MicroRNA-34a, Prostate Cancer Stem Cells, and Therapeutic Development
_Li.JPG)




Abstract
:Simple Summary
Abstract
1. Introduction: PCa Cell Heterogeneity, PCSCs and CRPC
2. miR-34a Expression Decreases with Increasing PCa Grade but Correlates with Better Patient Survival
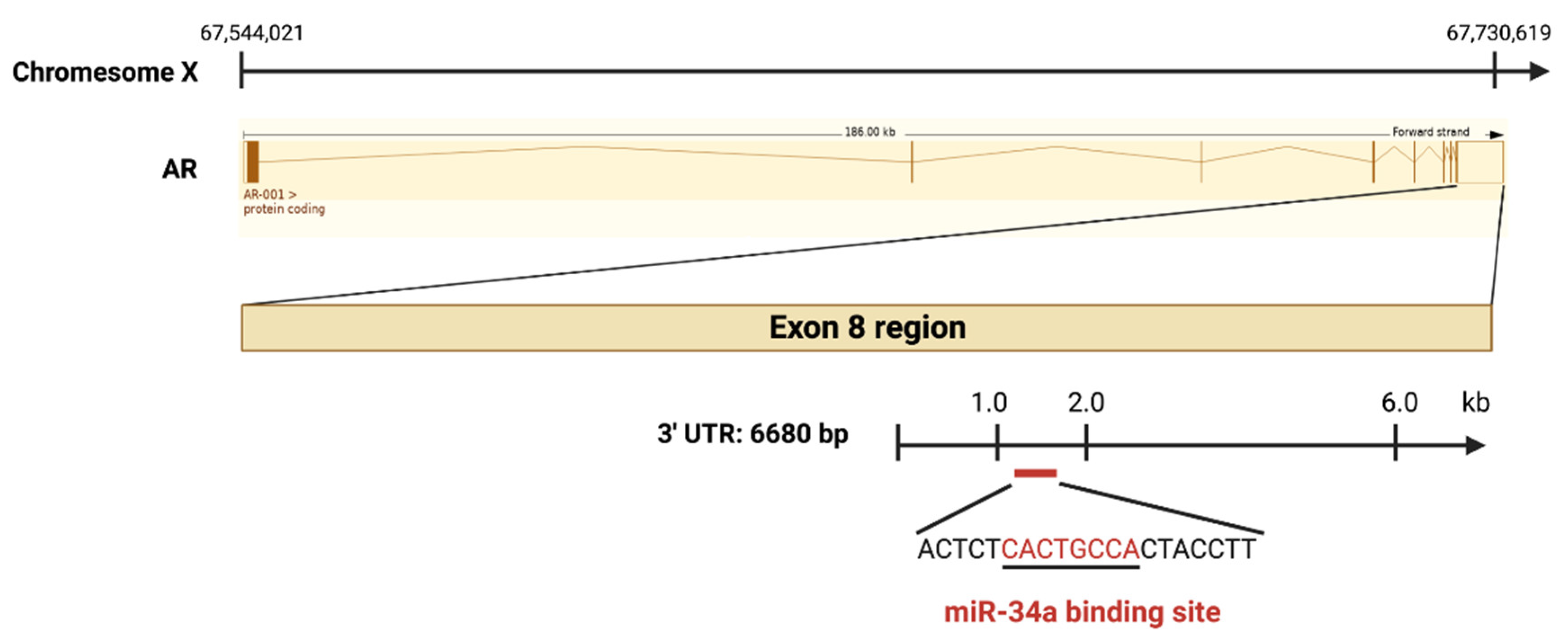
3. Mechanisms of miR-34a Regulation
3.1. Transcriptional Regulation of miR-34a Expression
3.2. Epigenetic Regulation of miR-34a Expression
4. Tumor Suppressive Role of miR-34a in PCa
4.1. Targeting Invasiveness and Metastasis
4.2. Targeting Stemness
4.3. Targeting Epigenome
4.4. Targeting Cell Survival
5. miR-34a Therapeutic Development for Aggressive PCa
5.1. miR-34a Replacement Therapy
5.2. Preclinical Studies of miR-34a in PCa

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Delivery System | Mouse Model | Route of Administration | Dose | Dose Schedule | Reference |
|---|---|---|---|---|---|
| Lipid-based transfection reagent | Orthotopic PC3 and DU145 xenografts | i.v. | 1 mg/kg | Every 2 days for 5 times | [15] |
| Chitosan nanoparticle | s.c PC3MM2 xenograft; intra-femoral model | i.v. | 250 μg/kg | Every 3 days for three weeks | [47] |
| Cationic polypeptide-based micelles | s.c DU145 xenograft | i.v. | 2 mg/kg | Every 4 days for 4 times | [84] |
| PEG-PCD micelles | Orthotopic PC3-TXR xenograft | i.v. | 10 mg/kg (Rubone) | Every 2 days for 5 times | [61] |
| pH and GSH responsive micelles | Orthotopic PC3-TXR xenograft | i.v. | 25 mg/kg (Rubone) | Every 2 days for 9 times | [85] |
| mPEG-PLGA-PLL nanoparticles | s.c. PC3 xenograft | i.v. | 1.5 mg/kg | Every 3 days for 7 times | [82] |
| rAAV9-miR-34a vector | TRAMP genetic model | i.p. | 1 × 1011 genome copy | Single dose | [86] |
5.3. Novel miR-34a Delivery Systems for Targeting PCa
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
List of Abbreviations
| PCa | Prostate Cancer |
| AR | Androgen Receptor |
| CRPC | Castration Resistant Prostate Cancer |
| CSCs | Cancer Stem Cells |
| PCSCs | Prostate Cancer Stem Cells |
| miR-34a | microRNA-34a |
| AUA | American Urological Association |
| GS | Gleason Score |
| LN | Lymph Node |
| ARSIs | AR Signaling Inhibitors |
| ADT | Androgen-Deprivation Therapy |
| Enza | Enzalutamide |
| metastatic CRPC | mCRPC |
| miRNAs | microRNAs |
| nt | Nucleotide |
| pri-miRNA | primary miRNA |
| mRNAs | Messenger RNAs |
| 3’-UTR | 3’-Untranslated Regions |
| TCGA | The Cancer Genome Atlas |
| T | Tumor |
| RISC | RNA-Induced Silencing Complex |
| lncRNAs | Long non-coding RNAs |
| DOX | Doxorubicin |
| PRC2 | Polycomb Repressive Complex 2 |
| 5Aza-2′dC | 5-aza-2′deoxycytidine |
| EMT | Epithelial-to-Mesenchymal Transition |
| STMN1 | Stathmin 1 |
| TCF | T-Cell Factor |
| LEF | Lymphoid Enhancer Factor |
| SIRT1 | Sirtuin-1 |
| HuR | Human antigen R protein |
| AE | Adverse Events |
| UIMC | Ultrasound Induces Microbubble Cavitation |
| mPEG-PLGA-PLL | Methoxy polyethylene glycol-polylacticco-glycolic acid-polylysine |
| SHRss | Self-assembling disulfide cross-linked stearyl-peptide-based micellar system |
