
New Perspectives on the Role of Integrin-Linked Kinase (ILK) Signaling in Cancer Metastasis


Abstract
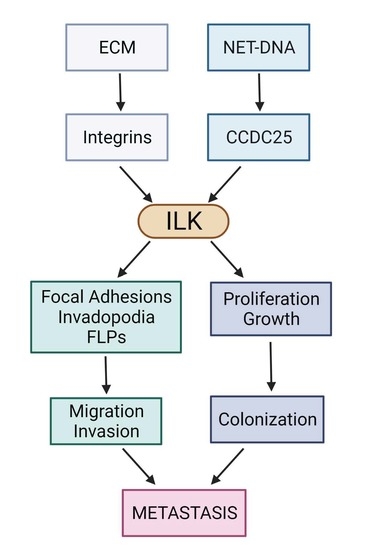
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. ILK Functions as a Signaling Node within Focal Adhesions
3. Role of ILK Signaling in Invasive Structures, Invasion, Colonization, and Dormancy
3.1. Invasive Structures
3.2. Mechanotransduction, Matrix Stiffness, and Invasion
3.3. Extravasation and Colonization
3.4. ILK as a Key Component of Neutrophil Extracellular Trap (NET) Mediated Invasion and Metastasis
3.5. ILK and Extracellular Vesicles
4. Role of ILK Signaling in EMT and the Hippo Pathway
4.1. Epithelial Mesenchymal Transition
4.2. Hippo Pathway
5. The Role of ILK Signaling in Therapeutic Resistance
6. The Role of ILK in Leukemia and Resistance to TKIs
7. Outstanding Questions
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nguyen, B.; Fong, C.; Luthra, A.; Smith, S.A.; DiNatale, R.G.; Nandakumar, S.; Walch, H.; Chatila, W.K.; Madupuri, R.; Kundra, R.; et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 2022, 185, 563–575.e11. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Ganesh, K. Metastasis-Initiating Cells and Ecosystems. Cancer Discov. 2021, 11, 971–994. [Google Scholar] [CrossRef] [PubMed]
- Legate, K.R.; Montañez, E.; Kudlacek, O.; Fässler, R. ILK, PINCH and parvin: The tIPP of integrin signalling. Nat. Rev. Mol. Cell Biol. 2006, 7, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Cabodi, S.; Camacho-Leal, M.D.P.; Di Stefano, P.; Defilippi, P. Integrin signalling adaptors: Not only figurants in the cancer story. Nat. Cancer 2010, 10, 858–870. [Google Scholar] [CrossRef]
- Attwell, S.; Roskelley, C.; Dedhar, S. The integrin-linked kinase (ILK) suppresses anoikis. Oncogene 2000, 19, 3811–3815. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Lin, L.; Sun, K.; Zhang, T.; Chen, W.; Li, L.; Xie, Y.; Wu, C.; Wei, Z.; Yu, C. Complex structures of Rsu1 and PINCH1 reveal a regulatory mechanism of the ILK/PINCH/Parvin complex for F-actin dynamics. elife 2021, 10, e64395. [Google Scholar] [CrossRef]
- Górska, A.; Mazur, A.J. Integrin-linked kinase (ILK): The known vs. the unknown and perspectives. Cell Mol. Life Sci. 2022, 79, 100. [Google Scholar] [CrossRef]
- McDonald, P.C.; Fielding, A.; Dedhar, S. Integrin-linked kinase—essential roles in physiology and cancer biology. J. Cell Sci. 2008, 121, 3121–3132. [Google Scholar] [CrossRef] [Green Version]
- Hannigan, G.E.; Troussard, A.A.; Dedhar, S. Integrin-linked kinase: A cancer therapeutic target unique among its ILK. Nat. Cancer 2005, 5, 51–63. [Google Scholar] [CrossRef]
- Kadry, Y.A.; Huet-Calderwood, C.; Simon, B.; Calderwood, D.A. Kindlin-2 interacts with a highly-conserved surface of ILK to regulate focal adhesion localization and cell spreading. J. Cell Sci. 2018, 131, jcs221184. [Google Scholar] [CrossRef] [Green Version]
