
The Potential Tumor-Suppressor DHRS7 Inversely Correlates with EGFR Expression in Prostate Cancer Cells and Tumor Samples

 , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Knockdown Using siRNA
2.4. Gene Expression Analysis by RT-qPCR
2.5. Protein Expression Analysis by Western Blotting
2.6. Proteome Analyses in LNCaP Cells
2.7. TCGA Data Analysis
2.8. Tissue Microarray and Immunohistochemistry
2.9. Statistical Analysis
3. Results
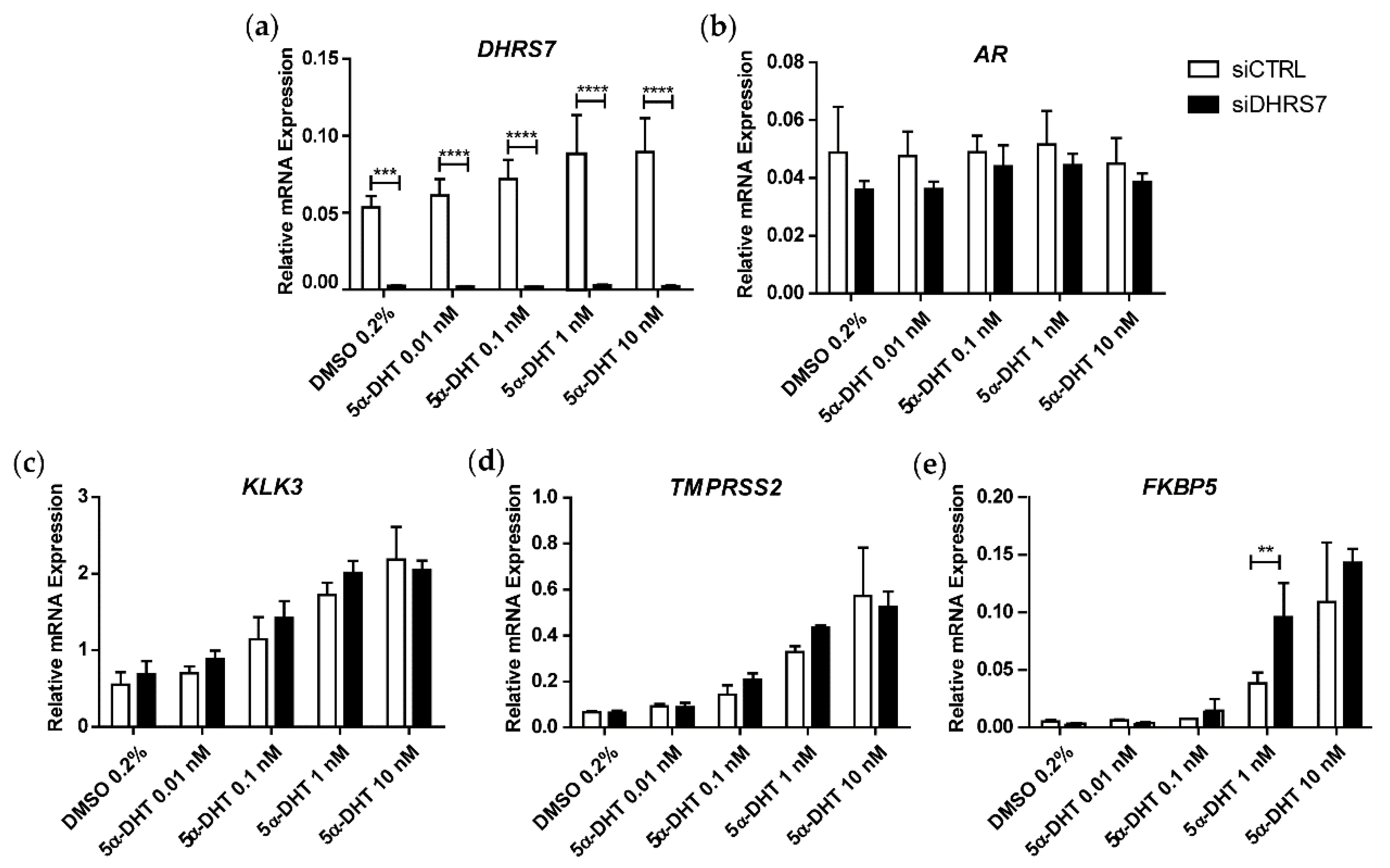
3.1. Knockdown of DHRS7 Does Not Alter AR-Regulated Gene Expression
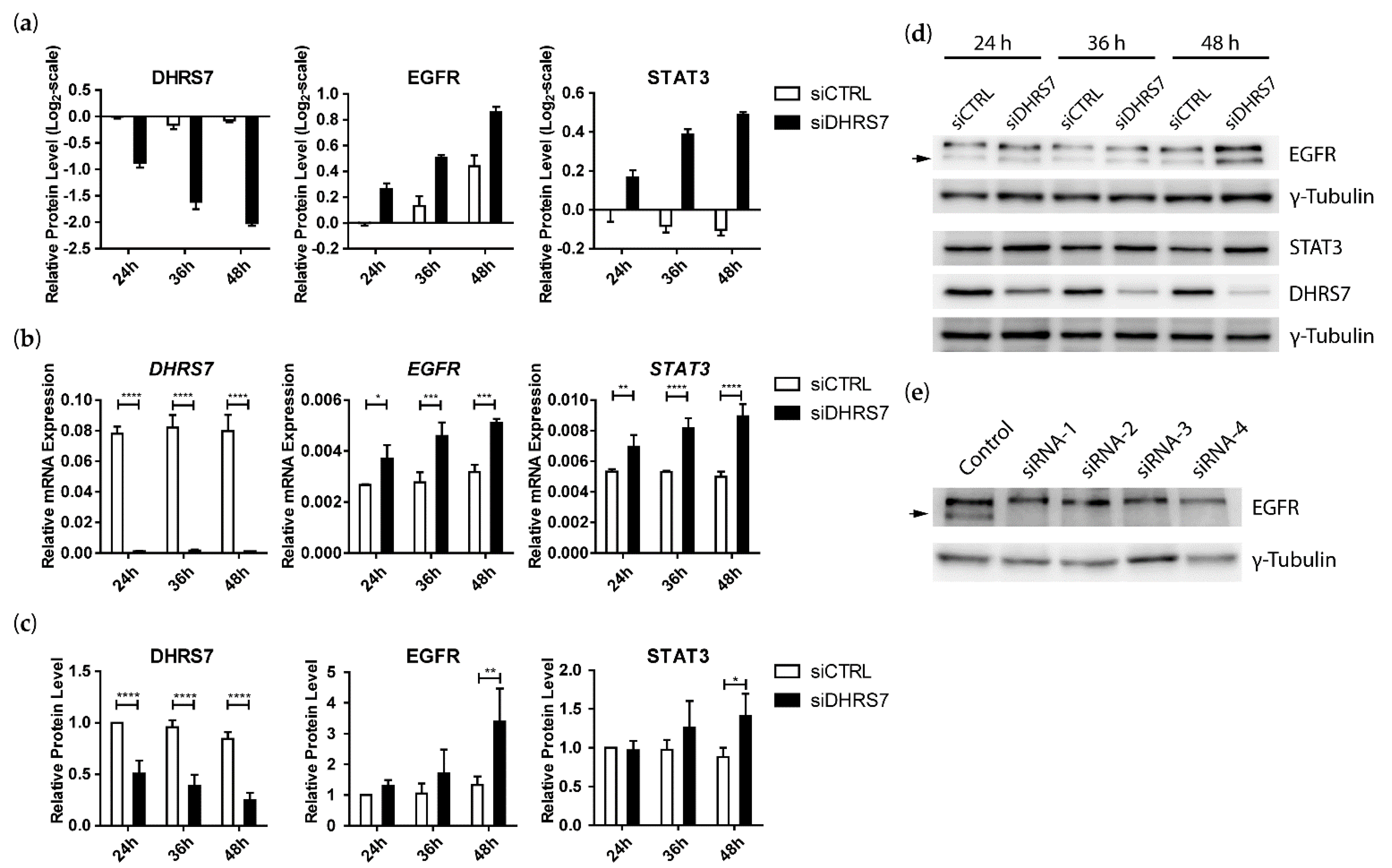
3.2. Proteomics Analyses Indicated That DHRS7 Knockdown Did Not Affect AR and AR Target Protein Expression but Did Increase EGFR and STAT3 Expression in LNCaP Cells
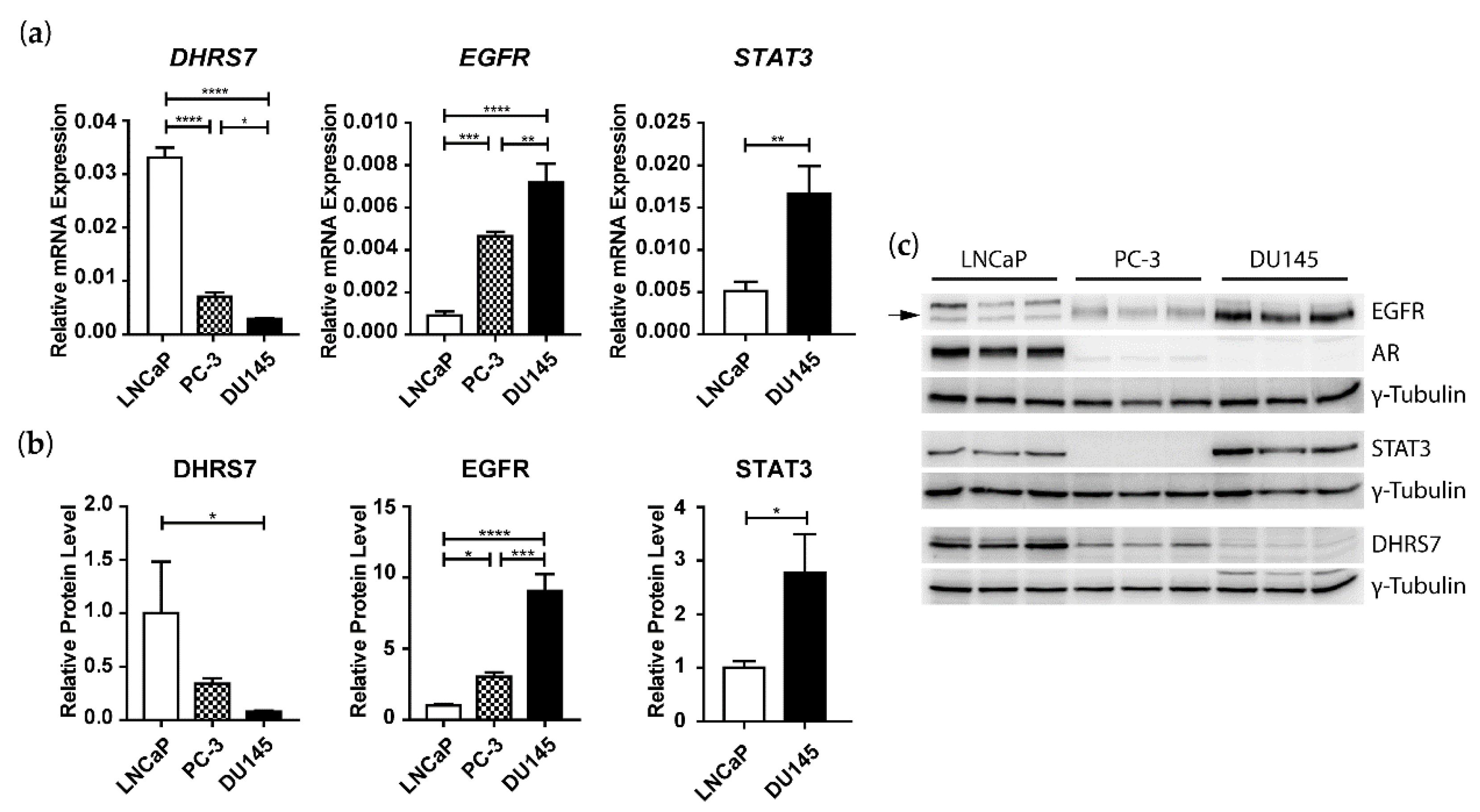
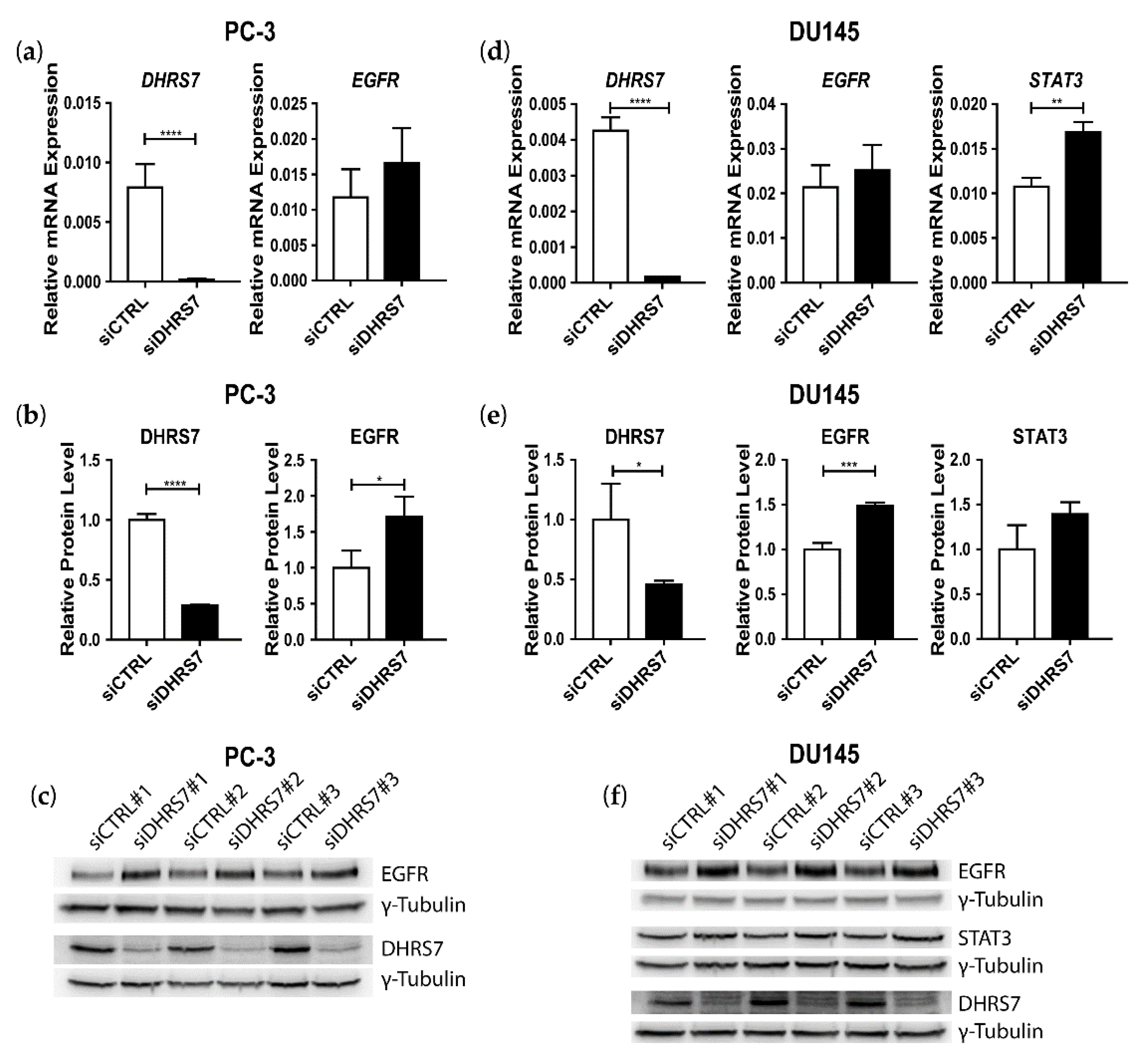
3.3. Inverse Correlation of DHRS7 and EGFR Expression in LNCaP, PC-3, and DU145 Cells
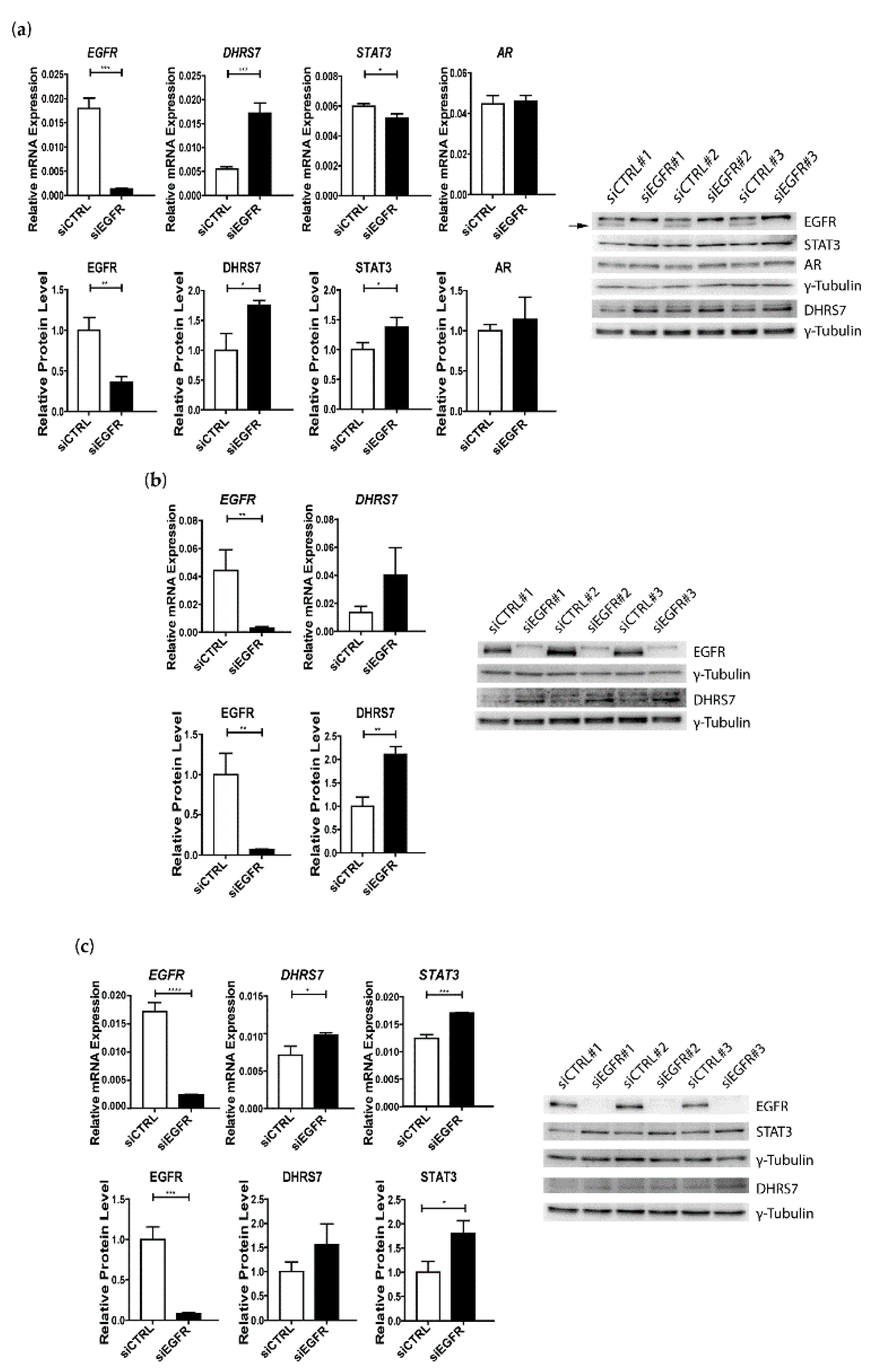
3.4. Knockdown of EGFR Leads to Increased Expression of DHRS7
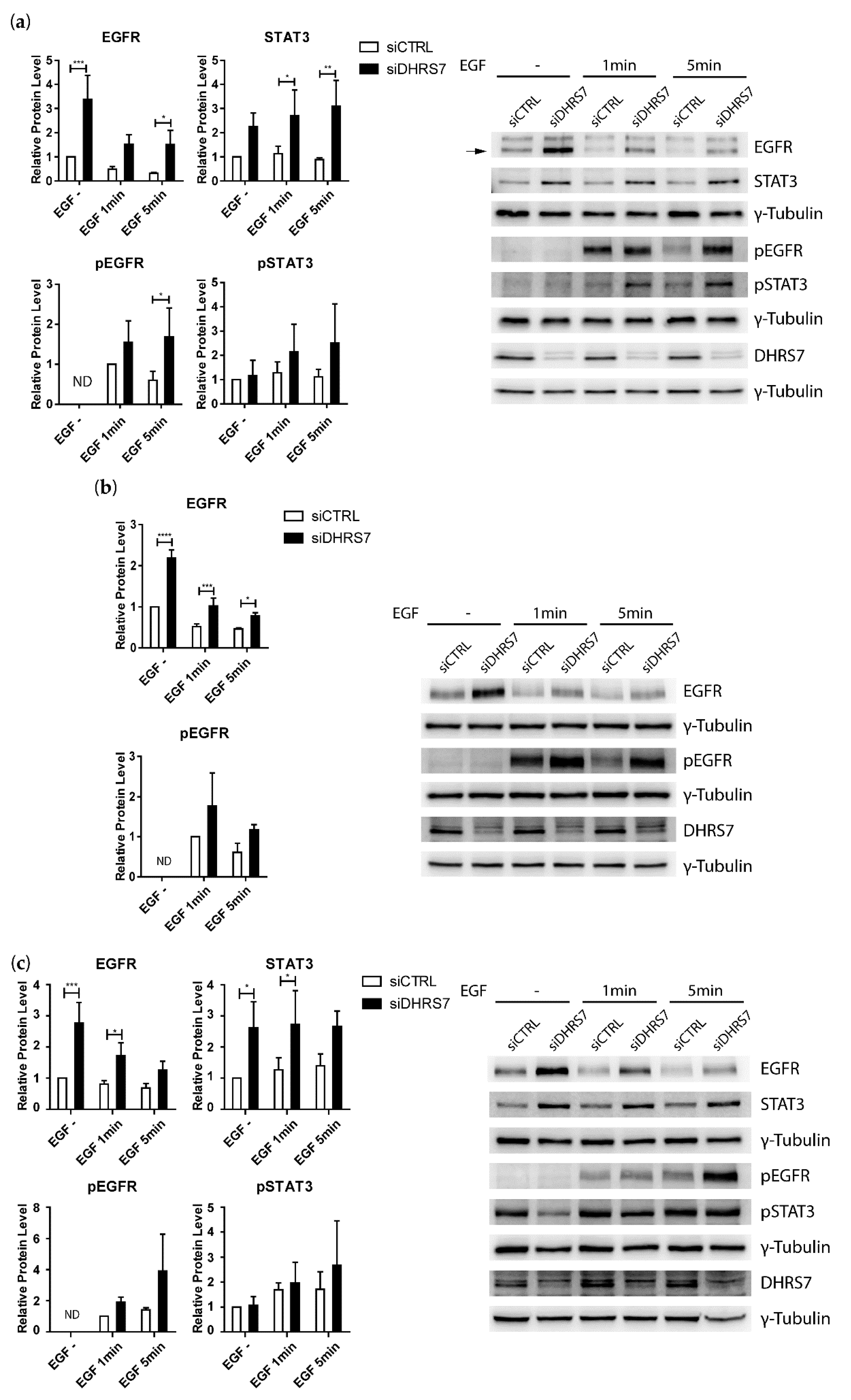
3.5. Increased EGFR Expression after DHRS7 Knockdown Is Accompanied by Increased Phosphorylation after EGF Treatment
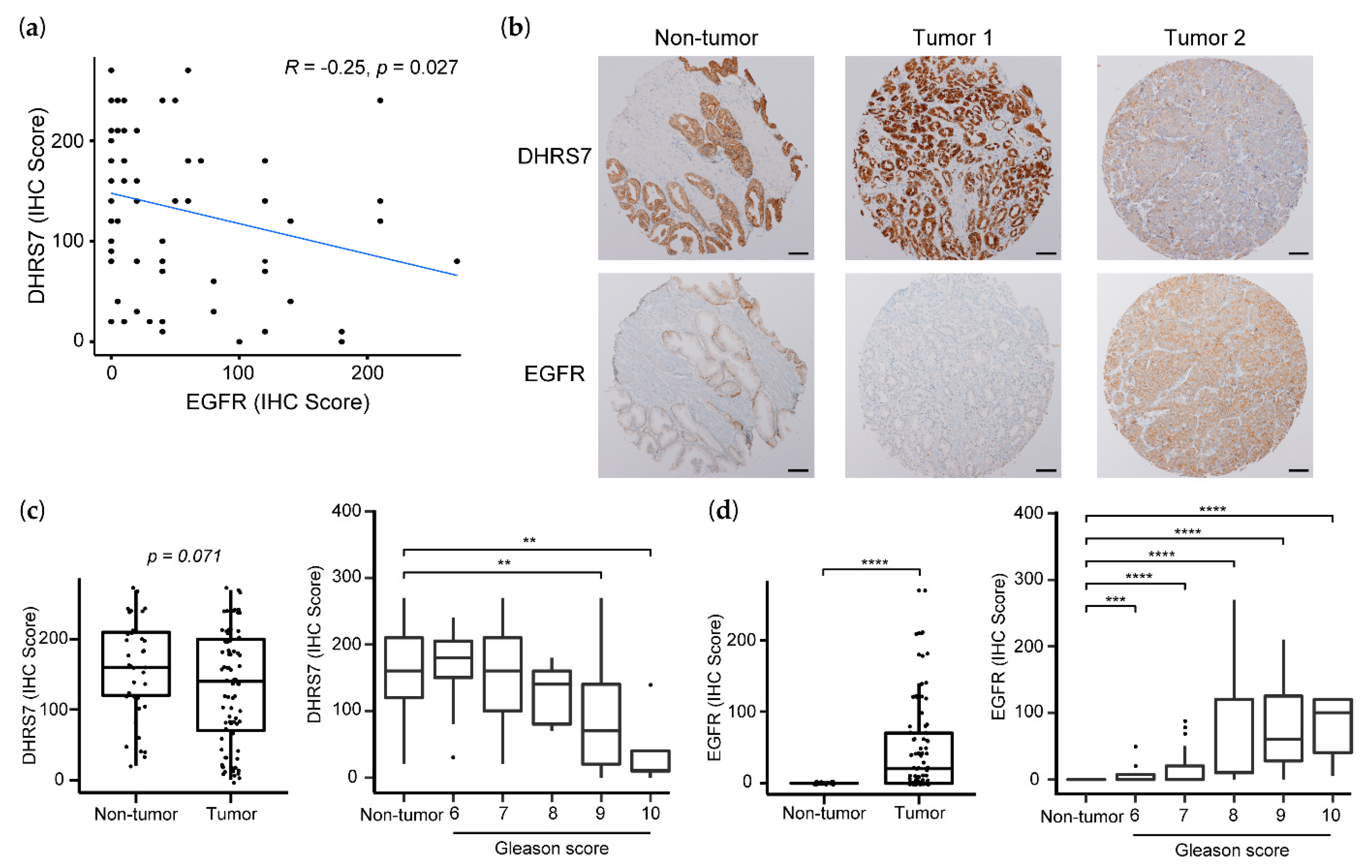
3.6. Analysis of the TCGA Database Revealed an Inverse Correlation between DHRS7 and EGFR Expression
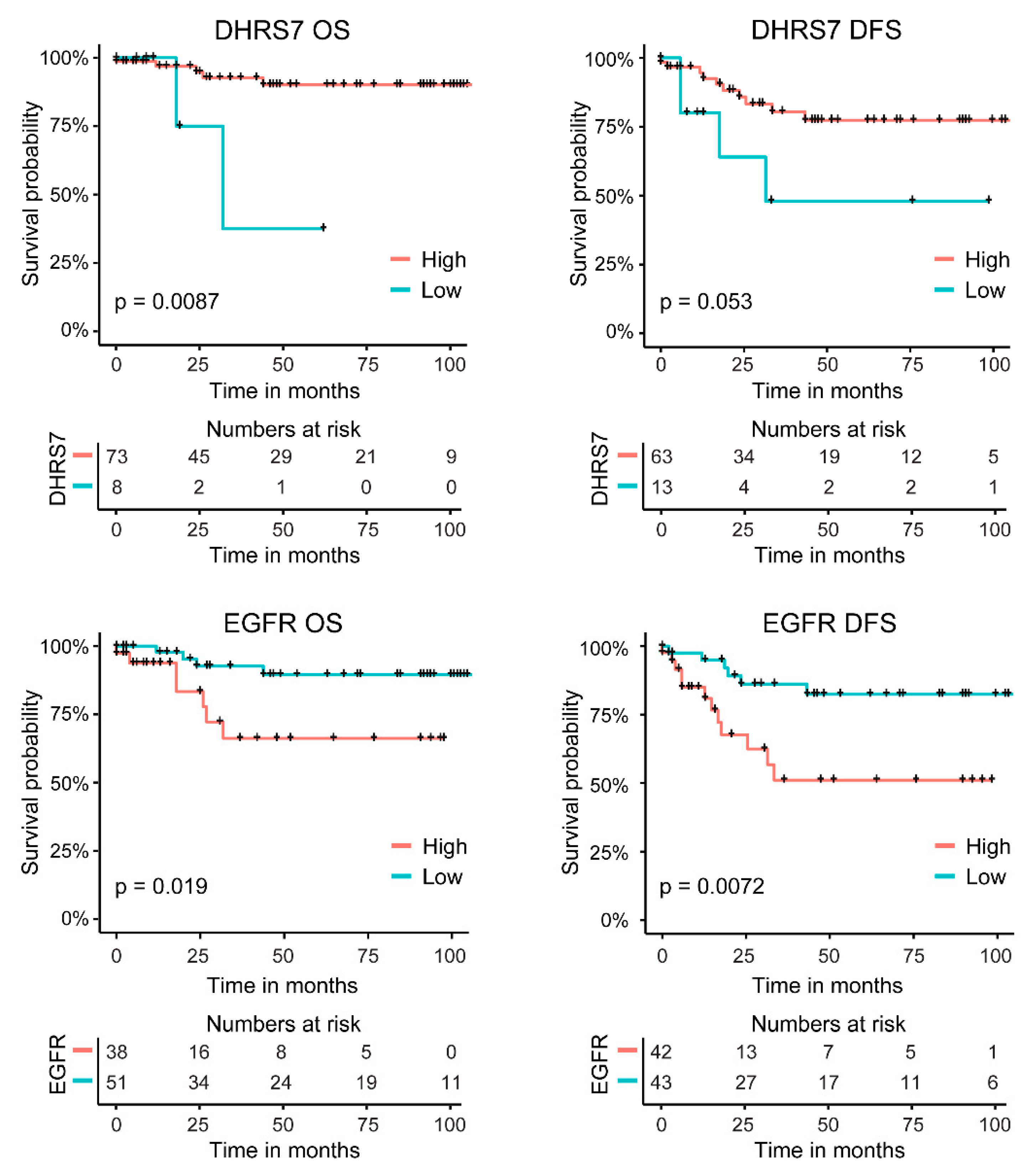
3.7. Correlation of Lower DHRS7 and Higher EGFR Expression with Gleason Score and Reduced Survival Probability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate cancer. Nat. Rev. Dis. Primers 2021, 7, 9. [Google Scholar] [CrossRef]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Andersson, S.; Geissler, W.M.; Wu, L.; Davis, D.L.; Grumbach, M.M.; New, M.I.; Schwarz, H.P.; Blethen, S.L.; Mendonca, B.B.; Bloise, W.; et al. Molecular genetics and pathophysiology of 17 beta-hydroxysteroid dehydrogenase 3 deficiency. J. Clin. Endocrinol. Metab. 