| GSH | Glutathione |
| rAAV | Recombinant Adeno-Associated Virus |
| TRAMP | Transgenic Adenocarcinoma Mouse Prostate |
| AP | Anterior Prostate |
| DLP | Dorsal Lateral Prostate |
| WT | Wild Type |
| PIN | Prostatic Intraepithelial Neoplasia |
| FR | Folate Receptor |
| PSMA | Prostate-Specific Membrane Antigen |
| FOLH1 | Folate Hydrolase 1 |
| PET | Positron Emission Tomography |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.G. Understanding and targeting prostate cancer cell heterogeneity and plasticity. Semin. Cancer Biol. 2021, 82, 68–93. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Li, Q.; Deng, Q.; Chao, H.-P.; Liu, X.; Lu, Y.; Lin, K.; Liu, B.; Tang, G.W.; Zhang, D.; Tracz, A.; et al. Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat. Commun. 2018, 9, 3600. [Google Scholar] [CrossRef]
- Liu, X.; Chen, X.; Rycaj, K.; Chao, H.-P.; Deng, Q.; Jeter, C.; Liu, C.; Honorio, S.; Li, H.; Davis, T.; et al. Systematic dissection of phenotypic, functional, and tumorigenic heterogeneity of human prostate cancer cells. Oncotarget 2015, 6, 23959–23986. [Google Scholar] [CrossRef]
- Qin, J.; Liu, X.; Laffin, B.; Chen, X.; Choy, G.; Jeter, C.R.; Calhoun-Davis, T.; Li, H.; Palapattu, G.S.; Pang, S.; et al. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell 2012, 10, 556–569. [Google Scholar] [CrossRef]
- Liu, X.; Li, W.; Puzanov, I.; Goodrich, D.W.; Chatta, G.; Tang, D.G. Prostate cancer as a dedifferentiated organ: Androgen receptor, cancer stem cells, and cancer stemness. Essays Biochem. 2022, 66, 291–303. [Google Scholar] [CrossRef]
- Lionetti, M.; Musto, P.; Di Martino, M.T.; Fabris, S.; Agnelli, L.; Todoerti, K.; Tuana, G.; Mosca, L.; Cantafio, M.E.G.; Grieco, V.; et al. Biological and Clinical Relevance of miRNA Expression Signatures in Primary Plasma Cell Leukemia. Clin. Cancer Res. 2013, 19, 3130–3142. [Google Scholar] [CrossRef]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [Green Version]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef]
- Bader, A.G. MiR-34—A microRNA replacement therapy is headed to the clinic. Front. Genet. 2012, 3, 120. [Google Scholar] [CrossRef]
- Li, Q.; Liu, B.; Chao, H.P.; Ji, Y.; Lu, Y.; Mehmood, R.; Jeter, C.; Chen, T.; Moore, J.R.; Li, W.; et al. LRIG1 is a pleiotropic androgen receptor-regulated feedback tumor suppressor in prostate cancer. Nat. Commun. 2019, 10, 5494. [Google Scholar] [CrossRef]
- Li, W.; Wang, Y.; Liu, R.; Kasinski, A.L.; Shen, H.; Slack, F.J.; Tang, D.G. MicroRNA-34a: Potent Tumor Suppressor, Cancer Stem Cell Inhibitor, and Potential Anticancer Therapeutic. Front. Cell Dev. Biol. 2021, 9, 640587. [Google Scholar] [CrossRef]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef]
- Okada, N.; Lin, C.P.; Ribeiro, M.C.; Biton, A.; Lai, G.; He, X.; Bu, P.; Vogel, H.; Jablons, D.M.; Keller, A.C. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev. 2014, 28, 438–450. [Google Scholar] [CrossRef]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [Green Version]
- Cotter, K.; Rubin, M.A. The evolving landscape of prostate cancer somatic mutations. Prostate 2022, 82, S13–S24. [Google Scholar] [CrossRef]