- Burridge, K. Focal adhesions: A personal perspective on a half century of progress. FEBS J. 2017, 284, 3355–3361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilinc, A.N.; Han, S.; Barrett, L.A.; Anandasivam, N.; Nelson, C.M. Integrin-linked kinase tunes cell–cell and cell-matrix adhesions to regulate the switch between apoptosis and EMT downstream of TGFβ1. Mol. Biol. Cell 2021, 32, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-O.; Shin, S.; Karreth, F.A.; Buel, G.R.; Jedrychowski, M.P.; Plas, D.R.; Dedhar, S.; Gygi, S.P.; Roux, P.; Dephoure, N.; et al. Focal Adhesion- and IGF1R-Dependent Survival and Migratory Pathways Mediate Tumor Resistance to mTORC1/2 Inhibition. Mol. Cell 2017, 67, 512–527.e4. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.C.; Oloumi, A.; Mills, J.; Dobreva, I.; Maidan, M.; Gray, V.; Wederell, E.D.; Bally, M.B.; Foster, L.J.; Dedhar, S. Rictor and Integrin-Linked Kinase Interact and Regulate Akt Phosphorylation and Cancer Cell Survival. Cancer Res. 2008, 68, 1618–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, W.S.; McDonald, P.C.; Nemirovsky, O.; Awrey, S.; Chafe, S.C.; Schaeffer, D.F.; Li, J.; Renouf, D.J.; Stanger, B.Z.; Dedhar, S. Overcoming Adaptive Resistance to KRAS and MEK Inhibitors by Co-targeting mTORC1/2 Complexes in Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100131. [Google Scholar] [CrossRef]
- Park, H.; Yamamoto, H.; Mohn, L.; Ambühl, L.; Kanai, K.; Schmidt, I.; Kim, K.-P.; Fraccaroli, A.; Feil, S.; Junge, H.J.; et al. Integrin-linked kinase controls retinal angiogenesis and is linked to Wnt signaling and exudative vitreoretinopathy. Nat. Commun. 2019, 10, 5243. [Google Scholar] [CrossRef]
- Cerutti, J.M.; Oler, G.; Michaluart, P.; Delcelo, R.; Beaty, R.M.; Shoemaker, J.; Riggins, G.J. Molecular Profiling of Matched Samples Identifies Biomarkers of Papillary Thyroid Carcinoma Lymph Node Metastasis. Cancer Res. 2007, 67, 7885–7892. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Talebzadeh-Farrooji, M.; Osborne, M.J.; Prokop, J.W.; McDonald, P.C.; Karar, J.; Hou, Z.; He, M.; Kebebew, E.; Orntoft, T.; et al. LIMD2 Is a Small LIM-Only Protein Overexpressed in Metastatic Lesions That Regulates Cell Motility and Tumor Progression by Directly Binding to and Activating the Integrin-Linked Kinase. Cancer Res. 2014, 74, 1390–1403. [Google Scholar] [CrossRef] [Green Version]
- Serrano, I.; McDonald, P.C.; Lock, F.; Muller, W.J.; Dedhar, S. Inactivation of the Hippo tumour suppressor pathway by integrin-linked kinase. Nat. Commun. 2013, 4, 2976. [Google Scholar] [CrossRef]
- Fan, Y.; Gong, Y.; Ghosh, P.K.; Graham, L.M.; Fox, P.L. Spatial Coordination of Actin Polymerization and ILK–Akt2 Activity during Endothelial Cell Migration. Dev. Cell 2009, 16, 661–674. [Google Scholar] [CrossRef] [Green Version]
