
Paving the Way to Solid Tumors: Challenges and Strategies for Adoptively Transferred Transgenic T Cells in the Tumor Microenvironment


Abstract
:Simple Summary
Abstract
1. T Cells: Essential Players across Immunotherapeutic Approaches
1.1. T Cells as Key Players of the Antitumor Immune Response
1.2. Targets for T Cell-Based Immunotherapeutic Approaches
2. T Cell-Based Adoptive Cellular Therapy for Solid Tumors: Where Are We in the Clinic?
2.1. TCR-Based Approaches
2.2. CAR-Based Approaches
3. Challenges for T Cells in the Tumor Microenvironment
3.1. Infiltrating the Tumor: Cold and Hot Tumor Microenvironment
3.1.1. Targeting Tumor Vessels
3.1.2. Application of Proinflammatory Stimuli
3.1.3. Engineering T Cells to Alter the Cytokine Milieu
3.2. Effectively Targeting and Eradicating Tumor Cells
3.2.1. TCR-T Cells and Peptide-MHC-Complexes
Target Identification
HLA-Expression
TCR Identification and Assessment
Influence of T Cell Stimulation Strength on T Cell Function and Presence within the Tumor
Administration of CD4-T Helper Cells
CAR-T Cells in Solid Tumors: Armored CARs
Receptor Specificity: On- and Off-Target Dose-Limiting Toxicity
3.3. Overcoming T Cell Exhaustion and Dysfunction
3.3.1. Hostile Environment for T Cells in the Tumor
3.3.2. T Cell Exhaustion due to Chronic Activation
3.3.3. T Cell Senescence
3.3.4. Strategies to Counteract T Cell Dysfunction
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Turner, D.C.; Felip, E.; Lena, H.; Cappuzzo, F.; Horn, L.; Garon, E.B.; Hui, R.; Arkenau, H.T.; Gubens, M.A.; et al. Systematic evaluation of pembrolizumab dosing in patients with advanced non-small-cell lung cancer. Ann. Oncol. 2016, 27, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Five-Year Outcomes with Pembrolizumab Versus Chemotherapy for Metastatic Non-Small-Cell Lung Cancer with PD-L1 Tumor Proportion Score ≥ 50. J. Clin. Oncol. 2021, 39, 2339–2349. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Mack, M.; Riethmüller, G.; Kufer, P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 7021–7025. [Google Scholar] [CrossRef]
- Viardot, A.; Goebeler, M.E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef]
- Garfall, A.L.; June, C.H. Trispecific antibodies offer a third way forward for anticancer immunotherapy. Nature 2019, 575, 450–451. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A Pilot Trial Using Lymphocytes Genetically Engineered with an NY-ESO-1-Reactive T-cell Receptor: Long-term Follow-up and Correlates with Response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef]
- Pagès, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [CrossRef]
- Fuchs, T.L.; Sioson, L.; Sheen, A.; Jafari-Nejad, K.; Renaud, C.J.; Andrici, J.; Ahadi, M.; Chou, A.; Gill, A.J. Assessment of Tumor-infiltrating Lymphocytes Using International TILs Working Group (ITWG) System Is a Strong Predictor of Overall Survival in Colorectal Carcinoma: A Study of 1034 Patients. Am. J. Surg. Pathol. 2020, 44, 536–544. [Google Scholar] [CrossRef]
- James, F.R.; Jiminez-Linan, M.; Alsop, J.; Mack, M.; Song, H.; Brenton, J.D.; Pharoah, P.D.P.; Ali, H.R. Association between tumour infiltrating lymphocytes, histotype and clinical outcome in epithelial ovarian cancer. BMC Cancer 2017, 17, 657. [Google Scholar] [CrossRef]
- Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Berger, A.; Bindea, G.; Meatchi, T.; Bruneval, P.; Trajanoski, Z.; Fridman, W.H.; Pagès, F.; et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J. Clin. Oncol. 2011, 29, 610–618. [Google Scholar] [CrossRef]
- Al-Shibli, K.I.; Donnem, T.; Al-Saad, S.; Persson, M.; Bremnes, R.M.; Busund, L.T. Prognostic effect of epithelial and stromal lymphocyte infiltration in non-small cell lung cancer. Clin. Cancer Res. 2008, 14, 5220–5227. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, S.M.; Paish, E.C.; Powe, D.G.; Macmillan, R.D.; Grainge, M.J.; Lee, A.H.; Ellis, I.O.; Green, A.R. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J. Clin. Oncol. 2011, 29, 1949–1955. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Hamann, D.; Baars, P.A.; Rep, M.H.; Hooibrink, B.; Kerkhof-Garde, S.R.; Klein, M.R.; van Lier, R.A. Phenotypic and functional separation of memory and effector human CD8+ T cells. J. Exp. Med. 1997, 186, 1407–1418. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Pasetto, A.; Jia, L.; Deniger, D.C.; Stevanović, S.; Robbins, P.F.; Rosenberg, S.A. Tumor-infiltrating human CD4(+) regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci. Immunol. 2019, 4, eaao4310. [Google Scholar] [CrossRef]
- Carrier, Y.; Yuan, J.; Kuchroo, V.K.; Weiner, H.L. Th3 cells in peripheral tolerance. I. Induction of Foxp3-positive regulatory T cells by Th3 cells derived from TGF-beta T cell-transgenic mice. J. Immunol. 2007, 178, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Roncarolo, M.G.; Gregori, S.; Bacchetta, R.; Battaglia, M. Tr1 cells and the counter-regulation of immunity: Natural mechanisms and therapeutic applications. Curr. Top. Microbiol. Immunol. 2014, 380, 39–68. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression-implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.M.; Boniface, J.J.; Reich, Z.; Lyons, D.; Hampl, J.; Arden, B.; Chien, Y. Ligand recognition by alpha beta T cell receptors. Annu. Rev. Immunol. 1998, 16, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Mak, T.W. Genomic organization of the T cell receptor. Cancer Detect. Prev. 1989, 14, 261–267. [Google Scholar]
- Nikolich-Zugich, J.; Slifka, M.K.; Messaoudi, I. The many important facets of T-cell repertoire diversity. Nat. Rev. Immunol. 2004, 4, 123–132. [Google Scholar] [CrossRef]
- Starr, T.K.; Jameson, S.C.; Hogquist, K.A. Positive and negative selection of T cells. Annu. Rev. Immunol. 2003, 21, 139–176. [Google Scholar] [CrossRef]
- König, R. Interactions between MHC molecules and co-receptors of the TCR. Curr. Opin. Immunol. 2002, 14, 75–83. [Google Scholar] [CrossRef]
- Marelli-Berg, F.M.; Okkenhaug, K.; Mirenda, V. A two-signal model for T cell trafficking. Trends Immunol. 2007, 28, 267–273. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Spiess, P.; Lafreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Sprent, J. Central tolerance of T cells. Int. Rev. Immunol. 1995, 13, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Nathan, P.; Hassel, J.C.; Rutkowski, P.; Baurain, J.F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2021, 385, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Orlando, D.; Miele, E.; De Angelis, B.; Guercio, M.; Boffa, I.; Sinibaldi, M.; Po, A.; Caruana, I.; Abballe, L.; Carai, A.; et al. Adoptive Immunotherapy Using PRAME-Specific T Cells in Medulloblastoma. Cancer Res. 2018, 78, 3337–3349. [Google Scholar] [CrossRef] [PubMed]
- Luk, S.J.; van der Steen, D.M.; Hagedoorn, R.S.; Jordanova, E.S.; Schilham, M.W.; Bovée, J.V.; Cleven, A.H.; Falkenburg, J.F.; Szuhai, K.; Heemskerk, M.H. PRAME and HLA Class I expression patterns make synovial sarcoma a suitable target for PRAME specific T-cell receptor gene therapy. Oncoimmunology 2018, 7, e1507600. [Google Scholar] [CrossRef]