1996, 81, 130–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geissler, W.M.; Davis, D.L.; Wu, L.; Bradshaw, K.D.; Patel, S.; Mendonca, B.B.; Elliston, K.O.; Wilson, J.D.; Russell, D.W.; Andersson, S. Male pseudohermaphroditism caused by mutations of testicular 17 beta-hydroxysteroid dehydrogenase 3. Nat. Genet. 1994, 7, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Biswas, M.G.; Russell, D.W. Expression cloning and characterization of oxidative 17beta- and 3alpha-hydroxysteroid dehydrogenases from rat and human prostate. J. Biol. Chem. 1997, 272, 15959–15966. [Google Scholar] [CrossRef] [Green Version]
- Penning, T.M.; Bauman, D.R.; Jin, Y.; Rizner, T.L. Identification of the molecular switch that regulates access of 5alpha-DHT to the androgen receptor. Mol. Cell Endocrinol. 2007, 265–266, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Bauman, D.R.; Steckelbroeck, S.; Williams, M.V.; Peehl, D.M.; Penning, T.M. Identification of the major oxidative 3alpha-hydroxysteroid dehydrogenase in human prostate that converts 5alpha-androstane-3alpha,17beta-diol to 5alpha-dihydrotestosterone: A potential therapeutic target for androgen-dependent disease. Mol. Endocrinol. 2006, 20, 444–458. [Google Scholar] [CrossRef] [Green Version]
- Hettel, D.; Sharifi, N. HSD3B1 status as a biomarker of androgen deprivation resistance and implications for prostate cancer. Nat. Rev. Urol. 2018, 15, 191–196. [Google Scholar] [CrossRef]
- Haeseleer, F.; Palczewski, K. Short-chain dehydrogenases/reductases in retina. Methods Enzymol. 2000, 316, 372–383. [Google Scholar] [CrossRef]
- Stambergova, H.; Zemanova, L.; Lundova, T.; Malcekova, B.; Skarka, A.; Safr, M.; Wsol, V. Human DHRS7, promising enzyme in metabolism of steroids and retinoids? J. Steroid Biochem. Mol. Biol. 2016, 155 Pt A, 112–119. [Google Scholar] [CrossRef]
- Seibert, J.K.; Quagliata, L.; Quintavalle, C.; Hammond, T.G.; Terracciano, L.; Odermatt, A. A role for the dehydrogenase DHRS7 (SDR34C1) in prostate cancer. Cancer Med. 2015, 4, 1717–1729. [Google Scholar] [CrossRef] [PubMed]