- Joshi, M.B.; Ivanov, D.; Philippova, M.; Erne, P.; Resink, T.J. Integrin-linked kinase is an essential mediator for T-cadherin-dependent signaling via Akt and GSK3{beta} in endothelial cells. FASEB J. 2007, 21, 3083–3095. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Brooks, M.W.; Weinberg, R.A. An Integrin-Linked Machinery of Cytoskeletal Regulation that Enables Experimental Tumor Initiation and Metastatic Colonization. Cancer Cell 2013, 24, 481–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibue, T.; Brooks, M.W.; Inan, M.F.; Reinhardt, F.; Weinberg, R.A. The Outgrowth of Micrometastases Is Enabled by the Formation of Filopodium-like Protrusions. Cancer Discov. 2012, 2, 706–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masi, I.; Caprara, V.; Spadaro, F.; Chellini, L.; Sestito, R.; Zancla, A.; Rainer, A.; Bagnato, A.; Rosanò, L. Endothelin-1 drives invadopodia and interaction with mesothelial cells through ILK. Cell Rep. 2021, 34, 108800. [Google Scholar] [CrossRef] [PubMed]
- Greco, M.; Moro, L.; Forciniti, S.; Alfarouk, K.; Cannone, S.; Cardone, R.; Reshkin, S. Integrin-Linked Kinase Links Integrin Activation to Invadopodia Function and Invasion via the p(T567)-Ezrin/NHERF1/NHE1 Pathway. Int. J. Mol. Sci. 2021, 22, 2162. [Google Scholar] [CrossRef]
- Branch, K.M.; Hoshino, D.; Weaver, A.M. Adhesion rings surround invadopodia and promote maturation. Biol. Open 2012, 1, 711–722. [Google Scholar] [CrossRef] [Green Version]
- Rabie, E.M.; Zhang, S.X.; Dunn, C.E.; Nelson, C.M. Substratum stiffness signals through integrin-linked kinase and β1-integrin to regulate midbody proteins and abscission during EMT. Mol. Biol. Cell 2021, 32, 1664–1676. [Google Scholar] [CrossRef]
- Naso, J.R.; Topham, J.T.; Karasinska, J.M.; Lee, M.K.; Kalloger, S.E.; Wong, H.; Nelson, J.; Moore, R.A.; Mungall, A.J.; Jones, S.J.; et al. Tumor infiltrating neutrophils and gland formation predict overall survival and molecular subgroups in pancreatic ductal adenocarcinoma. Cancer Med. 2020, 10, 1155–1165. [Google Scholar] [CrossRef]
- Anlaş, A.A.; Nelson, C.M. Soft Microenvironments Induce Chemoresistance by Increasing Autophagy Downstream of Integrin-Linked Kinase. Cancer Res. 2020, 80, 4103–4113. [Google Scholar] [CrossRef]
- Han, S.; Pang, M.-F.; Nelson, C.M. Substratum stiffness tunes proliferation downstream of Wnt3a in part by regulating integrin-linked kinase and frizzled-1. J. Cell Sci. 2018, 131, jcs210476. [Google Scholar] [CrossRef] [Green Version]
- Pang, M.-F.; Siedlik, M.J.; Han, S.; Stallings-Mann, M.; Radisky, D.C.; Nelson, C.M. Tissue Stiffness and Hypoxia Modulate the Integrin-Linked Kinase ILK to Control Breast Cancer Stem-like Cells. Cancer Res. 2016, 76, 5277–5287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Er, E.E.; Valiente, M.; Ganesh, K.; Zou, Y.; Agrawal, S.; Hu, J.; Griscom, B.; Rosenblum, M.; Boire, A.; Brogi, E.; et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat. Cell Biol. 2018, 20, 966–978. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, Q.; Zhang, X.; Liu, X.; Zhou, B.; Chen, J.; Huang, D.; Li, J.; Li, H.; Chen, F.; et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 2020, 583, 133–138. [Google Scholar] [CrossRef]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef] [Green Version]