- Argani, P.; Iacobuzio-Donahue, C.; Ryu, B.; Rosty, C.; Goggins, M.; Wilentz, R.E.; Murugesan, S.R.; Leach, S.D.; Jaffee, E.; Yeo, C.J.; et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: Identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin. Cancer Res. 2001, 7, 3862–3868. [Google Scholar]
- Chang, K.; Pastan, I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc. Natl. Acad. Sci. USA 1996, 93, 136–140. [Google Scholar] [CrossRef]
- Ordóñez, N.G. Application of mesothelin immunostaining in tumor diagnosis. Am. J. Surg. Pathol. 2003, 27, 1418–1428. [Google Scholar] [CrossRef]
- Morello, A.; Sadelain, M.; Adusumilli, P.S. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov. 2016, 6, 133–146. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef]
- Anderson, K.G.; Voillet, V.; Bates, B.M.; Chiu, E.Y.; Burnett, M.G.; Garcia, N.M.; Oda, S.K.; Morse, C.B.; Stromnes, I.M.; Drescher, C.W.; et al. Engineered Adoptive T-cell Therapy Prolongs Survival in a Preclinical Model of Advanced-Stage Ovarian Cancer. Cancer Immunol. Res. 2019, 7, 1412–1425. [Google Scholar] [CrossRef]
- Künkele, A.; Taraseviciute, A.; Finn, L.S.; Johnson, A.J.; Berger, C.; Finney, O.; Chang, C.A.; Rolczynski, L.S.; Brown, C.; Mgebroff, S.; et al. Preclinical Assessment of CD171-Directed CAR T-cell Adoptive Therapy for Childhood Neuroblastoma: CE7 Epitope Target Safety and Product Manufacturing Feasibility. Clin. Cancer Res. 2017, 23, 466–477. [Google Scholar] [CrossRef] [Green Version]
- Stadler, C.R.; Bähr-Mahmud, H.; Plum, L.M.; Schmoldt, K.; Kölsch, A.C.; Türeci, Ö.; Sahin, U. Characterization of the first-in-class T-cell-engaging bispecific single-chain antibody for targeted immunotherapy of solid tumors expressing the oncofetal protein claudin 6. Oncoimmunology 2016, 5, e1091555. [Google Scholar] [CrossRef]
- Mackensen, A.; Koenecke, C.; Haanen, J.; Alsdorf, W.; Desuki, A.; Wagner-Drouet, E.; Heudobler, D.; Borchmann, P.; Wiegert, E.; Schulz, C.; et al. 958 BNT211: A phase I/II trial to evaluate safety and efficacy of CLDN6 CAR-T cells and vaccine-mediated in vivo expansion in patients with CLDN6-positive advanced solid tumors. J. ImmunoTher. Cancer 2021, 9, A1008. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef]
- Liu, G.Y.; Fairchild, P.J.; Smith, R.M.; Prowle, J.R.; Kioussis, D.; Wraith, D.C. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity 1995, 3, 407–415. [Google Scholar] [CrossRef]
- Zehn, D.; Bevan, M.J. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity 2006, 25, 261–270. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Wolfl, M.; Kuball, J.; Ho, W.Y.; Nguyen, H.; Manley, T.J.; Bleakley, M.; Greenberg, P.D. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood 2007, 110, 201–210. [Google Scholar] [CrossRef]
- Yossef, R.; Tran, E.; Deniger, D.C.; Gros, A.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Cafri, G.; Robbins, P.F.; et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 2018, 3, e122467. [Google Scholar] [CrossRef] [PubMed]
- Inozume, T.; Hanada, K.; Wang, Q.J.; Ahmadzadeh, M.; Wunderlich, J.R.; Rosenberg, S.A.; Yang, J.C. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J. Immunother. 2010, 33, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, G.; Stromhaug, K.; Klaeger, S.; Kula, T.; Frederick, D.T.; Le, P.M.; Forman, J.; Huang, T.; Li, S.; Zhang, W.; et al. Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature 2021, 596, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Zheng, Z.; Lowery, F.J.; Gartner, J.J.; Prickett, T.D.; Robbins, P.F.; Rosenberg, S.A. Direct identification of neoantigen-specific TCRs from tumor specimens by high-throughput single-cell sequencing. J. Immunother. Cancer 2021, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Lowery, F.J.; Krishna, S.; Yossef, R.; Parikh, N.B.; Chatani, P.D.; Zacharakis, N.; Parkhurst, M.R.; Levin, N.; Sindiri, S.; Sachs, A.; et al. Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science 2022, 375, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Dembić, Z.; Haas, W.; Weiss, S.; McCubrey, J.; Kiefer, H.; von Boehmer, H.; Steinmetz, M. Transfer of specificity by murine alpha and beta T-cell receptor genes. Nature 1986, 320, 232–238. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Druta, M.; Liebner, D.A.; Schuetze, S.; Somaiah, N.; Tine, B.A.V.; Tap, W.D.; Pulham, T.; Chagin, K.; Norry, E.; et al. Pilot study of NY-ESO-1c259 T cells in advanced myxoid/round cell liposarcoma. J. Clin. Oncol. 2018, 36, 3005. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Parker, L.L.; Lu, T.; Zheng, Z.; Toomey, M.A.; White, D.E.; Yao, X.; Li, Y.F.; Robbins, P.F.; Feldman, S.A.; et al. Treatment of Patients with Metastatic Cancer Using a Major Histocompatibility Complex Class II–Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J. Clin. Oncol. 2017, 35, 3322–3329. [Google Scholar] [CrossRef]
- Hong, D.S.; Tine, B.A.V.; Olszanski, A.J.; Johnson, M.L.; Liebner, D.A.; Trivedi, T.; Lin, Q.; Elefant, E.; Dryer-Minnerly, R.; Navenot, J.-M.; et al. Phase I dose escalation and expansion trial to assess the safety and efficacy of ADP-A2M4 SPEAR T cells in advanced solid tumors. J. Clin. Oncol. 2020, 38, 102. [Google Scholar] [CrossRef]
- Blumenschein, G.R.; Devarakonda, S.; Johnson, M.; Moreno, V.; Gainor, J.; Edelman, M.J.; Heymach, J.V.; Govindan, R.; Bachier, C.; de Spéville, B.D.; et al. Phase I clinical trial evaluating the safety and efficacy of ADP-A2M10 SPEAR T cells in patients with MAGE-A10(+) advanced non-small cell lung cancer. J. Immunother. Cancer 2022, 10, e003581. [Google Scholar] [CrossRef]
- Kebriaei, P.; Klebanoff, C.A.; Creelan, B.C.; Hong, D.S.; Blumenschein, G.R.; Drakaki, A.; Tewari, A.; Thomrongsith, L.; Jiang, Y.; Jain, R.K. A phase 1 multicenter study evaluating the safety and efficacy of MHC class II-restricted MAGE-A3/A6 T-cell receptor engineered T cells (KITE-718) in patients with advanced cancers. J. Clin. Oncol. 2018, 36, TPS3104. [Google Scholar] [CrossRef]
- Blumenschein, G.R.; Bourgogne, A.; Reinhardt, C.; Ma, H.; Walter, S.; Weinschenk, T.; Kebriaei, P.; Hwu, P.; Singh, H. Phase I trial evaluating genetically modified autologous T cells (ACTengine IMA201) expressing a T-cell receptor recognizing a cancer/germline antigen in patients with squamous NSCLC or HNSCC. J. Clin. Oncol. 2018, 36, TPS78. [Google Scholar] [CrossRef]
- Wermke, M.; Tsimberidou, A.-M.; Mohamed, A.; Mayer-Mokler, A.; Satelli, A.; Reinhardt, C.; Araujo, D.; Maurer, D.; Blumenschein, G.J.; Singh, H.; et al. 959 Safety and anti-tumor activity of TCR-engineered autologous, PRAME-directed T cells across multiple advanced solid cancers at low doses–clinical update on the ACTengine® IMA203 trial. J. ImmunoTher. Cancer 2021, 9, A1009. [Google Scholar] [CrossRef]
- Chmielowski, B.; Ejadi, S.; Funke, R.; Stallings-Schmitt, T.; Denker, M.; Frohlich, M.W.; Franzusoff, A.J.; Abedi, M.; Cristea, M.C. A phase Ia/Ib, open-label first-in-human study of the safety, tolerability, and feasibility of gene-edited autologous NeoTCR-T cells (NeoTCR-P1) administered to patients with locally advanced or metastatic solid tumors. J. Clin. Oncol. 2020, 38, TPS3151. [Google Scholar] [CrossRef]