- Hamid, A.A.; Gray, K.P.; Shaw, G.; MacConaill, L.E.; Evan, C.; Bernard, B.; Loda, M.; Corcoran, N.M.; Van Allen, E.M.; Choudhury, A.D.; et al. Compound Genomic Alterations of TP53, PTEN, and RB1 Tumor Suppressors in Localized and Metastatic Prostate Cancer. Eur. Urol. 2019, 76, 89–97. [Google Scholar] [CrossRef]
- Mateo, J.; Seed, G.; Bertan, C.; Rescigno, P.; Dolling, D.; Figueiredo, I.; Miranda, S.; Nava Rodrigues, D.; Gurel, B.; Clarke, M.; et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J. Clin. Investig. 2020, 130, 1743–1751. [Google Scholar] [CrossRef]
- Teroerde, M.; Nientiedt, C.; Duensing, A.; Hohenfellner, M.; Stenzinger, A.; Duensing, S. Revisiting the Role of p53 in Prostate Cancer. In Prostate Cancer; Bott, S.R.J., Ng, K.L., Eds.; Exon Publications: Brisbane, Australia, 2021. [Google Scholar]
- Fujita, Y.; Kojima, K.; Hamada, N.; Ohhashi, R.; Akao, Y.; Nozawa, Y.; Deguchi, T.; Ito, M. Effects of miR-34a on cell growth and chemoresistance in prostate cancer PC3 cells. Biochem. Biophys. Res. Commun. 2008, 377, 114–119. [Google Scholar] [CrossRef]
- Lodygin, D.; Tarasov, V.; Epanchintsev, A.; Berking, C.; Knyazeva, T.; Körner, H.; Knyazev, P.; Diebold, J.; Hermeking, H. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 2008, 7, 2591–2600. [Google Scholar] [CrossRef]
- Concepcion, C.P.; Han, Y.-C.; Mu, P.; Bonetti, C.; Yao, E.; D’Andrea, A.; Vidigal, J.A.; Maughan, W.P.; Ogrodowski, P.; Ventura, A. Intact p53-dependent responses in miR-34-deficient mice. PLoS Genet. 2012, 8, e1002797. [Google Scholar] [CrossRef]
- Chang, T.C.; Yu, D.; Lee, Y.-S.; Wentzel, E.A.; Arking, D.E.; West, K.M.; Dang, C.V.; Thomas-Tikhonenko, A.; Mendell, J.T. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat. Genet. 2008, 40, 43–50. [Google Scholar] [CrossRef]
- Craig, V.J.; Cogliatti, S.B.; Imig, J.; Renner, C.; Neuenschwander, S.; Rehrauer, H.; Schlapbach, R.; Dirnhofer, S.; Tzankov, A.; Müller, A. Myc-mediated repression of microRNA-34a promotes high-grade transformation of B-cell lymphoma by dysregulation of FoxP1. Blood 2011, 117, 6227–6236. [Google Scholar] [CrossRef]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Rokhlin, O.W.; Scheinker, V.S.; Taghiyev, A.F.; Bumcrot, D.; Glover, R.A.; Cohen, M.B. MicroRNA-34 mediates AR-dependent p53-induced apoptosis in prostate cancer. Cancer Biol. Ther. 2008, 7, 1288–1296. [Google Scholar] [CrossRef] [Green Version]
- Ding, M.; Lin, B.; Li, T.; Liu, Y.; Li, Y.; Zhou, X.; Miao, M.; Gu, J.; Pan, H.; Yang, F.; et al. A dual yet opposite growth-regulating function of miR-204 and its target XRN1 in prostate adenocarcinoma cells and neuroendocrine-like prostate cancer cells. Oncotarget 2015, 6, 7686–7700. [Google Scholar] [CrossRef]