- Rayes, R.F.; Mouhanna, J.G.; Nicolau, I.; Bourdeau, F.; Giannias, B.; Rousseau, S.; Quail, D.; Walsh, L.; Sangwan, V.; Bertos, N.; et al. Primary tumors induce neutrophil extracellular traps with targetable metastasis-promoting effects. JCI Insight 2019, 5, e128008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohme, S.; Yazdani, H.O.; Al-Khafaji, A.B.; Chidi, A.P.; Loughran, P.; Mowen, A.K.; Wang, Y.; Simmons, R.L.; Huang, H.; Tsung, A. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016, 76, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- Demers, M.; Wong, S.L.; Martinod, K.; Gallant, M.; Cabral, J.E.; Wang, Y.; Wagner, D.D. Priming of neutrophils toward NETosis promotes tumor growth. OncoImmunology 2016, 5, e1134073. [Google Scholar] [CrossRef] [Green Version]
- Demers, M.; Wagner, D.D. Neutrophil extracellular traps. OncoImmunology 2013, 2, e22946. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Cong, M.; Li, J.; He, D.; Wu, Q.; Tian, P.; Wang, Y.; Yang, S.; Liang, C.; Liang, Y.; et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell 2021, 39, 423–437.e7. [Google Scholar] [CrossRef]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, 4227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahangiri, A.; Nguyen, A.; Chandra, A.; Sidorov, M.K.; Yagnik, G.; Rick, J.; Han, S.W.; Chen, W.; Flanigan, P.M.; Schneidman-Duhovny, D.; et al. Cross-activating c-Met/β1 integrin complex drives metastasis and invasive resistance in cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E8685–E8694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolini, I.; Ghosh, J.C.; Kossenkov, A.V.; Mulugu, S.; Krishn, S.R.; Vaira, V.; Qin, J.; Plow, E.F.; Languino, L.R.; Altieri, D.C. Small Extracellular Vesicle Regulation of Mitochondrial Dynamics Reprograms a Hypoxic Tumor Microenvironment. Dev. Cell 2020, 55, 163–177.e6. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, I.; Perego, M.; Ghosh, J.C.; Kossenkov, A.V.; Altieri, D.C. NFκB activation by hypoxic small extracellular vesicles drives oncogenic reprogramming in a breast cancer microenvironment. Oncogene 2022, 41, 2520–2525. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Serrano, I.; McDonald, P.C.; Lock, F.E.; Dedhar, S. Role of the integrin-linked kinase (ILK)/Rictor complex in TGFβ-1-induced epithelial–mesenchymal transition (EMT). Oncogene 2012, 32, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Yang, K.; Tang, X.-F.; Lin, N.-N.; Liu, J.-Y. Expression of integrin-linked kinase in adenoid cystic carcinoma of salivary glands correlates with epithelial–mesenchymal transition markers and tumor progression. Med. Oncol. 2013, 30, 619. [Google Scholar] [CrossRef]
- Zhao, D.; Tang, X.-F.; Yang, K.; Liu, J.-Y.; Ma, X.-R. Over-expression of integrin-linked kinase correlates with aberrant expression of Snail, E-cadherin and N-cadherin in oral squamous cell carcinoma: Implications in tumor progression and metastasis. Clin. Exp. Metastasis 2012, 29, 957–969. [Google Scholar] [CrossRef]