- Bishop, M.R.; Maziarz, R.T.; Waller, E.K.; Jäger, U.; Westin, J.R.; McGuirk, J.P.; Fleury, I.; Holte, H.; Borchmann, P.; Del Corral, C.; et al. Tisagenlecleucel in relapsed/refractory diffuse large B-cell lymphoma patients without measurable disease at infusion. Blood Adv. 2019, 3, 2230–2236. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Wallin, E.; von Heijne, G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998, 7, 1029–1038. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Osman, N.; Lucas, S.; Cantrell, D. The role of tyrosine phosphorylation in the interaction of cellular tyrosine kinases with the T cell receptor zeta chain tyrosine-based activation motif. Eur. J. Immunol. 1995, 25, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Hwu, P.; Shafer, G.E.; Treisman, J.; Schindler, D.G.; Gross, G.; Cowherd, R.; Rosenberg, S.A.; Eshhar, Z. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain. J. Exp. Med. 1993, 178, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Hwu, P.; Yang, J.C.; Cowherd, R.; Treisman, J.; Shafer, G.E.; Eshhar, Z.; Rosenberg, S.A. In vivo antitumor activity of T cells redirected with chimeric antibody/T-cell receptor genes. Cancer Res. 1995, 55, 3369–3373. [Google Scholar] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Camus, M.; Tosolini, M.; Mlecnik, B.; Pagès, F.; Kirilovsky, A.; Berger, A.; Costes, A.; Bindea, G.; Charoentong, P.; Bruneval, P.; et al. Coordination of intratumoral immune reaction and human colorectal cancer recurrence. Cancer Res. 2009, 69, 2685–2693. [Google Scholar] [CrossRef]
- Kather, J.N.; Suarez-Carmona, M.; Charoentong, P.; Weis, C.A.; Hirsch, D.; Bankhead, P.; Horning, M.; Ferber, D.; Kel, I.; Herpel, E.; et al. Topography of cancer-associated immune cells in human solid tumors. eLife 2018, 7, e36967. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, J.K.; Day, T.A.; Gillespie, M.B.; Rosenzweig, S.A.; Young, M.R. Secretion of vascular endothelial growth factor by oral squamous cell carcinoma cells skews endothelial cells to suppress T-cell functions. Hum. Immunol. 2009, 70, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef]
- Dirkx, A.E.; Egbrink, M.G.O.; Kuijpers, M.J.; van der Niet, S.T.; Heijnen, V.V.; Steege, J.C.B.-t.; Wagstaff, J.; Griffioen, A.W. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res. 2003, 63, 2322–2329. [Google Scholar]
- Bouzin, C.; Brouet, A.; De Vriese, J.; Dewever, J.; Feron, O. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: Identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J. Immunol. 2007, 178, 1505–1511. [Google Scholar] [CrossRef]
- Buckanovich, R.J.; Facciabene, A.; Kim, S.; Benencia, F.; Sasaroli, D.; Balint, K.; Katsaros, D.; O’Brien-Jenkins, A.; Gimotty, P.A.; Coukos, G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat. Med. 2008, 14, 28–36. [Google Scholar] [CrossRef]
- Zhang, J.; Endres, S.; Kobold, S. Enhancing tumor T cell infiltration to enable cancer immunotherapy. Immunotherapy 2019, 11, 201–213. [Google Scholar] [CrossRef]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef]
- Bourhis, M.; Palle, J.; Galy-Fauroux, I.; Terme, M. Direct and Indirect Modulation of T Cells by VEGF-A Counteracted by Anti-Angiogenic Treatment. Front. Immunol. 2021, 12, 616837. [Google Scholar] [CrossRef]
- Johansson, A.; Hamzah, J.; Payne, C.J.; Ganss, R. Tumor-targeted TNFα stabilizes tumor vessels and enhances active immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 7841–7846. [Google Scholar] [CrossRef]
- Johansson-Percival, A.; Li, Z.J.; Lakhiani, D.D.; He, B.; Wang, X.; Hamzah, J.; Ganss, R. Intratumoral LIGHT Restores Pericyte Contractile Properties and Vessel Integrity. Cell Rep. 2015, 13, 2687–2698. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Jabouille, A.; Steri, V.; Johansson-Percival, A.; Michael, I.P.; Kotamraju, V.R.; Junckerstorff, R.; Nowak, A.K.; Hamzah, J.; Lee, G.; et al. Vascular targeting of LIGHT normalizes blood vessels in primary brain cancer and induces intratumoural high endothelial venules. J. Pathol. 2018, 245, 209–221. [Google Scholar] [CrossRef]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9, eaak9679. [Google Scholar] [CrossRef]
- Shigeta, K.; Datta, M.; Hato, T.; Kitahara, S.; Chen, I.X.; Matsui, A.; Kikuchi, H.; Mamessier, E.; Aoki, S.; Ramjiawan, R.R.; et al. Dual Programmed Death Receptor-1 and Vascular Endothelial Growth Factor Receptor-2 Blockade Promotes Vascular Normalization and Enhances Antitumor Immune Responses in Hepatocellular Carcinoma. Hepatology 2020, 71, 1247–1261. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Y.; Jia, W.; Deng, H.; Li, G.; Deng, W.; Chen, J.; Kim, B.Y.S.; Jiang, W.; Liu, Q.; et al. Low-Dose Anti-Angiogenic Therapy Sensitizes Breast Cancer to PD-1 Blockade. Clin. Cancer Res. 2020, 26, 1712–1724. [Google Scholar] [CrossRef]
- Hack, S.P.; Spahn, J.; Chen, M.; Cheng, A.L.; Kaseb, A.; Kudo, M.; Lee, H.C.; Yopp, A.; Chow, P.; Qin, S. IMbrave 050: A Phase III trial of atezolizumab plus bevacizumab in high-risk hepatocellular carcinoma after curative resection or ablation. Future Oncol. 2020, 16, 975–989. [Google Scholar] [CrossRef]
- Wilky, B.A.; Trucco, M.M.; Subhawong, T.K.; Florou, V.; Park, W.; Kwon, D.; Wieder, E.D.; Kolonias, D.; Rosenberg, A.E.; Kerr, D.A.; et al. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: A single-centre, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 837–848. [Google Scholar] [CrossRef]
- Ganss, R.; Ryschich, E.; Klar, E.; Arnold, B.; Hämmerling, G.J. Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. 2002, 62, 1462–1470. [Google Scholar]
- Makarevic, A.; Rapp, C.; Dettling, S.; Reuss, D.; Jungk, C.; Abdollahi, A.; von Deimling, A.; Unterberg, A.; Herold-Mende, C.; Warta, R. Increased Radiation-Associated T-Cell Infiltration in Recurrent IDH-Mutant Glioma. Int. J. Mol. Sci. 2020, 21, 7801. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.Z.; Zhu, Y.Y.; Ruan, M.; Chen, L.; Zhang, Q.Y. Local Irradiation Sensitized Tumors to Adoptive T Cell Therapy via Enhancing the Cross-Priming, Homing, and Cytotoxicity of Antigen-Specific CD8 T Cells. Front. Immunol. 2019, 10, 2857. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Ilovitsh, T.; Feng, Y.; Foiret, J.; Kheirolomoom, A.; Zhang, H.; Ingham, E.S.; Ilovitsh, A.; Tumbale, S.K.; Fite, B.Z.; Wu, B.; et al. Low-frequency ultrasound-mediated cytokine transfection enhances T cell recruitment at local and distant tumor sites. Proc. Natl. Acad. Sci. USA 2020, 117, 12674–12685. [Google Scholar] [CrossRef]
- Zhang, N.; Song, J.; Liu, Y.; Liu, M.; Zhang, L.; Sheng, D.; Deng, L.; Yi, H.; Wu, M.; Zheng, Y.; et al. Photothermal therapy mediated by phase-transformation nanoparticles facilitates delivery of anti-PD1 antibody and synergizes with antitumor immunotherapy for melanoma. J. Control Release 2019, 306, 15–28. [Google Scholar] [CrossRef]
- Qi, X.; Yang, M.; Ma, L.; Sauer, M.; Avella, D.; Kaifi, J.T.; Bryan, J.; Cheng, K.; Staveley-O’Carroll, K.F.; Kimchi, E.T.; et al. Synergizing sunitinib and radiofrequency ablation to treat hepatocellular cancer by triggering the antitumor immune response. J. Immunother. Cancer 2020, 8, e001038. [Google Scholar] [CrossRef]