- Ostling, P.; Leivonen, S.-K.; Aakula, A.; Kohonen, P.; Mäkelä, R.; Hagman, Z.; Edsjö, A.; Kangaspeska, S.; Edgren, H.; Nicorici, D.; et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res. 2011, 71, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Öner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J. Clin. Investig. 2014, 124, 1853–1867. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Xiao, Y.; Hu, Y.; Xiao, Y.; Yin, Z.; Liu, L.; Kang, X.; Chen, Y. Methylation-induced silencing of miR-34a enhances chemoresistance by directly upregulating ATG4B-induced autophagy through AMPK/mTOR pathway in prostate cancer. Oncol. Rep. 2016, 35, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bu, P.; Ai, Y.; Srinivasan, T.; Chen, H.J.; Xiang, K.; Lipkin, S.M.; Shen, X. A long non-coding RNA targets microRNA miR-34a to regulate colon cancer stem cell asymmetric division. eLife 2016, 5, e14620. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Heath, E.; Chen, W.; Cher, M.; Powell, I.; Heilbrun, L.; Li, Y.; Ali, S.; Sethi, S.; Hassan, O.; et al. Epigenetic silencing of miR-34a in human prostate cancer cells and tumor tissue specimens can be reversed by BR-DIM treatment. Am. J. Transl. Res. 2012, 4, 14–23. [Google Scholar] [PubMed]
- Nanni, S.; Aiello, A.; Re, A.; Guffanti, A.; Benvenuti, V.; Colussi, C.; Castro-Vega, L.J.; Felsani, A.; Londono-Vallejo, A.; Capogrossi, M.C.; et al. Estrogen-dependent dynamic profile of eNOS-DNA associations in prostate cancer. PLoS ONE 2013, 8, e62522. [Google Scholar] [CrossRef]
- Choi, Y.J.; Lin, C.-P.; Ho, J.J.; He, X.; Okada, N.; Bu, P.; Zhong, Y.; Kim, S.Y.; Bennett, M.J.; Chen, C.; et al. MiR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat. Cell Biol. 2011, 13, 1353–1360. [Google Scholar] [CrossRef]
- Wang, L.; Wang, E.; Wang, Y.; Mines, R.; Xiang, K.; Sun, Z.; Zhou, G.; Chen, K.-Y.; Rakhilin, N.; Chao, S.; et al. MiR-34a is a microRNA safeguard for Citrobacter-induced inflammatory colon oncogenesis. eLife 2018, 7, e39479. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Hwang, C.-I.; Corney, D.C.; Flesken-Nikitin, A.; Jiang, L.; Öner, G.M.; Munroe, R.J.; Schimenti, J.C.; Hermeking, H.; Nikitin, A.Y. MiR-34 cooperates with p53 in suppression of prostate cancer by joint regulation of stem cell compartment. Cell Rep. 2014, 6, 1000–1007. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Kelnar, K.; Vlassov, A.V.; Brown, D.; Wang, J.; Tang, D.G. Distinct microRNA expression profiles in prostate cancer stem/progenitor cells and tumor-suppressive functions of let-7. Cancer Res. 2012, 72, 3393–3404. [Google Scholar] [CrossRef]
- Boccaccio, C.; Comoglio, P.M. Comoglio, Invasive growth: A MET-driven genetic programme for cancer and stem cells. Nat. Rev. Cancer 2006, 6, 637–645. [Google Scholar] [CrossRef]
- Rajasekhar, V.K.; Studer, L.; Gerald, W.; Socci, N.D.; Scher, H.I. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. Nat. Commun. 2011, 2, 162. [Google Scholar] [CrossRef]
- Corney, D.C.; Hwang, C.-I.; Matoso, A.; Vogt, M.; Flesken-Nikitin, A.; Godwin, A.K.; Kamat, A.A.; Sood, A.K.; Ellenson, L.H.; Hermeking, H.; et al. Frequent downregulation of miR-34 family in human ovarian cancers. Clin. Cancer Res. 2010, 16, 1119–1128. [Google Scholar] [CrossRef]