- Zhao, M.; Gao, Y.; Wang, L.; Liu, S.; Han, B.; Ma, L.; Ling, Y.; Mao, S.; Wang, X. Overexpression of Integrin-linked Kinase Promotes Lung Cancer Cell Migration and Invasion via NF-κB-mediated Upregulation of Matrix Metalloproteinase-9. Int. J. Med. Sci. 2013, 10, 995–1002. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Yin, H.; Wang, R.; Wu, D.; Sun, W.; Liu, B.; Su, Q. Overexpression of integrin-linked kinase (ILK) promotes migration and invasion of colorectal cancer cells by inducing epithelial–mesenchymal transition via NF-κB signaling. Acta Histochem. 2013, 116, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Xiong, D.; Liou, Y.; Shu, J.; Li, D.; Zhang, L.; Chen, J. Down-regulating ribonuclease inhibitor enhances metastasis of bladder cancer cells through regulating epithelial–mesenchymal transition and ILK signaling pathway. Exp. Mol. Pathol. 2014, 96, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Li, D.; Xiong, D.-M.; Li, L.; Jiang, R.; Chen, J.-X. A novel role of ribonuclease inhibitor in regulation of epithelial-to-mesenchymal transition and ILK signaling pathway in bladder cancer cells. Cell Tissue Res. 2013, 353, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Han, K.S.; Li, N.; Raven, P.A.; Fazli, L.; Ettinger, S.; Hong, S.J.; Gleave, M.E.; So, A.I. Targeting Integrin-Linked Kinase Suppresses Invasion and Metastasis through Downregulation of Epithelial-to-Mesenchymal Transition in Renal Cell Carcinoma. Mol. Cancer Ther. 2015, 14, 1024–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Que, L.; Zhao, D.; Tang, X.-F.; Liu, J.-Y.; Zhang, X.-Y.; Zhan, Y.-H.; Zhang, L. Effects of lentivirus-mediated shRNA targeting integrin-linked kinase on oral squamous cell carcinoma in vitro and in vivo. Oncol. Rep. 2015, 35, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Gil, D.; Zarzycka, M.; Ciołczyk-Wierzbicka, D.; Laidler, P. Integrin linked kinase regulates endosomal recycling of N-cadherin in melanoma cells. Cell. Signal. 2020, 72, 109642. [Google Scholar] [CrossRef]
- Lin, L.; Luo, X.; Wang, L.; Xu, F.; He, Y.; Wang, Q.; Yuan, C.; Xu, J.; Yan, L.; Hao, H. BML-111 inhibits EMT, migration and metastasis of TAMs-stimulated triple-negative breast cancer cells via ILK pathway. Int. Immunopharmacol. 2020, 85, 106625. [Google Scholar] [CrossRef]
- Feng, Y.; Le, F.; Tian, P.; Zhong, Y.; Zhan, F.; Huang, G.; Hu, H.; Chen, T.; Tan, B. GTW inhibits the Epithelial to Mesenchymal Transition of Epithelial Ovarian Cancer via ILK/AKT/GSK3β/Slug Signalling Pathway. J. Cancer 2021, 12, 1386–1397. [Google Scholar] [CrossRef]
- Lu, J.; Xu, Y.; Zhao, Z.; Ke, X.; Wei, X.; Kang, J.; Zong, X.; Mao, H.; Liu, P. Emodin suppresses proliferation, migration and invasion in ovarian cancer cells by down regulating ILK in vitro and in vivo. OncoTargets Ther. 2017, 10, 3579–3589. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Zhou, X.; Hao, J.; Dai, H.; Zhang, J.; He, Y.; Hao, H. Lipoxin A(4) and its analog suppress hepatocarcinoma cell epithelial-mesenchymal transition, migration and metastasis via regulating integrin-linked kinase axis. Prostaglandins Other Lipid Mediat 2018, 137, 9–19. [Google Scholar] [CrossRef]