- Pichler, R.; Fritz, J.; Zavadil, C.; Schäfer, G.; Culig, Z.; Brunner, A. Tumor-infiltrating immune cell subpopulations influence the oncologic outcome after intravesical Bacillus Calmette-Guérin therapy in bladder cancer. Oncotarget 2016, 7, 39916–39930. [Google Scholar] [CrossRef]
- Boccafoschi, C.; Montefiore, F.; Pavesi, M.; Pastormerlo, M.; Annoscia, S.; Lozzi, C.; Betta, P.G. Immunophenotypic characterization of the bladder mucosa infiltrating lymphocytes after intravesical BCG treatment for superficial bladder carcinoma. Eur. Urol. 1992, 21, 304–308. [Google Scholar] [CrossRef]
- Srivastava, S.; Furlan, S.N.; Jaeger-Ruckstuhl, C.A.; Sarvothama, M.; Berger, C.; Smythe, K.S.; Garrison, S.M.; Specht, J.M.; Lee, S.M.; Amezquita, R.A.; et al. Immunogenic Chemotherapy Enhances Recruitment of CAR-T Cells to Lung Tumors and Improves Antitumor Efficacy when Combined with Checkpoint Blockade. Cancer Cell 2021, 39, 193–208.e110. [Google Scholar] [CrossRef]
- Chang, L.; Zhang, Z.; Chen, F.; Zhang, W.; Song, S.; Song, S. Irradiation enhances dendritic cell potential antitumor activity by inducing tumor cell expressing TNF-α. Med. Oncol. 2017, 34, 44. [Google Scholar] [CrossRef]
- Chen, H.Y.; Xu, L.; Li, L.F.; Liu, X.X.; Gao, J.X.; Bai, Y.R. Inhibiting the CD8(+) T cell infiltration in the tumor microenvironment after radiotherapy is an important mechanism of radioresistance. Sci. Rep. 2018, 8, 11934. [Google Scholar] [CrossRef]
- Bian, Z.; Shi, L.; Kidder, K.; Zen, K.; Garnett-Benson, C.; Liu, Y. Intratumoral SIRPα-deficient macrophages activate tumor antigen-specific cytotoxic T cells under radiotherapy. Nat. Commun. 2021, 12, 3229. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Li, W.; Lu, L.; Lu, J.; Wang, X.; Yang, C.; Jin, J.; Wu, L.; Hong, X.; Li, F.; Cao, D.; et al. cGAS-STING-mediated DNA sensing maintains CD8(+) T cell stemness and promotes antitumor T cell therapy. Sci. Transl. Med. 2020, 12, eaay9013. [Google Scholar] [CrossRef]
- Weiss, J.M.; Guérin, M.V.; Regnier, F.; Renault, G.; Galy-Fauroux, I.; Vimeux, L.; Feuillet, V.; Peranzoni, E.; Thoreau, M.; Trautmann, A.; et al. The STING agonist DMXAA triggers a cooperation between T lymphocytes and myeloid cells that leads to tumor regression. Oncoimmunology 2017, 6, e1346765. [Google Scholar] [CrossRef]
- Xu, N.; Palmer, D.C.; Robeson, A.C.; Shou, P.; Bommiasamy, H.; Laurie, S.J.; Willis, C.; Dotti, G.; Vincent, B.G.; Restifo, N.P.; et al. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J. Exp. Med. 2021, 218, e20200844. [Google Scholar] [CrossRef]
- Zaidi, A.H.; Kelly, R.J.; Gorbunova, A.; Omstead, A.N.; Salvitti, M.S.; Zheng, P.; Kosovec, J.E.; Lee, S.; Ayazi, S.; Babar, L.; et al. Intratumoral immunotherapy with STING agonist, ADU-S100, induces CD8+ T-cell mediated anti-tumor immunity in an esophageal adenocarcinoma model. Oncotarget 2021, 12, 292–303. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Sweis, R.F.; Hodi, F.S.; Messersmith, W.A.; Andtbacka, R.H.I.; Ingham, M.; Lewis, N.; Chen, X.; Pelletier, M.; Chen, X.; et al. Phase I Dose-Escalation Trial of MIW815 (ADU-S100), an Intratumoral STING Agonist, in Patients with Advanced/Metastatic Solid Tumors or Lymphomas. Clin. Cancer Res. 2022, 28, 677–688. [Google Scholar] [CrossRef]
- Flood, B.A.; Higgs, E.F.; Li, S.; Luke, J.J.; Gajewski, T.F. STING pathway agonism as a cancer therapeutic. Immunol. Rev. 2019, 290, 24–38. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef]
- Chen, H.Y.; Xie, H.Y.; Liu, X.X.; Li, L.F.; Bai, Y.R.; Gao, J.X. Splenic irradiation combined with tumor irradiation promotes T cell infiltration in the tumor microenvironment and helps in tumor control. Biochem. Biophys. Res. Commun. 2019, 510, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Bule, P.; Aguiar, S.I.; Aires-Da-Silva, F.; Dias, J.N.R. Chemokine-Directed Tumor Microenvironment Modulation in Cancer Immunotherapy. Int. J. Mol. Sci. 2021, 22, 9804. [Google Scholar] [CrossRef] [PubMed]
- Dangaj, D.; Bruand, M.; Grimm, A.J.; Ronet, C.; Barras, D.; Duttagupta, P.A.; Lanitis, E.; Duraiswamy, J.; Tanyi, J.L.; Benencia, F.; et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019, 35, 885–900.10. [Google Scholar] [CrossRef] [PubMed]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [Green Version]
- Lesch, S.; Blumenberg, V.; Stoiber, S.; Gottschlich, A.; Ogonek, J.; Cadilha, B.L.; Dantes, Z.; Rataj, F.; Dorman, K.; Lutz, J.; et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat. Biomed. Eng. 2021, 5, 1246–1260. [Google Scholar] [CrossRef]
- Whilding, L.M.; Halim, L.; Draper, B.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Maher, J. CAR T-Cells Targeting the Integrin αvβ6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674. [Google Scholar] [CrossRef]
- Idorn, M.; Skadborg, S.K.; Kellermann, L.; Halldórsdóttir, H.R.; Olofsson, G.; Met, Ö.; Straten, P.T. Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model. Oncoimmunology 2018, 7, e1450715. [Google Scholar] [CrossRef]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.; Kapoor, V.; Predina, J.; Powell, D.J., Jr.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef]
- Cadilha, B.L.; Benmebarek, M.R.; Dorman, K.; Oner, A.; Lorenzini, T.; Obeck, H.; Vänttinen, M.; Di Pilato, M.; Pruessmann, J.N.; Stoiber, S.; et al. Combined tumor-directed recruitment and protection from immune suppression enable CAR T cell efficacy in solid tumors. Sci. Adv. 2021, 7, eabi5781. [Google Scholar] [CrossRef]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, S.; Chen, X.; Cheng, S.; Zhang, Z.; Li, F.; Huang, L.; Yang, Y.; Zhou, B.; Yue, D.; et al. Cancer-cell-secreted CXCL11 promoted CD8(+) T cells infiltration through docetaxel-induced-release of HMGB1 in NSCLC. J. Immunother. Cancer 2019, 7, 42. [Google Scholar] [CrossRef]
- Pegram, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Curran, K.J.; Giralt, S.A.; Barker, J.N.; Brentjens, R.J. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia 2015, 29, 415–422. [Google Scholar] [CrossRef]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Di, S.; Shi, B.; Zhang, H.; Wang, Y.; Wu, X.; Luo, H.; Wang, H.; Li, Z.; Jiang, H. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3-Targeted Chimeric Antigen Receptor-Engineered T Cells in Hepatocellular Carcinoma. J. Immunol. 2019, 203, 198–207. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, C.; Landoni, E.; Metelitsa, L.; Dotti, G.; Savoldo, B. Eradication of Neuroblastoma by T Cells Redirected with an Optimized GD2-Specific Chimeric Antigen Receptor and Interleukin-15. Clin. Cancer Res. 2019, 25, 2915–2924. [Google Scholar] [CrossRef]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef]
- Golumba-Nagy, V.; Kuehle, J.; Hombach, A.A.; Abken, H. CD28-ζ CAR T Cells Resist TGF-β Repression through IL-2 Signaling, Which Can Be Mimicked by an Engineered IL-7 Autocrine Loop. Mol. Ther. 2018, 26, 2218–2230. [Google Scholar] [CrossRef]
- Brand, T.; MacLellan, W.R.; Schneider, M.D. A dominant-negative receptor for type beta transforming growth factors created by deletion of the kinase domain. J. Biol. Chem. 1993, 268, 11500–11503. [Google Scholar] [CrossRef]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef]