- Hwang, C.I.; Matoso, A.; Corney, D.C.; Flesken-Nikitin, A.; Körner, S.; Wang, W.; Boccaccio, C.; Thorgeirsson, S.S.; Comoglio, P.M.; Hermeking, H.; et al. Wild-type p53 controls cell motility and invasion by dual regulation of MET expression. Proc. Natl. Acad. Sci. USA 2011, 108, 14240–14245. [Google Scholar] [CrossRef]
- Gaur, S.; Wen, Y.; Song, J.H.; Parikh, N.U.; Mangala, L.S.; Blessing, A.M.; Ivan, C.; Wu, S.Y.; Varkaris, A.; Shi, Y.; et al. Chitosan nanoparticle-mediated delivery of miRNA-34a decreases prostate tumor growth in the bone and its expression induces non-canonical autophagy. Oncotarget 2015, 6, 29161–29177. [Google Scholar] [CrossRef]
- Yamamura, S.; Saini, S.; Majid, S.; Hirata, H.; Ueno, K.; Deng, G.; Dahiya, R. MicroRNA-34a modulates c-Myc transcriptional complexes to suppress malignancy in human prostate cancer cells. PLoS ONE 2012, 7, e29722. [Google Scholar] [CrossRef]
- Zhang, G.; Tian, X.; Li, Y.; Wang, Z.; Li, X.; Zhu, C. MiR-27b and miR-34a enhance docetaxel sensitivity of prostate cancer cells through inhibiting epithelial-to-mesenchymal transition by targeting ZEB1. Biomed. Pharm. 2018, 97, 736–744. [Google Scholar] [CrossRef]
- Chakravarthi, B.; Chandrashekar, D.S.; Agarwal, S.; Balasubramanya, S.A.H.; Pathi, S.S.; Goswami, M.T.; Jing, X.; Wang, R.; Mehra, R.; Asangani, I.A.; et al. miR-34a Regulates Expression of the Stathmin-1 Oncoprotein and Prostate Cancer Progression. Mol. Cancer Res. 2018, 16, 1125–1137. [Google Scholar] [CrossRef]
- Civenni, G.; Malek, A.; Albino, D.; Garcia-Escudero, R.; Napoli, S.; Di Marco, S.; Pinton, S.; Sarti, M.; Carbone, G.M.; Catapano, C.V. RNAi-mediated silencing of Myc transcription inhibits stem-like cell maintenance and tumorigenicity in prostate cancer. Cancer Res. 2013, 73, 6816–6827. [Google Scholar] [CrossRef] [Green Version]
- Dong, B.; Xu, G.C.; Liu, S.T.; Liu, T.; Geng, B. MiR-34a affects G2 arrest in prostate cancer PC3 cells via Wnt pathway and inhibits cell growth and migration. Eur. Rev. Med. Pharm. Sci. 2020, 24, 8349–8358. [Google Scholar]
- Chen, W.Y.; Liu, S.-Y.; Chang, Y.-S.; Yin, J.J.; Yeh, H.-L.; Mouhieddine, T.H.; Hadadeh, O.; Abou-Kheir, W.; Liu, Y.-N. MicroRNA-34a regulates WNT/TCF7 signaling and inhibits bone metastasis in Ras-activated prostate cancer. Oncotarget 2015, 6, 441–457. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Chiang, A.C.; Zhang, X.H.F.; Kim, J.Y.; Kris, M.G.; Ladanyi, M.; Gerald, W.L.; Massagué, J. WNT/TCF Signaling through LEF1 and HOXB9 Mediates Lung Adenocarcinoma Metastasis. Cell 2009, 138, 51–62. [Google Scholar] [CrossRef]
- Liang, J.; Li, Y.; Daniels, G.; Sfanos, K.; De Marzo, A.; Wei, J.; Li, X.; Chen, W.; Wang, J.; Zhong, X.; et al. LEF1 Targeting EMT in Prostate Cancer Invasion Is Regulated by miR-34a. Mol. Cancer Res. 2015, 13, 681–688. [Google Scholar] [CrossRef]
- Farah, E.; Li, C.; Cheng, L.; Kong, Y.; Lanman, N.A.; Pascuzzi, P.; Lorenz, G.R.; Zhang, Y.; Ahmad, N.; Li, L.; et al. NOTCH signaling is activated in and contributes to resistance in enzalutamide-resistant prostate cancer cells. J. Biol. Chem. 2019, 294, 8543–8554. [Google Scholar] [CrossRef]
