; Gu, W. Insluin and epithelial growth factor (EGF) promote programmed death ligand 1(PD-L1) production and transport in colon cancer stem cells. BMC Cancer 2019, 19, 153. [Google Scholar] [CrossRef]
- Chida, K.; Kawazoe, A.; Kawazu, M.; Suzuki, T.; Nakamura, Y.; Nakatsura, T.; Kuwata, T.; Ueno, T.; Kuboki, Y.; Kotani, D.; et al. A Low Tumor Mutational Burden and Mutations Are Predictors of a Negative Response to PD-1 Blockade in MSI-H/dMMR Gastrointestinal Tumors. Clin. Cancer Res. 2021, 27, 3714–3724. [Google Scholar] [CrossRef]
- Song, M.; Chen, D.; Lu, B.; Wang, C.; Zhang, J.; Huang, L.; Wang, X.; Timmons, C.L.; Hu, J.; Liu, B.; et al. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PLoS ONE 2013, 8, e65821. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.A.; de Carné Trécesson, S.; Rana, S.; Zecchin, D.; Moore, C.; Molina-Arcas, M.; East, P.; Spencer-Dene, B.; Nye, E.; Barnouin, K.; et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 2017, 47, 1083–1099.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, S.; Matrone, N.; Muddassir, A.L.; Martini, G.; Sorokin, A.; De Falco, V.; Giunta, E.F.; Ciardiello, D.; Martinelli, E.; Belli, V.; et al. Triple blockade of EGFR, MEK and PD-L1 has antitumor activity in colorectal cancer models with constitutive activation of MAPK signaling and PD-L1 overexpression. J. Exp. Clin. Cancer Res. 2019, 38, 492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, A.; Littman, D.R. Signal transduction by lymphocyte antigen receptors. Cell 1994, 76, 263–274. [Google Scholar] [CrossRef]
- Terheyden, P.; Schrama, D.; Pedersen, L.Ø.; Andersen, M.H.; Kämpgen, E.; thor Straten, P.; Becker, J.C. Longitudinal analysis of MART-1/HLA-A2-reactive T cells over the course of melanoma progression. Scand. J. Immunol. 2003, 58, 566–571. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Heath, V.; O’Garra, A.; Johnston, J.; McMahon, M. Sustained activation of the raf-MEK-ERK pathway elicits cytokine unresponsiveness in T cells. J. Immunol. 1999, 163, 5796–5805. [Google Scholar]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.-Y.; Hopson, C.; et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef] [Green Version]
- Vella, L.J.; Pasam, A.; Dimopoulos, N.; Andrews, M.; Knights, A.; Puaux, A.-L.; Louahed, J.; Chen, W.; Woods, K.; Cebon, J.S. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer Immunol. Res. 2014, 2, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.-N.J.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef] [Green Version]
- Kist, M.; Vucic, D. Cell death pathways: Intricate connections and disease implications. EMBO J. 2021, 40, e106700. [Google Scholar] [CrossRef]
- Scaffidi, C.; Fulda, S.; Srinivasan, A.; Friesen, C.; Li, F.; Tomaselli, K.J.; Debatin, K.M.; Krammer, P.H.; Peter, M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998, 17, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.M.; Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007, 26, 1324–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yu, J.; Park, B.H.; Kinzler, K.W.; Vogelstein, B. Role of BAX in the apoptotic response to anticancer agents. Science 2000, 290, 989–992. [Google Scholar] [CrossRef]