- Yang, J.; Hou, Y.; Zhou, M.; Wen, S.; Zhou, J.; Xu, L.; Tang, X.; Du, Y.-E.; Hu, P.; Liu, M. Twist induces epithelial-mesenchymal transition and cell motility in breast cancer via ITGB1-FAK/ILK signaling axis and its associated downstream network. Int. J. Biochem. Cell Biol. 2015, 71, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Expression of EMT inducers integrin-linked kinase (ILK) and ZEB1 in phyllodes breast tumors is associated with aggressive phenotype. Histol. Histopathol. 2018, 33, 937–949. [CrossRef]
- Shen, H.; Ma, J.-L.; Zhang, Y.; Deng, G.-L.; Qu, Y.-L.; Wu, X.-L.; He, J.-X.; Zhang, S.; Zeng, S. Integrin-linked kinase overexpression promotes epithelial-mesenchymal transition via nuclear factor-κB signaling in colorectal cancer cells. World J. Gastroenterol. 2016, 22, 3969–3977. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-L.; Li, J.; Gong, S.-Y.; Huang, M.; Li, R.; Xiong, G.-X.; Wang, F.; Zou, Q.-M.; Qi, Q.; Yin, X.-X. Targeting the ILK/YAP axis by LFG-500 blocks epithelial–mesenchymal transition and metastasis. Acta Pharmacol. Sin. 2021, 42, 1847–1859. [Google Scholar] [CrossRef]
- Qin, X.; Lv, X.; Li, P.; Yang, R.; Xia, Q.; Chen, Y.; Peng, Y.; Li, L.; Li, S.; Li, T.; et al. Matrix stiffness modulates ILK-mediated YAP activation to control the drug resistance of breast cancer cells. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2020, 1866, 165625. [Google Scholar] [CrossRef]
- Wang, Q.; Sang, W.; Xue, J.; Zhai, Y.; Hu, Y.; Su, L.; Zhang, W. The Expression and Prognostic Value of ILK and YAP1 in Glioma. Appl. Immunohistochem. Mol. Morphol. 2022, 30, e21–e29. [Google Scholar] [CrossRef] [PubMed]
- Sestito, R.; Tocci, P.; Roman, C.; Di Castro, V.; Bagnato, A. Functional interaction between endothelin-1 and ZEB1/YAP signaling regulates cellular plasticity and metastasis in high-grade serous ovarian cancer. J. Exp. Clin. Cancer Res. 2022, 41, 157. [Google Scholar] [CrossRef]
- Beetham, H.; Griffith, B.G.; Murina, O.; Loftus, A.E.; Parry, D.A.; Temps, C.; Culley, J.; Muir, M.; Unciti-Broceta, A.; Sims, A.H.; et al. Loss of Integrin-Linked Kinase Sensitizes Breast Cancer to SRC Inhibitors. Cancer Res. 2021, 82, 632–647. [Google Scholar] [CrossRef] [PubMed]
- Juratli, M.A.; Zhou, H.; Oppermann, E.; Bechstein, W.O.; Pascher, A.; Chun, F.K.-H.; Juengel, E.; Rutz, J.; Blaheta, R.A. Integrin α2 and β1 Cross-Communication with mTOR/AKT and the CDK-Cyclin Axis in Hepatocellular Carcinoma Cells. Cancers 2022, 14, 2430. [Google Scholar] [CrossRef]
- Tsoumas, D.; Nikou, S.; Giannopoulou, E.; Champeris Tsaniras, S.; Sirinian, C.; Maroulis, I.; Taraviras, S.; Zolota, V.; Kalofonos, H.P.; Bravou, V. ILK Expression in Colorectal Cancer Is Associated with EMT, Cancer Stem Cell Markers and Chemoresistance. J. Mol. Histol. 2018, 15, 127–141. [Google Scholar] [CrossRef] [Green Version]
- Nikou, S.; Arbi, M.; Dimitrakopoulos, F.