- Henry, G.H.; Malewska, A.; Joseph, D.B.; Malladi, V.S.; Lee, J.; Torrealba, J.; Mauck, R.J.; Gahan, J.C.; Raj, G.V.; Roehrborn, C.G.; et al. A Cellular Anatomy of the Normal Adult Human Prostate and Prostatic Urethra. Cell Rep. 2018, 25, 3530–3542.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stambergova, H.; Skarydova, L.; Dunford, J.E.; Wsol, V. Biochemical properties of human dehydrogenase/reductase (SDR family) member 7. Chem. Biol. Interact 2014, 207, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Araya, S.; Kratschmar, D.V.; Tsachaki, M.; Stucheli, S.; Beck, K.R.; Odermatt, A. DHRS7 (SDR34C1)—A new player in the regulation of androgen receptor function by inactivation of 5alpha-dihydrotestosterone? J. Steroid Biochem. Mol. Biol. 2017, 171, 288–295. [Google Scholar] [CrossRef]
- Zemanova, L.; Kirubakaran, P.; Pato, I.H.; Stambergova, H.; Vondrasek, J. The identification of new substrates of human DHRS7 by molecular modeling and in vitro testing. Int. J. Biol. Macromol. 2017, 105 Pt 1, 171–182. [Google Scholar] [CrossRef]
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006, 66, 2815–2825. [Google Scholar] [CrossRef] [Green Version]
- Ciardiello, F.; Tortora, G. EGFR antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [Green Version]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Song, L.; Turkson, J.; Karras, J.G.; Jove, R.; Haura, E.B. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene 2003, 22, 4150–4165. [Google Scholar] [CrossRef] [Green Version]
- Garcia, R.; Bowman, T.L.; Niu, G.; Yu, H.; Minton, S.; Muro-Cacho, C.A.; Cox, C.E.; Falcone, R.; Fairclough, R.; Parsons, S.; et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 2001, 20, 2499–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Taha-Mehlitz, S.; Bianco, G.; Coto-Llerena, M.; Kancherla, V.; Bantug, G.R.; Gallon, J.; Ercan, C.; Panebianco, F.; Eppenberger-Castori, S.; von Strauss, M.; et al. Adenylosuccinate lyase is oncogenic in colorectal cancer by causing mitochondrial dysfunction and independent activation of NRF2 and mTOR-MYC-axis. Theranostics 2021, 11, 4011–4029. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2010. [Google Scholar]