- van Delft, M.F.; Wei, A.H.; Mason, K.D.; Vandenberg, C.J.; Chen, L.; Czabotar, P.E.; Willis, S.N.; Scott, C.L.; Day, C.L.; Cory, S.; et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006, 10, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.-X.; Sneed, T.; et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Joudeh, J.; Claxton, D. Obatoclax mesylate: Pharmacology and potential for therapy of hematological neoplasms. Expert Opin. Investig. Drugs 2012, 21, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Timme, C.R.; Gruidl, M.; Yeatman, T.J. Gamma-secretase inhibition attenuates oxaliplatin-induced apoptosis through increased Mcl-1 and/or Bcl-xL in human colon cancer cells. Apoptosis 2013, 18, 1163–1174. [Google Scholar] [CrossRef]
- Hunter, A.M.; LaCasse, E.C.; Korneluk, R.G. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 2007, 12, 1543–1568. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef]
- Connolly, K.; Mitter, R.; Muir, M.; Jodrell, D.; Guichard, S. Stable XIAP knockdown clones of HCT116 colon cancer cells are more sensitive to TRAIL, taxanes and irradiation in vitro. Cancer Chemother. Pharmacol. 2009, 64, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Lippa, M.S.; Strockbine, L.D.; Le, T.T.; Branstetter, D.G.; Strathdee, C.A.; Holland, P.M. Expression of anti-apoptotic factors modulates Apo2L/TRAIL resistance in colon carcinoma cells. Apoptosis 2007, 12, 1465–1478. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Fu, Z.; Shi, J.; Jiang, X.; Wan, H. HtrA1 Down-regulation Induces Cisplatin Resistance in Colon Cancer by Increasing XIAP and Activating PI3K/Akt Pathway. Ann. Clin. Lab. Sci. 2017, 47, 264–270. [Google Scholar] [PubMed]
- Zhang, Y.; Talmon, G.; Wang, J. MicroRNA-587 antagonizes 5-FU-induced apoptosis and confers drug resistance by regulating PPP2R1B expression in colorectal cancer. Cell Death Dis. 2015, 6, e1845. [Google Scholar] [CrossRef] [Green Version]
- Gyrd-Hansen, M.; Meier, P. IAPs: From caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef]
- Enoch, T.; Norbury, C. Cellular responses to DNA damage: Cell-cycle checkpoints, apoptosis and the roles of p53 and ATM. Trends Biochem. Sci. 1995, 20, 426–430. [Google Scholar] [CrossRef]
- Yao, J.; Huang, A.; Zheng, X.; Liu, T.; Lin, Z.; Zhang, S.; Yang, Q.; Zhang, T.; Ma, H. 53BP1 loss induces chemoresistance of colorectal cancer cells to 5-fluorouracil by inhibiting the ATM-CHK2-P53 pathway. J. Cancer Res. Clin. Oncol. 2017, 143, 419–431. [Google Scholar] [CrossRef]
- Elsaleh, H.; Powell, B.; McCaul, K.; Grieu, F.; Grant, R.; Joseph, D.; Iacopetta, B. P53 alteration and microsatellite instability have predictive value for survival benefit from chemotherapy in stage III colorectal carcinoma. Clin. Cancer Res. 2001, 7, 1343–1349. [Google Scholar]
- Bunz, F.; Hwang, P.M.; Torrance, C.; Waldman, T.; Zhang, Y.; Dillehay, L.; Williams, J.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J. Clin. Investig. 1999, 104, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; Bazan, V.; Iacopetta, B.; Kerr, D.; Soussi, T.; Gebbia, N. The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: Influence of tumor site, type of mutation, and adjuvant treatment. J. Clin. Oncol. 2005, 23, 7518–7528. [Google Scholar] [CrossRef]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Nadler-Milbauer, M.; Apter, L.; Haupt, Y.; Haupt, S.; Barenholz, Y.; Minko, T.; Rubinstein, A. Synchronized release of Doxil and Nutlin-3 by remote degradation of polysaccharide matrices and its possible use in the local treatment of colorectal cancer. J. Drug Target. 