- Liu, X.; Luo, X.; Wu, Y.; Xia, D.; Chen, W.; Fang, Z.; Deng, J.; Hao, Y.; Yang, X.; Zhang, T.; et al. MicroRNA-34a Attenuates Paclitaxel Resistance in Prostate Cancer Cells via Direct Suppression of JAG1/Notch1 Axis. Cell. Physiol. Biochem. 2018, 50, 261–276. [Google Scholar] [CrossRef]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef]
- Liu, T.F.; McCall, C.E. Deacetylation by SIRT1 Reprograms Inflammation and Cancer. Genes Cancer 2013, 4, 135–147. [Google Scholar] [CrossRef]
- Kojima, K.; Fujita, Y.; Nozawa, Y.; Deguchi, T.; Ito, M. MiR-34a attenuates paclitaxel-resistance of hormone-refractory prostate cancer PC3 cells through direct and indirect mechanisms. Prostate 2010, 70, 1501–1512. [Google Scholar] [CrossRef]
- Wen, D.; Peng, Y.; Lin, F.; Singh, R.K.; Mahato, R.I. Micellar Delivery of miR-34a Modulator Rubone and Paclitaxel in Resistant Prostate Cancer. Cancer Res. 2017, 77, 3244–3254. [Google Scholar] [CrossRef]
- Corcoran, C.; Rani, S.; O’Driscoll, L. miR-34a is an intracellular and exosomal predictive biomarker for response to docetaxel with clinical relevance to prostate cancer progression. Prostate 2014, 74, 1320–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Ozpolat, B.; Sood, A.K.; Lopez-Berestein, G. Liposomal siRNA nanocarriers for cancer therapy. Adv. Drug Deliv. Rev. 2014, 66, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Orellana, E.A.; Tenneti, S.; Rangasamy, L.; Lyle, L.T.; Low, P.S.; Kasinski, A.L. FolamiRs: Ligand-targeted, vehicle-free delivery of microRNAs for the treatment of cancer. Sci. Transl. Med. 2017, 9, eaam9327. [Google Scholar] [CrossRef]
- Abdelaal, A.M.; Kasinski, A.L. Ligand-mediated delivery of RNAi-based therapeutics for the treatment of oncological diseases. NAR Cancer 2021, 3, zcab030. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.-K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 12, 1630–1637. [Google Scholar] [CrossRef]
- Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Patrawala, L.; Brown, D.; Bader, A.G. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010, 70, 5923–5930. [Google Scholar] [CrossRef]
- Daige, C.L.; Wiggins, J.F.; Priddy, L.; Nelligan-Davis, T.; Zhao, J.; Brown, D. Systemic delivery of a miR34a mimic as a potential therapeutic for liver cancer. Mol. Cancer Ther. 2014, 13, 2352–2360. [Google Scholar] [CrossRef]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108, djv303. [Google Scholar]
- Kasinski, A.L.; Kelnar, K.; Stahlhut, C.; Orellana, E.; Zhao, J.; Shimer, E.; Dysart, S.; Chen, X.; Bader, A.G.; Slack, F.J. A combinatorial microRNA therapeutics approach to suppressing non-small cell lung cancer. Oncogene 2015, 34, 3547–3555. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, X.; Zhang, X.; Liu, B.; Huang, L. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol. Ther. 2010, 18, 1650–1656. [Google Scholar] [CrossRef] [Green Version]
- Trang, P.; Wiggins, J.F.; Daige, C.L.; Cho, C.; Omotola, M.; Brown, D.; Weidhaas, J.B.; Bader, A.G.; Slack, F.J. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther. 2011, 19, 1116–1122. [Google Scholar] [CrossRef]