- Kassambara, A. ggpubr: ‘Ggplot2’ Based Publication Ready Plots; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawing Survival Curves Using ‘ggplot2’; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Makkonen, H.; Kauhanen, M.; Paakinaho, V.; Jaaskelainen, T.; Palvimo, J.J. Long-range activation of FKBP51 transcription by the androgen receptor via distal intronic enhancers. Nucleic Acids Res. 2009, 37, 4135–4148. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, G.D.; Bianco, R.; Tortora, G.; Ciardiello, F. Involvement of growth factor receptors of the epidermal growth factor receptor family in prostate cancer development and progression to androgen independence. Clin. Prostate Cancer 2003, 2, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Rusthoven, C.G.; Carlson, J.A.; Waxweiler, T.V.; Yeh, N.; Raben, D.; Flaig, T.W.; Kavanagh, B.D. The prognostic significance of Gleason scores in metastatic prostate cancer. Urol. Oncol. 2014, 32, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Labrie, F. Combined blockade of testicular and locally made androgens in prostate cancer: A highly significant medical progress based upon intracrinology. J. Steroid Biochem. Mol. Biol. 2015, 145, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Zannas, A.S.; Wiechmann, T.; Gassen, N.C.; Binder, E.B. Gene-Stress-Epigenetic Regulation of FKBP5: Clinical and Translational Implications. Neuropsychopharmacology 2016, 41, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Hernes, E.; Fossa, S.D.; Berner, A.; Otnes, B.; Nesland, J.M. Expression of the epidermal growth factor receptor family in prostate carcinoma before and during androgen-independence. Br. J. Cancer 2004, 90, 449–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlomm, T.; Kirstein, P.; Iwers, L.; Daniel, B.; Steuber, T.; Walz, J.; Chun, F.H.; Haese, A.; Kollermann, J.; Graefen, M.; et al. Clinical significance of epidermal growth factor receptor protein overexpression and gene copy number gains in prostate cancer. Clin. Cancer Res. 2007, 13 Pt 1, 6579–6584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, K.H.; Hong, M.E.; Jung, Y.Y.; Lee, C.H.; Lee, T.J.; Park, E.S.; Kim, M.K.; Yoo, J.H.; Lee, S.W. Correlation of AR, EGFR, and HER2 Expression Levels in Prostate Cancer: Immunohistochemical Analysis and Chromogenic In Situ Hybridization. Cancer Res. Treat. 