- Curran, K.J.; Seinstra, B.A.; Nikhamin, Y.; Yeh, R.; Usachenko, Y.; van Leeuwen, D.G.; Purdon, T.; Pegram, H.J.; Brentjens, R.J. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol. Ther. 2015, 23, 769–778. [Google Scholar] [CrossRef]
- Pang, N.; Shi, J.; Qin, L.; Chen, A.; Tang, Y.; Yang, H.; Huang, Y.; Wu, Q.; Li, X.; He, B.; et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J. Hematol. Oncol. 2021, 14, 118. [Google Scholar] [CrossRef]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef]
- Luo, H.; Su, J.; Sun, R.; Sun, Y.; Wang, Y.; Dong, Y.; Shi, B.; Jiang, H.; Li, Z. Coexpression of IL7 and CCL21 Increases Efficacy of CAR-T Cells in Solid Tumors without Requiring Preconditioned Lymphodepletion. Clin. Cancer Res. 2020, 26, 5494–5505. [Google Scholar] [CrossRef]
- Garber, K. Driving T-cell immunotherapy to solid tumors. Nat. Biotechnol. 2018, 36, 215–219. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Chen, Y.T.; Scanlan, M.J.; Sahin, U.; Türeci, O.; Gure, A.O.; Tsang, S.; Williamson, B.; Stockert, E.; Pfreundschuh, M.; Old, L.J. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc. Natl. Acad. Sci. USA 1997, 94, 1914–1918. [Google Scholar] [CrossRef]
- Aung, P.P.; Liu, Y.C.; Ballester, L.Y.; Robbins, P.F.; Rosenberg, S.A.; Lee, C.C. Expression of New York esophageal squamous cell carcinoma-1 in primary and metastatic melanoma. Hum. Pathol. 2014, 45, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.P.; Robbins, P.F.; Raffeld, M.; Aung, P.P.; Tsokos, M.; Rosenberg, S.A.; Miettinen, M.M.; Lee, C.C. NY-ESO-1 expression in synovial sarcoma and other mesenchymal tumors: Significance for NY-ESO-1-based targeted therapy and differential diagnosis. Mod. Pathol. 2012, 25, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Gnjatic, S.; Jungbluth, A.A.; Williamson, B.; Herr, H.; Stockert, E.; Dalbagni, G.; Donat, S.M.; Reuter, V.E.; Santiago, D.; et al. Frequency of NY-ESO-1 and LAGE-1 expression in bladder cancer and evidence of a new NY-ESO-1 T-cell epitope in a patient with bladder cancer. Cancer Immun. 2003, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.L.; Jungbluth, A.A.; Patel, S.G.; Iversen, K.; Hoshaw-Woodard, S.; Busam, K.J. Expression and significance of cancer testis antigens in primary mucosal melanoma of the head and neck. Head Neck 2004, 26, 1053–1057. [Google Scholar] [CrossRef]
- Odunsi, K.; Jungbluth, A.A.; Stockert, E.; Qian, F.; Gnjatic, S.; Tammela, J.; Intengan, M.; Beck, A.; Keitz, B.; Santiago, D.; et al. NY-ESO-1 and LAGE-1 cancer-testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer Res. 2003, 63, 6076–6083. [Google Scholar]
- Oba-Shinjo, S.M.; Caballero, O.L.; Jungbluth, A.A.; Rosemberg, S.; Old, L.J.; Simpson, A.J.; Marie, S.K. Cancer-testis (CT) antigen expression in medulloblastoma. Cancer Immun. 2008, 8, 7. [Google Scholar]
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018, 9, 947. [Google Scholar] [CrossRef]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Worning, P.; Lauemøller, S.L.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003, 12, 1007–1017. [Google Scholar] [CrossRef]
- Caron, E.; Kowalewski, D.J.; Koh, C.C.; Sturm, T.; Schuster, H.; Aebersold, R. Analysis of Major Histocompatibility Complex (MHC) Immunopeptidomes Using Mass Spectrometry. Mol. Cell. Proteom. 2015, 14, 3105–3117. [Google Scholar] [CrossRef]
- Stopfer, L.E.; Mesfin, J.M.; Joughin, B.A.; Lauffenburger, D.A.; White, F.M. Multiplexed relative and absolute quantitative immunopeptidomics reveals MHC I repertoire alterations induced by CDK4/6 inhibition. Nat. Commun. 2020, 11, 2760. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef]
- Losic, B.; Craig, A.J.; Villacorta-Martin, C.; Martins-Filho, S.N.; Akers, N.; Chen, X.; Ahsen, M.E.; von Felden, J.; Labgaa, I.; D’Avola, D.; et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 2020, 11, 291. [Google Scholar] [CrossRef]
- Jiménez-Sánchez, A.; Memon, D.; Pourpe, S.; Veeraraghavan, H.; Li, Y.; Vargas, H.A.; Gill, M.B.; Park, K.J.; Zivanovic, O.; Konner, J.; et al. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell 2017, 170, 927–938.e20. [Google Scholar] [CrossRef]
- Zhang, A.W.; McPherson, A.; Milne, K.; Kroeger, D.R.; Hamilton, P.T.; Miranda, A.; Funnell, T.; Little, N.; de Souza, C.P.E.; Laan, S.; et al. Interfaces of Malignant and Immunologic Clonal Dynamics in Ovarian Cancer. Cell 2018, 173, 1755–1769.e22. [Google Scholar] [CrossRef]
- Zhu, Y.; Meng, X.; Ruan, X.; Lu, X.; Yan, F.; Wang, F. Characterization of Neoantigen Load Subgroups in Gynecologic and Breast Cancers. Front. Bioeng. Biotechnol. 2020, 8, 702. [Google Scholar] [CrossRef]
- Chandran, S.S.; Ma, J.; Klatt, M.G.; Dündar, F.; Bandlamudi, C.; Razavi, P.; Wen, H.Y.; Weigelt, B.; Zumbo, P.; Fu, S.N.; et al. Immunogenicity and therapeutic targeting of a public neoantigen derived from mutated PIK3CA. Nat. Med. 2022, 28, 946–957. [Google Scholar] [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e1211. [Google Scholar] [CrossRef]
- Montesion, M.; Murugesan, K.; Jin, D.X.; Sharaf, R.; Sanchez, N.; Guria, A.; Minker, M.; Li, G.; Fisher, V.; Sokol, E.S.; et al. Somatic HLA Class I Loss Is a Widespread Mechanism of Immune Evasion Which Refines the Use of Tumor Mutational Burden as a Biomarker of Checkpoint Inhibitor Response. Cancer Discov. 2021, 11, 282–292. [Google Scholar] [CrossRef]
- Paulson, K.G.; Voillet, V.; McAfee, M.S.; Hunter, D.S.; Wagener, F.D.; Perdicchio, M.; Valente, W.J.; Koelle, S.J.; Church, C.D.; Vandeven, N.; et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat. Commun. 2018, 9, 3868. [Google Scholar] [CrossRef]
- Fangazio, M.; Ladewig, E.; Gomez, K.; Garcia-Ibanez, L.; Kumar, R.; Teruya-Feldstein, J.; Rossi, D.; Filip, I.; Pan-Hammarström, Q.; Inghirami, G.; et al. Genetic mechanisms of HLA-I loss and immune escape in diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2021, 118, e2104504118. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Cabrera, T.; Aptsiauri, N. “Hard” and "soft" lesions underlying the HLA class I alterations in cancer cells: Implications for immunotherapy. Int. J. Cancer 2010, 127, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, A.B.; Carretero, J.; Muñoz, J.A.; Zinchenko, S.; Ruiz-Cabello, F.; González-Aseguinolaza, G.; Garrido, F.; Aptsiauri, N. Adenovirus expressing β2-microglobulin recovers HLA class I expression and antitumor immunity by increasing T-cell recognition. Cancer Gene Ther. 2014, 21, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Ljunggren, H.G.; Kärre, K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef]
- Watson, N.F.; Ramage, J.M.; Madjd, Z.; Spendlove, I.; Ellis, I.O.; Scholefield, J.H.; Durrant, L.G. Immunosurveillance is active in colorectal cancer as downregulation but not complete loss of MHC class I expression correlates with a poor prognosis. Int. J. Cancer 2006, 118, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Méndez, R.; Rodríguez, T.; Del Campo, A.; Monge, E.; Maleno, I.; Aptsiauri, N.; Jiménez, P.; Pedrinaci, S.; Pawelec, G.; Ruiz-Cabello, F.; et al. Characterization of HLA class I altered phenotypes in a panel of human melanoma cell lines. Cancer Immunol. Immunother. 2008, 57, 719–729. [Google Scholar] [CrossRef]