- Craig, V.J.; Tzankov, A.; Flori, M.; Schmid, C.; Bader, A.G.; Müller, A. Systemic microRNA-34a delivery induces apoptosis and abrogates growth of diffuse large B-cell lymphoma in vivo. Leukemia 2012, 26, 2421–2424. [Google Scholar] [CrossRef]
- Hu, Q.L.; Jiang, Q.Y.; Jin, X.; Shen, J.; Wang, K.; Li, Y.B.; Xu, F.J.; Tang, G.P.; Li, Z.H. Cationic microRNA-delivering nanovectors with bifunctional peptides for efficient treatment of PANC-1 xenograft model. Biomaterials 2013, 34, 2265–2276. [Google Scholar] [CrossRef]
- Lin, X.; Chen, W.; Wei, F.; Zhou, B.P.; Hung, M.-C.; Xie, X. Nanoparticle Delivery of miR-34a Eradicates Long-term-cultured Breast Cancer Stem Cells via Targeting C22ORF28 Directly. Theranostics 2017, 7, 4805–4824. [Google Scholar] [CrossRef]
- Gibori, H.; Eliyahu, S.; Krivitsky, A.; Ben-Shushan, D.; Epshtein, Y.; Tiram, G.; Blau, R.; Ofek, P.; Lee, J.S.; Ruppin, E.; et al. Amphiphilic nanocarrier-induced modulation of PLK1 and miR-34a leads to improved therapeutic response in pancreatic cancer. Nat. Commun. 2018, 9, 16. [Google Scholar] [CrossRef]
- Jiang, G.; Chen, H.; Huang, J.; Song, Q.; Chen, Y.; Gu, X.; Jiang, Z.; Huang, Y.; Lin, Y.; Feng, J.; et al. Tailored Lipoprotein-Like miRNA Delivery Nanostructure Suppresses Glioma Stemness and Drug Resistance through Receptor-Stimulated Macropinocytosis. Adv. Sci. 2020, 7, 1903290. [Google Scholar] [CrossRef]
- Lu, C.; Han, H.D.; Mangala, L.S.; Ali-Fehmi, R.; Newton, C.S.; Ozbun, L.; Armaiz-Pena, G.N.; Hu, W.; Stone, R.L.; Munkarah, A.; et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell 2010, 18, 185–197. [Google Scholar] [CrossRef]
- Han, H.D.; Mangala, L.S.; Lee, J.W.; Shahzad, M.M.; Kim, H.S.; Shen, D.; Nam, E.J.; Mora, E.M.; Stone, R.L.; Lu, C.; et al. Targeted gene silencing using RGD-labeled chitosan nanoparticles. Clin. Cancer Res. 2010, 16, 3910–3922. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, C.; Tao, Y.; Zou, P.; Gao, F.; Jia, C.; Liu, L.; Li, G.; Zhang, G.; Duan, Y.; et al. Ultrasound-Induced Microbubble Cavitation Combined with miR-34a-Loaded Nanoparticles for the Treatment of Castration-Resistant Prostate Cancer. J. Biomed. Nanotechnol. 2021, 17, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, S.; Mitragotri, S. Ultrasound-induced cavitation: Applications in drug and gene delivery. Expert Opin. Drug Deliv. 2006, 3, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Liu, J.; Wu, X.; Tai, Z.; Gao, Y.; Zhu, Q.; Li, J.; Zhang, L.; Hu, C.; Gu, F.; et al. Reducible self-assembling cationic polypeptide-based micelles mediate co-delivery of doxorubicin and microRNA-34a for androgen-independent prostate cancer therapy. J. Control. Release 2016, 232, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Wen, D.; Wang, X.; Mahato, R.I. Dual responsive micelles capable of modulating miRNA-34a to combat taxane resistance in prostate cancer. Biomaterials 2019, 192, 95–108. [Google Scholar] [CrossRef]