-I.D.; Sirinian, C.; Chadla, P.; Pappa, I.; Ntaliarda, G.; Stathopoulos, G.T.; Papadaki, H.; Zolota, V.; et al. Integrin-linked kinase (ILK) regulates KRAS, IPP complex and Ras suppressor-1 (RSU1) promoting lung adenocarcinoma progression and poor survival. Histochem. J. 2020, 51, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Cardona, A.F.; Bracht, J.W.P.; Aldeguer, E.; Drozdowskyj, A.; Fernandez-Bruno, M.; Chaib, I.; Berenguer, J.; Santarpia, M.; Ito, M.; et al. Integrin-linked kinase (ILK) and src homology 2 domain-containing phosphatase 2 (SHP2): Novel targets in EGFR-mutation positive non-small cell lung cancer (NSCLC). EBioMedicine 2019, 39, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausmann, C.; Temme, A.; Cordes, N.; Eke, I. ILKAP, ILK and PINCH1 control cell survival of p53-wildtype glioblastoma cells after irradiation. Oncotarget 2015, 6, 34592–34605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghazi, A.; Le Corre, D.; Pilati, C.; Taieb, J.; Aparicio, T.; Didelot, A.; Dedhar, S.; Mulot, C.; Le Malicot, K.; Djouadi, F.; et al. Prognostic value of the PrPC-ILK-IDO1 axis in the mesenchymal colorectal cancer subtype. OncoImmunology 2021, 10, 1940674. [Google Scholar] [CrossRef]
- Rothe, K.; Babaian, A.; Nakamichi, N.; Chen, M.; Chafe, S.C.; Watanabe, A.; Forrest, D.L.; Mager, D.L.; Eaves, C.J.; Dedhar, S.; et al. Integrin-Linked Kinase Mediates Therapeutic Resistance of Quiescent CML Stem Cells to Tyrosine Kinase Inhibitors. Cell Stem Cell 2020, 27, 110–124.e9. [Google Scholar] [CrossRef]
- Kumar, R.; Pereira, R.S.; Zanetti, C.; Minciacchi, V.R.; Merten, M.; Meister, M.; Niemann, J.; Dietz, M.S.; Rüssel, N.; Schnütgen, F.; et al. Specific, targetable interactions with the microenvironment influence imatinib-resistant chronic myeloid leukemia. Leukemia 2020, 34, 2087–2101. [Google Scholar] [CrossRef]
- Alasseiri, M.; Ahmed, A.U.; Williams, B.R. Mechanisms and consequences of constitutive activation of integrin-linked kinase in acute myeloid leukemia. Cytokine Growth Factor Rev. 2018, 43, 1–7. [Google Scholar] [CrossRef]
- Maydan, M.; McDonald, P.C.; Sanghera, J.; Yan, J.; Rallis, C.; Pinchin, S.; Hannigan, G.E.; Foster, L.J.; Ish-Horowicz, D.; Walsh, M.P.; et al. Integrin-Linked Kinase Is a Functional Mn2+-Dependent Protein Kinase that Regulates Glycogen Synthase Kinase-3β (GSK-3β) Phosphorylation. PLoS ONE 2010, 5, e12356. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Wu, C. ILK: A pseudokinase in the center stage of cell-matrix adhesion and signaling. Curr. Opin. Cell Biol. 2012, 24, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Hannigan, G.E.; McDonald, P.C.; Walsh, M.P.; Dedhar, S. Integrin-linked kinase: Not so ‘pseudo’ after all. Oncogene 2011, 30, 4375–4385. [Google Scholar] [CrossRef] [Green Version]
- Erdődi, F.; Kiss, E.; Walsh, M.P.; Stefansson, B.; Deng, J.T.; Eto, M.; Brautigan, D.L.; Hartshorne, D.J. Phosphorylation of protein phosphatase type-1 inhibitory proteins by integrin-linked kinase and cyclic nucleotide-dependent protein kinases. Biochem. Biophys. Res. Commun. 2003, 306, 382–387. [Google Scholar] [CrossRef]