- Pfizenmaier, K.; Scheurich, P.; Schlüter, C.; Krönke, M. Tumor necrosis factor enhances HLA-A,B,C and HLA-DR gene expression in human tumor cells. J. Immunol. 1987, 138, 975–980. [Google Scholar]
- Propper, D.J.; Chao, D.; Braybrooke, J.P.; Bahl, P.; Thavasu, P.; Balkwill, F.; Turley, H.; Dobbs, N.; Gatter, K.; Talbot, D.C.; et al. Low-dose IFN-gamma induces tumor MHC expression in metastatic malignant melanoma. Clin. Cancer Res. 2003, 9, 84–92. [Google Scholar]
- Kidman, J.; Principe, N.; Watson, M.; Lassmann, T.; Holt, R.A.; Nowak, A.K.; Lesterhuis, W.J.; Lake, R.A.; Chee, J. Characteristics of TCR Repertoire Associated With Successful Immune Checkpoint Therapy Responses. Front. Immunol. 2020, 11, 587014. [Google Scholar] [CrossRef]
- Roth, A.; Khattra, J.; Yap, D.; Wan, A.; Laks, E.; Biele, J.; Ha, G.; Aparicio, S.; Bouchard-Côté, A.; Shah, S.P. PyClone: Statistical inference of clonal population structure in cancer. Nat. Methods 2014, 11, 396–398. [Google Scholar] [CrossRef]
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; Shamalov, K.; Trebska-McGowan, K.; Bliskovsky, V.V.; Parkhurst, M.R.; Ankri, C.; Prickett, T.D.; Crystal, J.S.; et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J. Clin. Investig. 2015, 125, 3981–3991. [Google Scholar] [CrossRef]
- Paria, B.C.; Levin, N.; Lowery, F.J.; Pasetto, A.; Deniger, D.C.; Parkhurst, M.R.; Yossef, R.; Kim, S.P.; Florentin, M.; Ngo, L.T.; et al. Rapid Identification and Evaluation of Neoantigen-reactive T-Cell Receptors From Single Cells. J. Immunother. 2021, 44, 1–8. [Google Scholar] [CrossRef]
- Li, Y.; Moysey, R.; Molloy, P.E.; Vuidepot, A.L.; Mahon, T.; Baston, E.; Dunn, S.; Liddy, N.; Jacob, J.; Jakobsen, B.K.; et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 2005, 23, 349–354. [Google Scholar] [CrossRef]
- Robbins, P.F.; Li, Y.F.; El-Gamil, M.; Zhao, Y.; Wargo, J.A.; Zheng, Z.; Xu, H.; Morgan, R.A.; Feldman, S.A.; Johnson, L.A.; et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J. Immunol. 2008, 180, 6116–6131. [Google Scholar] [CrossRef]
- Zeh, H.J., 3rd; Perry-Lalley, D.; Dudley, M.E.; Rosenberg, S.A.; Yang, J.C. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J. Immunol. 1999, 162, 989–994. [Google Scholar]
- Lövgren, T.; Baumgaertner, P.; Wieckowski, S.; Devêvre, E.; Guillaume, P.; Luescher, I.; Rufer, N.; Speiser, D.E. Enhanced cytotoxicity and decreased CD8 dependence of human cancer-specific cytotoxic T lymphocytes after vaccination with low peptide dose. Cancer Immunol. Immunother. 2012, 61, 817–826. [Google Scholar] [CrossRef]
- Aranda, F.; Llopiz, D.; Díaz-Valdés, N.; Riezu-Boj, J.I.; Bezunartea, J.; Ruiz, M.; Martínez, M.; Durantez, M.; Mansilla, C.; Prieto, J.; et al. Adjuvant combination and antigen targeting as a strategy to induce polyfunctional and high-avidity T-cell responses against poorly immunogenic tumors. Cancer Res. 2011, 71, 3214–3224. [Google Scholar] [CrossRef]
- Derby, M.; Alexander-Miller, M.; Tse, R.; Berzofsky, J. High-avidity CTL exploit two complementary mechanisms to provide better protection against viral infection than low-avidity CTL. J. Immunol. 2001, 166, 1690–1697. [Google Scholar] [CrossRef]
- Sim, M.J.W.; Lu, J.; Spencer, M.; Hopkins, F.; Tran, E.; Rosenberg, S.A.; Long, E.O.; Sun, P.D. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc. Natl. Acad. Sci. USA 2020, 117, 12826–12835. [Google Scholar] [CrossRef]
- Bräunlein, E.; Lupoli, G.; Füchsl, F.; Abualrous, E.T.; de Andrade Krätzig, N.; Gosmann, D.; Wietbrock, L.; Lange, S.; Engleitner, T.; Lan, H.; et al. Functional analysis of peripheral and intratumoral neoantigen-specific TCRs identified in a patient with melanoma. J. ImmunoTherapy Cancer 2021, 9, e002754. [Google Scholar] [CrossRef]
- Presotto, D.; Erdes, E.; Duong, M.N.; Allard, M.; Regamey, P.O.; Quadroni, M.; Doucey, M.A.; Rufer, N.; Hebeisen, M. Fine-Tuning of Optimal TCR Signaling in Tumor-Redirected CD8 T Cells by Distinct TCR Affinity-Mediated Mechanisms. Front. Immunol. 2017, 8, 1564. [Google Scholar] [CrossRef]
- Schmid, D.A.; Irving, M.B.; Posevitz, V.; Hebeisen, M.; Posevitz-Fejfar, A.; Sarria, J.C.; Gomez-Eerland, R.; Thome, M.; Schumacher, T.N.; Romero, P.; et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J. Immunol. 2010, 184, 4936–4946. [Google Scholar] [CrossRef]
- Slansky, J.E.; Jordan, K.R. The Goldilocks model for TCR-too much attraction might not be best for vaccine design. PLoS Biol. 2010, 8, e1000482. [Google Scholar] [CrossRef]
- Shakiba, M.; Zumbo, P.; Espinosa-Carrasco, G.; Menocal, L.; Dündar, F.; Carson, S.E.; Bruno, E.M.; Sanchez-Rivera, F.J.; Lowe, S.W.; Camara, S.; et al. TCR signal strength defines distinct mechanisms of T cell dysfunction and cancer evasion. J. Exp. Med. 2022, 219, e20201966. [Google Scholar] [CrossRef] [PubMed]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef]
- Hoogi, S.; Eisenberg, V.; Mayer, S.; Shamul, A.; Barliya, T.; Cohen, C.J. A TIGIT-based chimeric co-stimulatory switch receptor improves T-cell anti-tumor function. J. Immunother. Cancer 2019, 7, 243. [Google Scholar] [CrossRef]
- Olguín-Contreras, L.F.; Mendler, A.N.; Popowicz, G.; Hu, B.; Noessner, E. Double Strike Approach for Tumor Attack: Engineering T Cells Using a CD40L:CD28 Chimeric Co-Stimulatory Switch Protein for Enhanced Tumor Targeting in Adoptive Cell Therapy. Front. Immunol. 2021, 12, 750478. [Google Scholar] [CrossRef]
- Oda, S.K.; Daman, A.W.; Garcia, N.M.; Wagener, F.; Schmitt, T.M.; Tan, X.; Chapuis, A.G.; Greenberg, P.D. A CD200R-CD28 fusion protein appropriates an inhibitory signal to enhance T-cell function and therapy of murine leukemia. Blood 2017, 130, 2410–2419. [Google Scholar] [CrossRef]
- Omer, B.; Cardenas, M.G.; Pfeiffer, T.; Daum, R.; Huynh, M.; Sharma, S.; Nouraee, N.; Xie, C.; Tat, C.; Perconti, S.; et al. A Co-stimulatory CAR Improves TCR-based Cancer Immunotherapy. Cancer Immunol. Res. 2022, 10, 512–524. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A.; Simpson, T.R.; Peggs, K.S.; Merghoub, T.; Vider, J.; Fan, X.; Blasberg, R.; Yagita, H.; Muranski, P.; Antony, P.A.; et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 2010, 207, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy—new insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Kwek, S.S.; Raju, S.S.; Li, T.; McCarthy, E.; Chow, E.; Aran, D.; Ilano, A.; Pai, C.S.; Rancan, C.; et al. Intratumoral CD4(+) T Cells Mediate Anti-tumor Cytotoxicity in Human Bladder Cancer. Cell 2020, 181, 1612–1625.e13. [Google Scholar] [CrossRef]
- Muranski, P.; Boni, A.; Antony, P.A.; Cassard, L.; Irvine, K.R.; Kaiser, A.; Paulos, C.M.; Palmer, D.C.; Touloukian, C.E.; Ptak, K.; et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 2008, 112, 362–373. [Google Scholar] [CrossRef]
- Cachot, A.; Bilous, M.; Liu, Y.C.; Li, X.; Saillard, M.; Cenerenti, M.; Rockinger, G.A.; Wyss, T.; Guillaume, P.; Schmidt, J.; et al. Tumor-specific cytolytic CD4 T cells mediate immunity against human cancer. Sci. Adv. 2021, 7, eabe3348. [Google Scholar] [CrossRef]