2011, 19, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, Y.; Huang, J.; Wong, C.C.; Zhai, J.; Li, C.; Wei, G.; Zhao, L.; Wang, G.; Wei, H.; et al. TRIM67 Activates p53 to Suppress Colorectal Cancer Initiation and Progression. Cancer Res. 2019, 79, 4086–4098. [Google Scholar] [CrossRef] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Aredia, F.; Scovassi, A.I. Manipulation of autophagy in cancer cells: An innovative strategy to fight drug resistance. Future Med. Chem. 2013, 5, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Singh, U.K.; Chaudhary, A. Targeting autophagy to overcome drug resistance in cancer therapy. Future Med. Chem. 2015, 7, 1535–1542. [Google Scholar] [CrossRef]
- Koustas, E.; Sarantis, P.; Theoharis, S.; Saetta, A.A.; Chatziandreou, I.; Kyriakopoulou, G.; Giannopoulou, I.; Michelli, M.; Schizas, D.; Papavassiliou, A.G.; et al. Autophagy-related Proteins as a Prognostic Factor of Patients With Colorectal Cancer. Am. J. Clin. Oncol. 2019, 42, 767–776. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.; Song, D.; Yan, Y.; Huang, C.; Shen, C.; Lan, J.; Chen, Y.; Liu, A.; Wu, Q.; Sun, L.; et al. IL-6 regulates autophagy and chemotherapy resistance by promoting BECN1 phosphorylation. Nat. Commun. 2021, 12, 3651. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, C.; Liu, X.; Wang, Y.; Zhao, R.; Yang, Y.; Zheng, X.; Zhang, Y.; Zhang, X. circHIPK3 promotes oxaliplatin-resistance in colorectal cancer through autophagy by sponging miR-637. EBioMedicine 2019, 48, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lan, Z.; He, J.; Lai, Q.; Yao, X.; Li, Q.; Liu, Y.; Lai, H.; Gu, C.; Yan, Q.; et al. LncRNA SNHG6 promotes chemoresistance through ULK1-induced autophagy by sponging miR-26a-5p in colorectal cancer cells. Cancer Cell Int. 2019, 19, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Han, D.; Yuan, Z.; Hu, H.; Zhao, Z.; Yang, R.; Jin, Y.; Zou, C.; Chen, Y.; Wang, G.; et al. Long non-coding RNA H19 confers 5-Fu resistance in colorectal cancer by promoting SIRT1-mediated autophagy. Cell Death Dis. 2018, 9, 1149. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ai, F.-Y.; Zhang, D.-C.; Tian, L.; Yang, Z.-Y.; Liu, S.-J. LncRNA NEAT1 knockdown attenuates autophagy to elevate 5-FU sensitivity in colorectal cancer via targeting miR-34a. Cancer Med. 2020, 9, 1079–1091. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Zhou, S.; Wang, X.; Mao, E.; Huang, L. SNHG14 stimulates cell autophagy to facilitate cisplatin resistance of colorectal cancer by regulating miR-186/ATG14 axis. Biomed. Pharmacother. 2020, 121, 109580. [Google Scholar] [CrossRef]
- Guo, G.-F.; Jiang, W.-Q.; Zhang, B.; Cai, Y.-C.; Xu, R.-H.; Chen, X.-X.; Wang, F.; Xia, L.-P. Autophagy-related proteins Beclin-1 and LC3 predict cetuximab efficacy in advanced colorectal cancer. World J. Gastroenterol. 2011, 17, 4779–4786. [Google Scholar] [CrossRef]
- Clarke, A.J.; Simon, A.K. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat. Rev. Immunol. 2019, 19, 170–183. [Google Scholar] [CrossRef]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Zhang, Y.; Lin, S.; Liu, Y.; Li, W. Suppressing the KIF20A/NUAK1/Nrf2/GPX4 signaling pathway induces ferroptosis and enhances the sensitivity of colorectal cancer to oxaliplatin. Aging 2021, 13, 13515–13534. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Choudhary, B.S.; Shah, S.G.; Khapare, N.; Dwivedi, N.; Gaikwad, A.; Joshi, N.; Raichanna, J.; Basu, S.; Gurjar, M.; et al. Lipocalin 2 expression promotes tumor progression and therapy resistance by inhibiting ferroptosis in colorectal cancer. Int. J. Cancer 2021, 149, 1495–1511. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.A.; Piao, M.J.; Kim, K.C.; Kang, H.K.; Chang, W.Y.; Park, I.C.; Keum, Y.S.; Surh, Y.J.; Hyun, J.W. Epigenetic modification of Nrf2 in 5-fluorouracil-resistant colon cancer cells: Involvement of TET-dependent DNA demethylation. Cell Death Dis. 2014, 5, e1183. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Mo, J.; Dai, J.; Ye, C.; Cen, W.; Zheng, X.; Jiang, L.; Ye, L. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis. 2021, 12, 1079. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8 T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Chan, C.; Han, W.; Guo, N.; Weichselbaum, R.R.; Lin, W. Immunostimulatory nanomedicines synergize with checkpoint blockade immunotherapy to eradicate colorectal tumors. Nat. Commun. 2019, 10, 1899. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Duan, X.; Ni, K.; Li, Y.; Chan, C.; Lin, W. Co-delivery of dihydroartemisinin and pyropheophorbide-iron elicits ferroptosis to potentiate cancer immunotherapy. Biomaterials 2021, 280, 121315. [Google Scholar] [CrossRef]
- Katz, O.B.; Shaked, Y. Host effects contributing to cancer therapy resistance. Drug Resist. Updates 2015, 19, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Schiavoni, G.; Gabriele, L.; Mattei, F. The tumor microenvironment: A pitch for multiple players. Front. Oncol. 2013, 3, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Li, J.; Xie, K.; Zhang, T.; Lei, Y.; Chen, Y.; Zhang, L.; Huang, K.; Wang, K.; Wu, H.; et al. FGFR4 promotes stroma-induced epithelial-to-mesenchymal transition in colorectal cancer. Cancer Res. 2013, 73, 5926–5935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillén Díaz-Maroto, N.; Sanz-Pamplona, R.; Berdiel-Acer, M.; Cimas, F.J.; García, E.; Gonçalves-Ribeiro, S.; Albert, N.; Garcia-Vicién, G.; Capella, G.; Moreno, V.; et al. Noncanonical TGFβ Pathway Relieves the Blockade of IL1β/TGFβ-Mediated Crosstalk between Tumor and Stroma: TGFBR1 and TAK1 Inhibition in Colorectal Cancer. Clin. Cancer Res. 2019, 25, 4466–4479. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.-A.; Chen, Y.-F.; Bao, Y.; Mahara, S.; Yatim, S.M.J.M.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E5990–E5999. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Chan, K.; Qi, Y.; Lu, L.; Ning, F.; Wu, M.; Wang, H.; Wang, Y.; Cai, S.; Du, J. Participation of CCL1 in Snail-Positive Fibroblasts in Colorectal Cancer Contribute to 5-Fluorouracil/Paclitaxel Chemoresistance. Cancer Res. Treat. 2018, 50, 894–907. [Google Scholar] [CrossRef] [Green Version]
- Lotti, F.; Jarrar, A.M.; Pai, R.K.; Hitomi, M.; Lathia, J.; Mace, A.; Gantt, G.A.; Sukhdeo, K.; DeVecchio, J.; Vasanji, A.; et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J. Exp. Med. 2013, 210, 2851–2872. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Ding, L.; Zhang, D.; Shi, G.; Xu, Q.; Shen, S.; Wang, Y.; Wang, T.; Hou, Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics 2018, 8, 3932–3948. [Google Scholar] [CrossRef]
- Deng, X.; Ruan, H.; Zhang, X.; Xu, X.; Zhu, Y.; Peng, H.; Zhang, X.; Kong, F.; Guan, M. Long noncoding RNA CCAL transferred from fibroblasts by exosomes promotes chemoresistance of colorectal cancer cells. Int. J. Cancer 2020, 146, 1700–1716. [Google Scholar] [CrossRef] [PubMed]