- Ai, J.; Li, J.; Su, Q.; Ma, H.; He, R.; Wei, Q.; Li, H.; Gao, G. rAAV-based and intraprostatically delivered miR-34a therapeutics for efficient inhibition of prostate cancer progression. Gene Ther. 2021, 29, 418–424. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.M.; Beattie, S.G.; Gardner, P.J.; et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef]
- Leamon, C.; Low, P.S. Folate-mediated targeting: From diagnostics to drug and gene delivery. Drug Discov. Today 2001, 6, 44–51. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Kuratsukuri, K.; Landas, S.; Imaida, K.; Rovito, P.M.; Wang, C.Y.; Haas, G.P. Expression of prostate-specific membrane antigen in normal and malignant human tissues. World J. Surg. 2006, 30, 628–636. [Google Scholar] [CrossRef]
- Kularatne, S.A.; Wang, K.; Santhapuram, H.-K.R.; Low, P.S. Prostate-Specific Membrane Antigen Targeted Imaging and Therapy of Prostate Cancer Using a PSMA Inhibitor as a Homing Ligand. Mol. Pharm. 2009, 6, 780–789. [Google Scholar] [CrossRef]
- Bravaccini, S.; Puccetti, M.; Bocchini, M.; Ravaioli, S.; Celli, M.; Scarpi, E.; De Giorgi, U.; Tumedei, M.M.; Raulli, G.; Cardinale, L.; et al. PSMA expression: A potential ally for the pathologist in prostate cancer diagnosis. Sci. Rep. 2018, 8, 4254. [Google Scholar] [CrossRef]
- Sheehan, B.; Guo, C.; Neeb, A.; Paschalis, A.; Sandhu, S.; de Bono, J.S. Prostate-specific Membrane Antigen Biology in Lethal Prostate Cancer and its Therapeutic Implications. Eur. Urol. Focus 2021. [Google Scholar] [CrossRef]
- Wang, F.; Li, Z.; Feng, X.; Yang, D.; Lin, M. Advances in PSMA-targeted therapy for prostate cancer. Prostate Cancer Prostatic Dis. 2022, 25, 11–26. [Google Scholar] [CrossRef]
- Thomas, M.; Kularatne, S.A.; Qi, L.; Kleindl, P.; Leamon, C.P.; Hansen, M.J.; Low, P.S. Ligand-targeted delivery of small interfering RNAs to malignant cells and tissues. Ann. N. Y. Acad. Sci. 2009, 1175, 32–39. [Google Scholar] [CrossRef]
- Maurer, T.; Eiber, M.; Schwaiger, M.; Gschwend, J.E. Current use of PSMA–PET in prostate cancer management. Nat. Rev. Urol. 2016, 13, 226–235. [Google Scholar] [CrossRef]
- Staniszewska, M.; Costa, P.F.; Eiber, M.; Klose, J.; Wosniack, J.; Reis, H.; Szarvas, T.; Hadaschik, B.; Lückerath, K.; Herrmann, K.; et al. Enzalutamide Enhances PSMA Expression of PSMA-Low Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 7431. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Liu, X.; Dougherty, E.M.; Tang, D.G. MicroRNA-34a, Prostate Cancer Stem Cells, and Therapeutic Development. Cancers 2022, 14, 4538. https://doi.org/10.3390/cancers14184538
Li W, Liu X, Dougherty EM, Tang DG. MicroRNA-34a, Prostate Cancer Stem Cells, and Therapeutic Development. Cancers. 2022; 14(18):4538. https://doi.org/10.3390/cancers14184538
Chicago/Turabian StyleLi, Wen (Jess), Xiaozhuo Liu, Emily M. Dougherty, and Dean G. Tang. 2022. "MicroRNA-34a, Prostate Cancer Stem Cells, and Therapeutic Development" Cancers 14, no. 18: 4538. https://doi.org/10.3390/cancers14184538
APA StyleLi, W., Liu, X., Dougherty, E. M., & Tang, D. G. (2022). MicroRNA-34a, Prostate Cancer Stem Cells, and Therapeutic Development. Cancers, 14(18), 4538. https://doi.org/10.3390/cancers14184538