2012, 44, 50–56. [Google Scholar] [CrossRef]
- Whang, Y.E.; Armstrong, A.J.; Rathmell, W.K.; Godley, P.A.; Kim, W.Y.; Pruthi, R.S.; Wallen, E.M.; Crane, J.M.; Moore, D.T.; Grigson, G.; et al. A phase II study of lapatinib, a dual EGFR and HER-2 tyrosine kinase inhibitor, in patients with castration-resistant prostate cancer. Urol. Oncol. 2013, 31, 82–86. [Google Scholar] [CrossRef]
- Small, E.J.; Fontana, J.; Tannir, N.; DiPaola, R.S.; Wilding, G.; Rubin, M.; Iacona, R.B.; Kabbinavar, F.F. A phase II trial of gefitinib in patients with non-metastatic hormone-refractory prostate cancer. BJU Int. 2007, 100, 765–769. [Google Scholar] [CrossRef]
- Nabhan, C.; Lestingi, T.M.; Galvez, A.; Tolzien, K.; Kelby, S.K.; Tsarwhas, D.; Newman, S.; Bitran, J.D. Erlotinib has moderate single-agent activity in chemotherapy-naive castration-resistant prostate cancer: Final results of a phase II trial. Urology 2009, 74, 665–671. [Google Scholar] [CrossRef]
- Gravis, G.; Bladou, F.; Salem, N.; Goncalves, A.; Esterni, B.; Walz, J.; Bagattini, S.; Marcy, M.; Brunelle, S.; Viens, P. Results from a monocentric phase II trial of erlotinib in patients with metastatic prostate cancer. Ann. Oncol. 2008, 19, 1624–1628. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Matsuoka, Y.; Funahashi, A.; Kitano, H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol. 2005, 1, 2005.0010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 2018, 174, 433–447.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frattini, M.; Saletti, P.; Romagnani, E.; Martin, V.; Molinari, F.; Ghisletta, M.; Camponovo, A.; Etienne, L.L.; Cavalli, F.; Mazzucchelli, L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br. J. Cancer 2007, 97, 1139–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouali, S.; Chretien, A.S.; Ramacci, C.; Rouyer, M.; Becuwe, P.; Merlin, J.L. PTEN expression controls cellular response to cetuximab by mediating PI3K/AKT and RAS/RAF/MAPK downstream signaling in KRAS wild-type, hormone refractory prostate cancer cells. Oncol. Rep. 2009, 21, 731–735. [Google Scholar]