- Woolston, A.; Khan, K.; Spain, G.; Barber, L.J.; Griffiths, B.; Gonzalez-Exposito, R.; Hornsteiner, L.; Punta, M.; Patil, Y.; Newey, A.; et al. Genomic and Transcriptomic Determinants of Therapy Resistance and Immune Landscape Evolution during Anti-EGFR Treatment in Colorectal Cancer. Cancer Cell 2019, 36, 35–50.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luraghi, P.; Reato, G.; Cipriano, E.; Sassi, F.; Orzan, F.; Bigatto, V.; De Bacco, F.; Menietti, E.; Han, M.; Rideout, W.M.; et al. MET signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res. 2014, 74, 1857–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarthy, A.; Furness, A.; Joshi, K.; Ghorani, E.; Ford, K.; Ward, M.J.; King, E.V.; Lechner, M.; Marafioti, T.; Quezada, S.A.; et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat. Commun. 2018, 9, 3220. [Google Scholar] [CrossRef] [Green Version]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef] [PubMed]
- Ford, K.; Hanley, C.J.; Mellone, M.; Szyndralewiez, C.; Heitz, F.; Wiesel, P.; Wood, O.; Machado, M.; Lopez, M.-A.; Ganesan, A.-P.; et al. NOX4 Inhibition Potentiates Immunotherapy by Overcoming Cancer-Associated Fibroblast-Mediated CD8 T-cell Exclusion from Tumors. Cancer Res. 2020, 80, 1846–1860. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Katoh, H.; Wang, D.; Daikoku, T.; Sun, H.; Dey, S.K.; Dubois, R.N. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013, 24, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Chun, E.; Lavoie, S.; Michaud, M.; Gallini, C.A.; Kim, J.; Soucy, G.; Odze, R.; Glickman, J.N.; Garrett, W.S. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep. 2015, 12, 244–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cicco, P.; Sanders, T.; Cirino, G.; Maloy, K.J.; Ianaro, A. Hydrogen Sulfide Reduces Myeloid-Derived Suppressor Cell-Mediated Inflammatory Response in a Model of -Induced Colitis. Front. Immunol. 2018, 9, 499. [Google Scholar] [CrossRef] [Green Version]
- Toor, S.M.; Syed Khaja, A.S.; El Salhat, H.; Bekdache, O.; Kanbar, J.; Jaloudi, M.; Elkord, E. Increased Levels of Circulating and Tumor-Infiltrating Granulocytic Myeloid Cells in Colorectal Cancer Patients. Front. Immunol. 2016, 7, 560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiff, A.; Trikha, P.; Mundy-Bosse, B.; McMichael, E.; Mace, T.A.; Benner, B.; Kendra, K.; Campbell, A.; Gautam, S.; Abood, D.; et al. Nitric Oxide Production by Myeloid-Derived Suppressor Cells Plays a Role in Impairing Fc Receptor-Mediated Natural Killer Cell Function. Clin. Cancer Res. 2018, 24, 1891–1904. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Ramirez, Y.; Baltazar-Perez, I.; Martinez, Y.; Callejas, B.E.; Medina-Andrade, I.; Olguín, J.E.; Delgado-Buenrostro, N.L.; Chirino, Y.I.; Terrazas, L.I.; Leon-Cabrera, S. STAT1 Is Required for Decreasing Accumulation of Granulocytic Cells via IL-17 during Initial Steps of Colitis-Associated Cancer. Int. J. Mol. Sci. 2021, 22, 7695. [Google Scholar] [CrossRef]
- Wu, P.; Wu, D.; Ni, C.; Ye, J.; Chen, W.; Hu, G.; Wang, Z.; Wang, C.; Zhang, Z.; Xia, W.; et al. γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity 2014, 40, 785–800. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Cai, T.-T.; Wu, X.-J.; Liu, Y.-N.; He, J.; Zhang, X.-S.; Ma, G.; Li, J. Tumour YAP1 and PTEN expression correlates with tumour-associated myeloid suppressor cell expansion and reduced survival in colorectal cancer. Immunology 2018, 155, 263–272. [Google Scholar] [CrossRef]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572.e7. [Google Scholar] [CrossRef] [Green Version]