- Laugel, B.; van den Berg, H.A.; Gostick, E.; Cole, D.K.; Wooldridge, L.; Boulter, J.; Milicic, A.; Price, D.A.; Sewell, A.K. Different T cell receptor affinity thresholds and CD8 coreceptor dependence govern cytotoxic T lymphocyte activation and tetramer binding properties. J. Biol. Chem. 2007, 282, 23799–23810. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [Green Version]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef]
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Coyle, S.M.; Thomson, M.; Lim, W.A. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 2016, 164, 780–791. [Google Scholar] [CrossRef]
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378. [Google Scholar] [CrossRef]
- Salter, A.I.; Rajan, A.; Kennedy, J.J.; Ivey, R.G.; Shelby, S.A.; Leung, I.; Templeton, M.L.; Muhunthan, V.; Voillet, V.; Sommermeyer, D.; et al. Comparative analysis of TCR and CAR signaling informs CAR designs with superior antigen sensitivity and in vivo function. Sci. Signal. 2021, 14, eabe2606. [Google Scholar] [CrossRef]
- Helsen, C.W.; Hammill, J.A.; Lau, V.W.C.; Mwawasi, K.A.; Afsahi, A.; Bezverbnaya, K.; Newhook, L.; Hayes, D.L.; Aarts, C.; Bojovic, B.; et al. The chimeric TAC receptor co-opts the T cell receptor yielding robust anti-tumor activity without toxicity. Nat. Commun. 2018, 9, 3049. [Google Scholar] [CrossRef]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Amatya, C.; Pegues, M.A.; Lam, N.; Vanasse, D.; Geldres, C.; Choi, S.; Hewitt, S.M.; Feldman, S.A.; Kochenderfer, J.N. Development of CAR T Cells Expressing a Suicide Gene Plus a Chimeric Antigen Receptor Targeting Signaling Lymphocytic-Activation Molecule F7. Mol. Ther. 2021, 29, 702–717. [Google Scholar] [CrossRef]
- Jan, M.; Scarfò, I.; Larson, R.C.; Walker, A.; Schmidts, A.; Guirguis, A.A.; Gasser, J.A.; Słabicki, M.; Bouffard, A.A.; Castano, A.P.; et al. Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci. Transl. Med. 2021, 13, eabb6295. [Google Scholar] [CrossRef]
- Drent, E.; Poels, R.; Mulders, M.J.; van de Donk, N.; Themeli, M.; Lokhorst, H.M.; Mutis, T. Feasibility of controlling CD38-CAR T cell activity with a Tet-on inducible CAR design. PLoS ONE 2018, 13, e0197349. [Google Scholar] [CrossRef]
- Bachmann, M. The UniCAR system: A modular CAR T cell approach to improve the safety of CAR T cells. Immunol. Lett. 2019, 211, 13–22. [Google Scholar] [CrossRef]
- Kittel-Boselli, E.; Soto, K.E.G.; Loureiro, L.R.; Hoffmann, A.; Bergmann, R.; Arndt, C.; Koristka, S.; Mitwasi, N.; Kegler, A.; Bartsch, T.; et al. Targeting Acute Myeloid Leukemia Using the RevCAR Platform: A Programmable, Switchable and Combinatorial Strategy. Cancers 2021, 13, 4785. [Google Scholar] [CrossRef]
- Salzer, B.; Schueller, C.M.; Zajc, C.U.; Peters, T.; Schoeber, M.A.; Kovacic, B.; Buri, M.C.; Lobner, E.; Dushek, O.; Huppa, J.B.; et al. Engineering AvidCARs for combinatorial antigen recognition and reversible control of CAR function. Nat. Commun. 2020, 11, 4166. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Dehbashi, M.; Hojati, Z.; Motovali-Bashi, M.; Ganjalikhany, M.R.; Cho, W.C.; Shimosaka, A.; Navabi, P.; Ganjalikhani-Hakemi, M. A Novel CAR Expressing NK Cell Targeting CD25 With the Prospect of Overcoming Immune Escape Mechanism in Cancers. Front. Oncol. 2021, 11, 649710. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, e133977. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siska, P.J.; Beckermann, K.E.; Mason, F.M.; Andrejeva, G.; Greenplate, A.R.; Sendor, A.B.; Chiang, Y.J.; Corona, A.L.; Gemta, L.F.; Vincent, B.G.; et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2017, 2, 2379–3708. [Google Scholar] [CrossRef] [PubMed]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013.e20. [Google Scholar] [CrossRef]
- Beltra, J.C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825–841.e828. [Google Scholar] [CrossRef]
- Connolly, K.A.; Kuchroo, M.; Venkat, A.; Khatun, A.; Wang, J.; William, I.; Hornick, N.I.; Fitzgerald, B.L.; Damo, M.; Kasmani, M.Y.; et al. A reservoir of stem-like CD8(+) T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci. Immunol. 2021, 6, eabg7836. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Busch, D.H.; Fräßle, S.P.; Sommermeyer, D.; Buchholz, V.R.; Riddell, S.R. Role of memory T cell subsets for adoptive immunotherapy. Semin. Immunol. 2016, 28, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Hinrichs, C.S.; Borman, Z.A.; Cassard, L.; Gattinoni, L.; Spolski, R.; Yu, Z.; Sanchez-Perez, L.; Muranski, P.; Kern, S.J.; Logun, C.; et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 17469–17474. [Google Scholar] [CrossRef]
- Petersen, C.T.; Hassan, M.; Morris, A.B.; Jeffery, J.; Lee, K.; Jagirdar, N.; Staton, A.D.; Raikar, S.S.; Spencer, H.T.; Sulchek, T.; et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kδ inhibitors and VIP antagonists. Blood Adv. 2018, 2, 210–223. [Google Scholar] [CrossRef]
- Akbar, A.N.; Henson, S.M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 2011, 11, 289–295. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Motzer, R.J.; Rini, B.I.; Haanen, J.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Gravis-Mescam, G.; Uemura, M.; Lee, J.L.; et al. Updated efficacy results from the JAVELIN Renal 101 trial: First-line avelumab plus axitinib versus sunitinib in patients with advanced renal cell carcinoma. Ann. Oncol. 2020, 31, 1030–1039. [Google Scholar] [CrossRef]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andric, Z.; Geater, S.; et al. Atezolizumab for First-Line Treatment of PD-L1-Selected Patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Gao, J.; Ward, J.F.; Pettaway, C.A.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E.; et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555. [Google Scholar] [CrossRef]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef] [Green Version]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.J.; Powers, A.C.; Johnson, D.B. Endocrine toxicities of immune checkpoint inhibitors. Nat. Rev. Endocrinol. 2021, 17, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Tanoue, K.; Rosewell Shaw, A.; Watanabe, N.; Porter, C.; Rana, B.; Gottschalk, S.; Brenner, M.; Suzuki, M. Armed Oncolytic Adenovirus-Expressing PD-L1 Mini-Body Enhances Antitumor Effects of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2017, 77, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.E.; Rosewell Shaw, A.; Jung, Y.; Yip, T.; Castro, P.D.; Sandulache, V.C.; Sikora, A.; Gottschalk, S.; Ittman, M.M.; Brenner, M.K.; et al. Oncolytic Adenovirus Armed with BiTE, Cytokine, and Checkpoint Inhibitor Enables CAR T Cells to Control the Growth of Heterogeneous Tumors. Mol. Ther. 2020, 28, 1251–1262. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Zauderer, M.G.; Rusch, V.W.; O’Cearbhaill, R.; Zhu, A.; Ngai, D.; McGee, E.; Chintala, N.; Messinger, J.; Cheema, W.; et al. Regional delivery of mesothelin-targeted CAR T cells for pleural cancers: Safety and preliminary efficacy in combination with anti-PD-1 agent. J. Clin. Oncol. 2019, 37, 2511. [Google Scholar] [CrossRef]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef]