- Wilson, D.P.; Sutherland, C.; Borman, M.A.; Deng, J.T.; MacDonald, J.A.; Walsh, M.P. Integrin-linked kinase is responsible for Ca2+-independent myosin diphosphorylation and contraction of vascular smooth muscle. Biochem. J. 2005, 392, 641–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troussard, A.A.; McDonald, P.C.; Wederell, E.D.; Mawji, N.M.; Filipenko, N.R.; Gelmon, K.A.; Kucab, J.E.; Dunn, S.E.; Emerman, J.T.; Bally, M.B.; et al. Preferential Dependence of Breast Cancer Cells versus Normal Cells on Integrin-Linked Kinase for Protein Kinase B/Akt Activation and Cell Survival. Cancer Res. 2006, 66, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Yau, C.Y.; Wheeler, J.J.; Sutton, K.L.; Hedley, D.W. Inhibition of Integrin-Linked Kinase by a Selective Small Molecule Inhibitor, QLT0254, Inhibits the PI3K/PKB/mTOR, Stat3, and FKHR Pathways and Tumor Growth, and Enhances Gemcitabine-Induced Apoptosis in Human Orthotopic Primary Pancreatic Cancer Xenografts. Cancer Res. 2005, 65, 1497–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younes, M.N.; Yigitbasi, O.G.; Yazici, Y.D.; Jasser, S.A.; Bucana, C.D.; El-Naggar, A.K.; Mills, G.B.; Myers, J.N. Effects of the Integrin-Linked Kinase Inhibitor QLT0267 on Squamous Cell Carcinoma of the Head and Neck. Arch. Otolaryngol.-Head Neck Surg. 2007, 133, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-L.; Hsu, E.-C.; Chou, C.-C.; Chuang, H.-C.; Bai, L.-Y.; Kulp, S.K.; Chen, C.-S. Identification and Characterization of a Novel Integrin-Linked Kinase Inhibitor. J. Med. Chem. 2011, 54, 6364–6374. [Google Scholar] [CrossRef] [Green Version]
- Ning, Z.; Zhu, X.; Jiang, Y.; Gao, A.; Zou, S.; Gu, C.; He, C.; Chen, Y.; Ding, W.-Q.; Zhou, J. Integrin-Linked Kinase Is Involved In the Proliferation and Invasion of Esophageal Squamous Cell Carcinoma. J. Cancer 2020, 11, 324–333. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- van Bussel, M.T.J.; Awada, A.; de Jonge, M.J.A.; Mau-Sørensen, M.; Nielsen, D.; Schöffski, P.; Verheul, H.M.W.; Sarholz, B.; Berghoff, K.; El Bawab, S.; et al. A first-in-man phase 1 study of the DNA-dependent protein kinase inhibitor peposertib (formerly M3814) in patients with advanced solid tumours. Br. J. Cancer 2020, 124, 728–735. [Google Scholar] [CrossRef]
- Burke, M.R.; Smith, A.R.; Zheng, G. Overcoming Cancer Drug Resistance Utilizing PROTAC Technology. Front. Cell Dev. Biol. 2022, 10, 872729. [Google Scholar] [CrossRef]
- Hannigan, G.E.; Leung-Hagesteijn, C.; Fitz-Gibbon, L.; Coppolino, M.G.; Radeva, G.; Filmus, J.; Bell, J.C.; Dedhar, S. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature 1996, 379, 91–96. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McDonald, P.C.; Dedhar, S. New Perspectives on the Role of Integrin-Linked Kinase (ILK) Signaling in Cancer Metastasis. Cancers 2022, 14, 3209. https://doi.org/10.3390/cancers14133209
McDonald PC, Dedhar S. New Perspectives on the Role of Integrin-Linked Kinase (ILK) Signaling in Cancer Metastasis. Cancers. 2022; 14(13):3209. https://doi.org/10.3390/cancers14133209
Chicago/Turabian StyleMcDonald, Paul C., and Shoukat Dedhar. 2022. "New Perspectives on the Role of Integrin-Linked Kinase (ILK) Signaling in Cancer Metastasis" Cancers 14, no. 13: 3209. https://doi.org/10.3390/cancers14133209
APA StyleMcDonald, P. C., & Dedhar, S. (2022). New Perspectives on the Role of Integrin-Linked Kinase (ILK) Signaling in Cancer Metastasis. Cancers, 14(13), 3209. https://doi.org/10.3390/cancers14133209