- Condamine, T.; Kumar, V.; Ramachandran, I.R.; Youn, J.-I.; Celis, E.; Finnberg, N.; El-Deiry, W.S.; Winograd, R.; Vonderheide, R.H.; English, N.R.; et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J. Clin. Investig. 2014, 124, 2626–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, G.A.; Condamine, T.; Mony, S.; Hashimoto, A.; Wang, F.; Liu, Q.; Forero, A.; Bendell, J.; Witt, R.; Hockstein, N.; et al. Selective Targeting of Myeloid-Derived Suppressor Cells in Cancer Patients Using DS-8273a, an Agonistic TRAIL-R2 Antibody. Clin. Cancer Res. 2017, 23, 2942–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Zhang, X.; Li, Z.; Zhu, B. Metabolic regulatory crosstalk between tumor microenvironment and tumor-associated macrophages. Theranostics 2021, 11, 1016–1030. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.-B.; Lü, M.-H.; Fan, Y.-H.; Cao, Y.-L.; Zhang, Z.-R.; Yang, S.-M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef] [Green Version]
- Jinushi, M.; Chiba, S.; Yoshiyama, H.; Masutomi, K.; Kinoshita, I.; Dosaka-Akita, H.; Yagita, H.; Takaoka, A.; Tahara, H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 12425–12430. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Yao, S.; Hu, Y.; Feng, Y.; Li, M.; Bian, Z.; Zhang, J.; Qin, Y.; Qi, X.; Zhou, L.; et al. The Immune-microenvironment Confers Chemoresistance of Colorectal Cancer through Macrophage-Derived IL6. Clin. Cancer Res. 2017, 23, 7375–7387. [Google Scholar] [CrossRef] [Green Version]
- Malesci, A.; Bianchi, P.; Celesti, G.; Basso, G.; Marchesi, F.; Grizzi, F.; Di Caro, G.; Cavalleri, T.; Rimassa, L.; Palmqvist, R.; et al. Tumor-associated macrophages and response to 5-fluorouracil adjuvant therapy in stage III colorectal cancer. Oncoimmunology 2017, 6, e1342918. [Google Scholar] [CrossRef]
- Feng, Q.; Chang, W.; Mao, Y.; He, G.; Zheng, P.; Tang, W.; Wei, Y.; Ren, L.; Zhu, D.; Ji, M.; et al. Tumor-associated Macrophages as Prognostic and Predictive Biomarkers for Postoperative Adjuvant Chemotherapy in Patients with Stage II Colon Cancer. Clin. Cancer Res. 2019, 25, 3896–3907. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Stanley, E.R.; Chitu, V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 2014, 6, a021857. [Google Scholar] [CrossRef] [Green Version]
- Doleschel, D.; Hoff, S.; Koletnik, S.; Rix, A.; Zopf, D.; Kiessling, F.; Lederle, W. Regorafenib enhances anti-PD1 immunotherapy efficacy in murine colorectal cancers and their combination prevents tumor regrowth. J. Exp. Clin. Cancer Res. 2021, 40, 288. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, S.; Hara, H.; Takahashi, N.; Kojima, T.; Kawazoe, A.; Asayama, M.; Yoshii, T.; Kotani, D.; Tamura, H.; Mikamoto, Y.; et al. Regorafenib Plus Nivolumab in Patients With Advanced Gastric or Colorectal Cancer: An Open-Label, Dose-Escalation, and Dose-Expansion Phase Ib Trial (REGONIVO, EPOC1603). J. Clin. Oncol. 2020, 38, 2053–2061. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.; Raghav, K.P.S.; Chang, D.Z.; Bendell, J.C.; Larson, T.; Cohn, A.L.; Huyck, T.K.; Cosgrove, D.; Fiorillo, J.A.; Garbo, L.E.; et al. Single-arm, phase 2 study of regorafenib plus nivolumab in patients with mismatch repair-proficient (pMMR)/microsatellite stable (MSS) colorectal cancer (CRC). J. Clin. Oncol. 2021, 39, 3560. [Google Scholar] [CrossRef]