- Cathomas, R.; Rothermundt, C.; Klingbiel, D.; Bubendorf, L.; Jaggi, R.; Betticher, D.C.; Brauchli, P.; Cotting, D.; Droege, C.; Winterhalder, R.; et al. Efficacy of cetuximab in metastatic castration-resistant prostate cancer might depend on EGFR and PTEN expression: Results from a phase II trial (SAKK 08/07). Clin. Cancer Res. 2012, 18, 6049–6057. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Campbell, C.L.; Jiang, Z.; Savarese, D.M.; Savarese, T.M. Increased expression of the interleukin-11 receptor and evidence of STAT3 activation in prostate carcinoma. Am. J. Pathol. 2001, 158, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Mora, L.B.; Buettner, R.; Seigne, J.; Diaz, J.; Ahmad, N.; Garcia, R.; Bowman, T.; Falcone, R.; Fairclough, R.; Cantor, A.; et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: Direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002, 62, 6659–6666. [Google Scholar]
- Lake, D.; Correa, S.A.; Muller, J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol. Life Sci. 2016, 73, 4397–4413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nacerddine, K.; Beaudry, J.B.; Ginjala, V.; Westerman, B.; Mattiroli, F.; Song, J.Y.; van der Poel, H.; Ponz, O.B.; Pritchard, C.; Cornelissen-Steijger, P.; et al. Akt-mediated phosphorylation of Bmi1 modulates its oncogenic potential, E3 ligase activity, and DNA damage repair activity in mouse prostate cancer. J. Clin. Investig. 2012, 122, 1920–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persad, S.; Attwell, S.; Gray, V.; Delcommenne, M.; Troussard, A.; Sanghera, J.; Dedhar, S. Inhibition of integrin-linked kinase (ILK) suppresses activation of protein kinase B/Akt and induces cell cycle arrest and apoptosis of PTEN-mutant prostate cancer cells. Proc. Natl. Acad. Sci. USA 2000, 97, 3207–3212. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Cheville, J.C.; Pan, Y.; Roche, P.C.; Schmidt, L.J.; Tindall, D.J. PTEN induces chemosensitivity in PTEN-mutated prostate cancer cells by suppression of Bcl-2 expression. J. Biol. Chem. 2001, 276, 38830–38836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Shinriki, S.; Maeshiro, M.; Hirayama, M.; Jono, H.; Yoshida, R.; Nakayama, H.; Matsui, H. The Tumour Suppressor CYLD Is Required for Clathrin-Mediated Endocytosis of EGFR and Cetuximab-Induced Apoptosis in Head and Neck Squamous Cell Carcinoma. Cancers 2021, 14, 173. [Google Scholar] [CrossRef]
- Hiebert, B.M.; Janzen, B.W.; Sanjanwala, R.M.; Ong, A.D.; Feldman, R.D.; Kim, J.O. Impact of spironolactone exposure on prostate cancer incidence amongst men with heart failure: A Pharmacoepidemiological study. Br. J. Clin. Pharmacol. 2021, 87, 1801–1813. [Google Scholar] [CrossRef]
- Mackenzie, I.S.; Morant, S.V.; Wei, L.; Thompson, A.M.; MacDonald, T.M. Spironolactone use and risk of incident cancers: A retrospective, matched cohort study. Br. J. Clin. Pharmacol. 2017, 83, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Arriza, J.L.; Weinberger, C.; Cerelli, G.; Glaser, T.M.; Handelin, B.L.; Housman, D.E.; Evans, R.M. Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science 1987, 237, 268–275. [Google Scholar] [CrossRef]
- Krug, A.W.; Grossmann, C.; Schuster, C.; Freudinger, R.; Mildenberger, S.; Govindan, M.V.; Gekle, M. Aldosterone stimulates epidermal growth factor receptor expression. J. Biol. Chem. 2003, 278, 43060–43066. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, C.; Krug, A.W.; Freudinger, R.; Mildenberger, S.; Voelker, K.; Gekle, M. Aldosterone-induced EGFR expression: Interaction between the human mineralocorticoid receptor and the human EGFR promoter. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1790–E1800. [Google Scholar] [CrossRef] [Green Version]