- Poorebrahim, M.; Melief, J.; Pico de Coaña, Y.; Wickström, S.L.; Cid-Arregui, A.; Kiessling, R. Counteracting CAR T cell dysfunction. Oncogene 2021, 40, 421–435. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Cheng, C.; Cheng, A.W.; Zhang, X.; Li, N.; Xia, C.; Wei, X.; Liu, X.; Wang, H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017, 27, 154–157. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, X.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 2017, 8, 17002–17011. [Google Scholar] [CrossRef]
- Odorizzi, P.M.; Pauken, K.E.; Paley, M.A.; Sharpe, A.; Wherry, E.J. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 2015, 212, 1125–1137. [Google Scholar] [CrossRef]
- Huard, B.; Tournier, M.; Hercend, T.; Triebel, F.; Faure, F. Lymphocyte-activation gene 3/major histocompatibility complex class II interaction modulates the antigenic response of CD4+ T lymphocytes. Eur. J. Immunol. 1994, 24, 3216–3221. [Google Scholar] [CrossRef]
- Okazaki, T.; Okazaki, I.M.; Wang, J.; Sugiura, D.; Nakaki, F.; Yoshida, T.; Kato, Y.; Fagarasan, S.; Muramatsu, M.; Eto, T.; et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J. Exp. Med. 2011, 208, 395–407. [Google Scholar] [CrossRef]
- Schietinger, A.; Philip, M.; Krisnawan, V.E.; Chiu, E.Y.; Delrow, J.J.; Basom, R.S.; Lauer, P.; Brockstedt, D.G.; Knoblaugh, S.E.; Hämmerling, G.J.; et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 2016, 45, 389–401. [Google Scholar] [CrossRef]
- Dafni, U.; Michielin, O.; Lluesma, S.M.; Tsourti, Z.; Polydoropoulou, V.; Karlis, D.; Besser, M.J.; Haanen, J.; Svane, I.M.; Ohashi, P.S.; et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: A systematic review and meta-analysis. Ann. Oncol. 2019, 30, 1902–1913. [Google Scholar] [CrossRef] [Green Version]


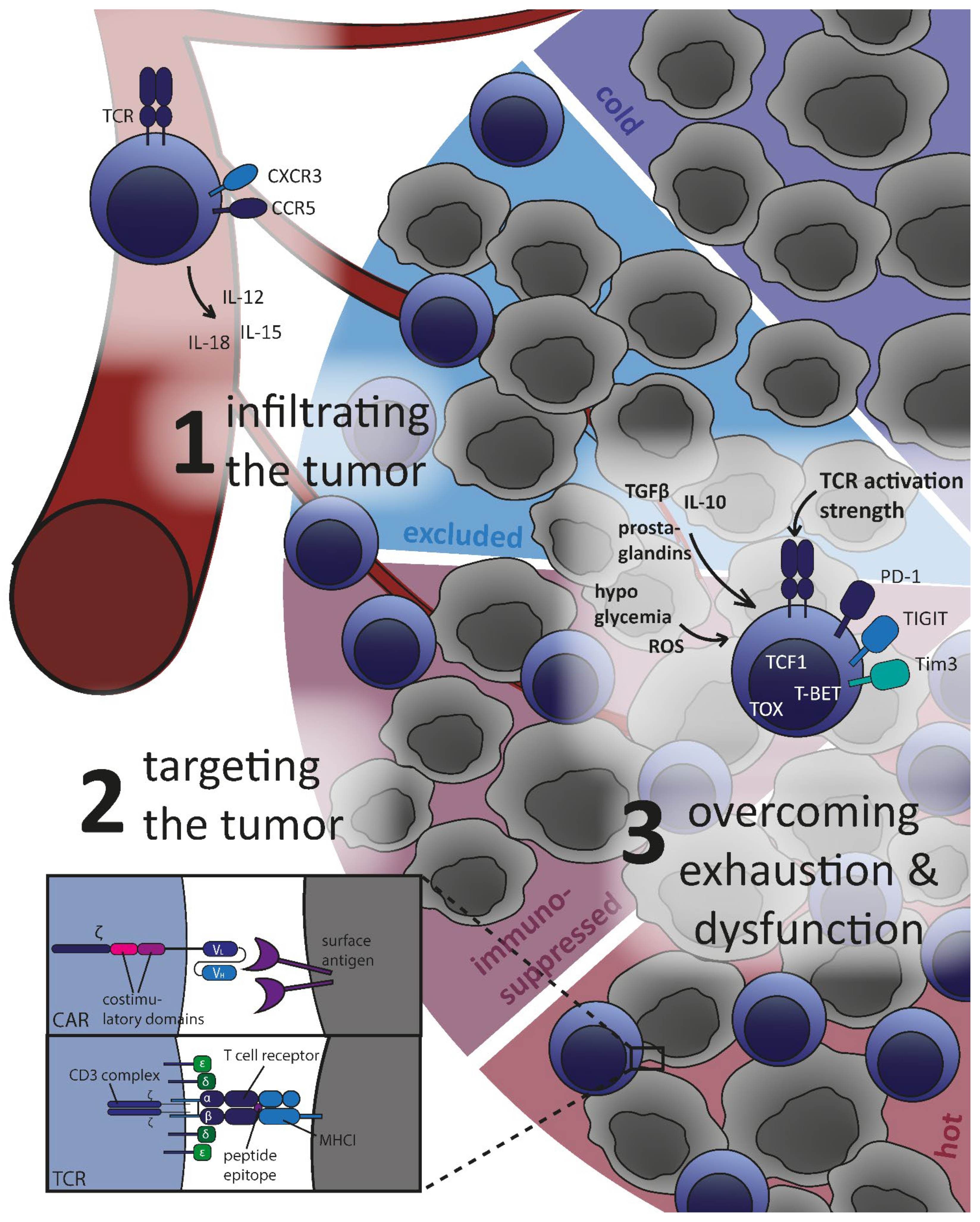{kind=link}
| Target | Target | TCR | HLA | Entities | Sponsor | Phase | n 1 | Start | Further Therapy | Study ID 2 | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TAA | WT-1 | WT1-TCRc4 | HLA-A*0201 | mesothelioma, NSCLC (both stage III-IV) | Fred Hutchinson Cancer Center/ Juno Therapeutics | I/II | 11 | 2015 | Aldesleukin | NCT02408016 | |
| MSLN | FH-TCR-Tᴍsʟɴ | HLA-A*0201 | pancreatic ductal adenocarcinoma | Fred Hutchinson Cancer Center/ Juno Therapeutics | I/II | 15 | 2021 | NCT04809766 | |||
| CTA/ Oncofetal proteins | NY-ESO-1 | Anti-NY ESO-1 mTCR PBL | HLA-A*0201 | melanoma, meningioma, breast CA, NSCLC, HCC | NCI | II | 11 | 2013 | Aldesleukin | NCT01967823 | |
| NY-ESO-1 | Anti-NY ESO-1 mTCR PBL | HLA-A*0201 | melanoma, renal cell cancer, metastatic cancer | NCI | II | 45 | 2008 | Aldesleukin | NCT00670748 | [55] | |
| NY-ESO-1 | NY-ESO-1c259T | HLA-A*0201, HLA-A*0205, and/or HLA-A*0206 | melanoma | Adapt immune/ Glaxo SmithKline | I/II | 4 | 2011 | NCT01350401 | |||
| NY-ESO-1 | NY-ESO-1c259T | HLA-A*0201, HLA-A*0205, and/or HLA-A*0206 | ovarian cancer | Adapt immune/ Glaxo SmithKline | I/II | 9 | 2013 | NCT01892293 | |||
| NY-ESO-1 | NY-ESO-1c259T/GSK3377794 | HLA-A*0201, HLA-A*0205, and/or HLA-A*0206 | liposarcoma | Adapt immune/ Glaxo SmithKline | II | 23 | 2016 | NCT02992743 | [67] | ||
| NY-ESO-1 | TBI-1301 | HLA-A*02:01 or HLA-A*02:06 | sarcoma, melanoma, esophageal, ovarian, lung, bladder, or liver cancer | University Health Network, Toronto | I | 22 | 2016 | NCT02869217 | |||
| MAGE-A3/12 | PG13-MAGE-A3 TCR9W11 | HLA-A*0201 | metastatic cancer, metastatic renal cancer, metastatic melanoma | NCI | I/II | 9 | 2010 | Aldesleukin | NCT01273181 | [56] | |
| MAGE-A3/12 | Anti-MAGE-A3-DP4 TCR | HLA-DPB1*0401 | melanoma, cervical, renal, urothelial, or breast cancer | NCI | I/II | 21 | 2014 | Aldesleukin | NCT02111850 | [68] | |
| MAGE-A4 | TBI-1201 | HLA-A*24:02 | various entities | Mie University | I | 18 | 2014 | NCT02096614 | |||
| MAGE-A4 | MAGE-A4c1032T | HLA-A*02 | bladder, head and neck, ovarian, esophageal, gastric cancer, melanoma, NSCLC, synovial sarcoma, liposarcoma | Adapt immune/ Glaxo SmithKline | I | 54 | 2017 | NCT03132922 | [69] | ||
| MAGE-A10 | MAGE A10c796T | HLA-A*0201 and/or HLA-A*0206 | NSCLC | Adapt immune/ Glaxo SmithKline | I | 28 | 2015 | NCT02592577 | [70] | ||
| MAGE-A3/A6 | KITE-718 | HLA-DPB1*0401 | various entities | Kite Pharma | I | 16 | 2017 | NCT03139370 | [71] | ||
| MAGE- A4/A8 | ACTengine IMA201-101 | HLA-A*0201 | various entities | Immatics | I | 22 | 2018 | NCT03247309 | [72] | ||
| MAGE- A1 | ACTengine IMA202-101 | N.A. | various entities | Immatics | I | 15 | 2019 | NCT03441100 | |||
| PRAME | IMA203-101 ACTengine | HLA-A*0201 | various entities | Immatics | I | 42 | 2019 | IL-2, Nivolumab (Cohort B) | NCT03686124 | [73] | |
| AFP | AFPc332T | HLA-A*02 | HCC | Adapt immune/ Glaxo SmithKline | I | 45 | 2017 | NCT03132792 | |||
| neoantigens | personalized | NEO-PTC-01 | Persona lized | melanoma | BioNTech | I | 52 | 2020 | NCT04625205 | ||
| NeoTCR-P1 | solid tumors | PACT Pharma, Inc. | Ia/Ib | 148 | 2019 | Aldesleukin, Nivolumab | NCT03970382 | [74] | |||
| N.A. | (neuro)endocrine tumors, NSCLC, ovarian, breast, GI cancers | NCI | II | 270 | 2018 | Aldesleukin, Pembrolizumab | NCT03412877 | ||||
| N.A. | malignant epithelial neoplasms | Providence Health & Services | I/Ib | 24 | 2022 | CDX-1140 (CD40 activation), Pembrolizumab | NCT04520711 |
| CAR | Target | CAR- Elements | Checkpoint Inhibitor | Entity | Sponsor | Phase | n 1 | Start | Study ID 2 | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| CART-EGFRvIII | EGFR | 41BB | Pembrolizumab | glioblastoma | University of Pennsylvania | I | 7 | 2019 | NCT03726515 | |
| iC9.GD2-CAR3 | GD-2 | CD28 OX40 + iCaspase9 | Pembrolizumab | neuroblastoma | Baylor College of Medicine | I | 11 | 2013 | NCT01822652 | [270] |
| HER2-CAR T | HER2 | CD28 | Pembrolizumab/Nivolumab | sarcoma | Baylor College of Medicine | I | 25 | 2021 | NCT0499500/HEROS 3.0 | |
| iCasp9M28z | MSLN | CD28 + iCaspase9 | Pembrolizumab | malignant pleural disease, mesothelioma, lung Cancer, breast Cancer | Memorial Sloan Kettering Cancer Center | I/II | 113 | 2015 | NCT02414269 | [275] |
| IL13Ra2-CAR | IL13Rα2 | 41BB + truncated CD19 | Nivolumab + Ipilimumab | glioblastoma | City of Hope Medical Center | I | 60 | 2019 | NCT04003649 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Füchsl, F.; Krackhardt, A.M. Paving the Way to Solid Tumors: Challenges and Strategies for Adoptively Transferred Transgenic T Cells in the Tumor Microenvironment. Cancers 2022, 14, 4192. https://doi.org/10.3390/cancers14174192
Füchsl F, Krackhardt AM. Paving the Way to Solid Tumors: Challenges and Strategies for Adoptively Transferred Transgenic T Cells in the Tumor Microenvironment. Cancers. 2022; 14(17):4192. https://doi.org/10.3390/cancers14174192
Chicago/Turabian StyleFüchsl, Franziska, and Angela M. Krackhardt. 2022. "Paving the Way to Solid Tumors: Challenges and Strategies for Adoptively Transferred Transgenic T Cells in the Tumor Microenvironment" Cancers 14, no. 17: 4192. https://doi.org/10.3390/cancers14174192
APA StyleFüchsl, F., & Krackhardt, A. M. (2022). Paving the Way to Solid Tumors: Challenges and Strategies for Adoptively Transferred Transgenic T Cells in the Tumor Microenvironment. Cancers, 14(17), 4192. https://doi.org/10.3390/cancers14174192