- Baer, C.; Squadrito, M.L.; Laoui, D.; Thompson, D.; Hansen, S.K.; Kiialainen, A.; Hoves, S.; Ries, C.H.; Ooi, C.-H.; De Palma, M. Suppression of microRNA activity amplifies IFN-γ-induced macrophage activation and promotes anti-tumour immunity. Nat. Cell Biol. 2016, 18, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-F.; Lin, C.-A.; Yuan, T.-H.; Yeh, H.-Y.; Su, S.-F.; Guo, C.-L.; Chang, G.-C.; Li, K.-C.; Ho, C.-C.; Chen, H.-W. The M1/M2 spectrum and plasticity of malignant pleural effusion-macrophage in advanced lung cancer. Cancer Immunol. Immunother. 2021, 70, 1435–1450. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Tumor-associated macrophages. Curr. Biol. 2020, 30, R246–R248. [Google Scholar] [CrossRef]
- Song, M.; Li, Z.-H.; Gu, H.-S.; Tang, R.-Y.; Zhang, R.; Zhu, Y.-L.; Liu, J.-L.; Zhang, J.-J.; Wang, L.-Y. Spore Polysaccharide Inhibits the Growth of Hepatocellular Carcinoma Cells by Altering Macrophage Polarity and Induction of Apoptosis. J. Immunol. Res. 2021, 2021, 6696606. [Google Scholar] [CrossRef]
- Bi, S.; Huang, W.; Chen, S.; Huang, C.; Li, C.; Guo, Z.; Yang, J.; Zhu, J.; Song, L.; Yu, R. Cordyceps militaris polysaccharide converts immunosuppressive macrophages into M1-like phenotype and activates T lymphocytes by inhibiting the PD-L1/PD-1 axis between TAMs and T lymphocytes. Int. J. Biol. Macromol. 2020, 150, 261–280. [Google Scholar] [CrossRef]
- Nie, W.; Wu, G.; Zhang, J.; Huang, L.-L.; Ding, J.; Jiang, A.; Zhang, Y.; Liu, Y.; Li, J.; Pu, K.; et al. Responsive Exosome Nano-bioconjugates for Synergistic Cancer Therapy. Angew. Chem. Int. Ed. Engl. 2020, 59, 2018–2022. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Hack, S.P.; Zhu, A.X.; Wang, Y. Augmenting Anticancer Immunity Through Combined Targeting of Angiogenic and PD-1/PD-L1 Pathways: Challenges and Opportunities. Front. Immunol. 2020, 11, 598877. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat Rev. 2020, 86, 102017. [Google Scholar] [CrossRef] [PubMed]
- Pogue-Geile, K.; Yothers, G.; Taniyama, Y.; Tanaka, N.; Gavin, P.; Colangelo, L.; Blackmon, N.; Lipchik, C.; Kim, S.R.; Sharif, S.; et al. Defective mismatch repair and benefit from bevacizumab for colon cancer: Findings from NSABP C-08. J. Natl. Cancer Inst. 2013, 105, 989–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, H.-J.; Parikh, A.R.; Spigel, D.R.; Cohn, A.L.; Yoshino, T.; Kochenderfer, M.D.; Elez, E.; Shao, S.H.; Deming, D.A.; Holdridge, R.C.; et al. Nivolumab (NIVO) + 5-fluorouracil/leucovorin/oxaliplatin (mFOLFOX6)/bevacizumab (BEV) versus mFOLFOX6/BEV for first-line (1L) treatment of metastatic colorectal cancer (mCRC): Phase 2 results from CheckMate 9X8. J. Clin. Oncol. 2022, 40, 8. [Google Scholar] [CrossRef]
- Vallböhmer, D.; Zhang, W.; Gordon, M.; Yang, D.Y.; Yun, J.; Press, O.A.; Rhodes, K.E.; Sherrod, A.E.; Iqbal, S.; Danenberg, K.D.; et al. Molecular determinants of cetuximab efficacy. J. Clin. Oncol. 2005, 23, 3536–3544. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Shen, X.; Chen, G.; Du, J. Drug Resistance in Colorectal Cancer: From Mechanism to Clinic. Cancers 2022, 14, 2928. https://doi.org/10.3390/cancers14122928
Wang Q, Shen X, Chen G, Du J. Drug Resistance in Colorectal Cancer: From Mechanism to Clinic. Cancers. 2022; 14(12):2928. https://doi.org/10.3390/cancers14122928
Chicago/Turabian StyleWang, Qianyu, Xiaofei Shen, Gang Chen, and Junfeng Du. 2022. "Drug Resistance in Colorectal Cancer: From Mechanism to Clinic" Cancers 14, no. 12: 2928. https://doi.org/10.3390/cancers14122928
APA StyleWang, Q., Shen, X., Chen, G., & Du, J. (2022). Drug Resistance in Colorectal Cancer: From Mechanism to Clinic. Cancers, 14(12), 2928. https://doi.org/10.3390/cancers14122928