- Madrigal-Martinez, A.; Constancio, V.; Lucio-Cazana, F.J.; Fernandez-Martinez, A.B. PROSTAGLANDIN E2 stimulates cancer-related phenotypes in prostate cancer PC3 cells through cyclooxygenase-2. J. Cell Physiol. 2019, 234, 7548–7559. [Google Scholar] [CrossRef] [PubMed]
- Pai, R.; Soreghan, B.; Szabo, I.L.; Pavelka, M.; Baatar, D.; Tarnawski, A.S. Prostaglandin E2 transactivates EGF receptor: A novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat. Med. 2002, 8, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Sharaf, A.; Mensching, L.; Keller, C.; Rading, S.; Scheffold, M.; Palkowitsch, L.; Djogo, N.; Rezgaoui, M.; Kestler, H.A.; Moepps, B.; et al. Systematic Affinity Purification Coupled to Mass Spectrometry Identified p62 as Part of the Cannabinoid Receptor CB2 Interactome. Front. Mol. Neurosci. 2019, 12, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stücheli, S.; Araya, S.; Ercan, C.; Moser, S.O.; Gallon, J.; Jenö, P.; Piscuoglio, S.; Terracciano, L.; Odermatt, A. The Potential Tumor-Suppressor DHRS7 Inversely Correlates with EGFR Expression in Prostate Cancer Cells and Tumor Samples. Cancers 2022, 14, 3074. https://doi.org/10.3390/cancers14133074
Stücheli S, Araya S, Ercan C, Moser SO, Gallon J, Jenö P, Piscuoglio S, Terracciano L, Odermatt A. The Potential Tumor-Suppressor DHRS7 Inversely Correlates with EGFR Expression in Prostate Cancer Cells and Tumor Samples. Cancers. 2022; 14(13):3074. https://doi.org/10.3390/cancers14133074
Chicago/Turabian StyleStücheli, Simon, Selene Araya, Caner Ercan, Seraina O. Moser, John Gallon, Paul Jenö, Salvatore Piscuoglio, Luigi Terracciano, and Alex Odermatt. 2022. "The Potential Tumor-Suppressor DHRS7 Inversely Correlates with EGFR Expression in Prostate Cancer Cells and Tumor Samples" Cancers 14, no. 13: 3074. https://doi.org/10.3390/cancers14133074
APA StyleStücheli, S., Araya, S., Ercan, C., Moser, S. O., Gallon, J., Jenö, P., Piscuoglio, S., Terracciano, L., & Odermatt, A. (2022). The Potential Tumor-Suppressor DHRS7 Inversely Correlates with EGFR Expression in Prostate Cancer Cells and Tumor Samples. Cancers, 14(13), 3074. https://doi.org/10.3390/cancers14133074