
Development of the Novel Bifunctional Fusion Protein BR102 That Simultaneously Targets PD-L1 and TGF-β for Anticancer Immunotherapy
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibody Generation and Purification
2.2. Cell Lines and Cell Culture
2.3. Antibody Affinity Measurement
2.4. Binding ELISA Assay
2.5. Competition ELISA Assay
2.6. PD-1/PD-L1 Reporter Gene Assay
2.7. TGF-β Reporter Gene Assay
2.8. TF-1 Cell Proliferation
2.9. Allogeneic Mixed Lymphocyte Reaction
2.10. Mouse Tumor Xenograft Models
2.11. Toxicity Study in Non-Human Primates
2.12. Statistics
3. Results
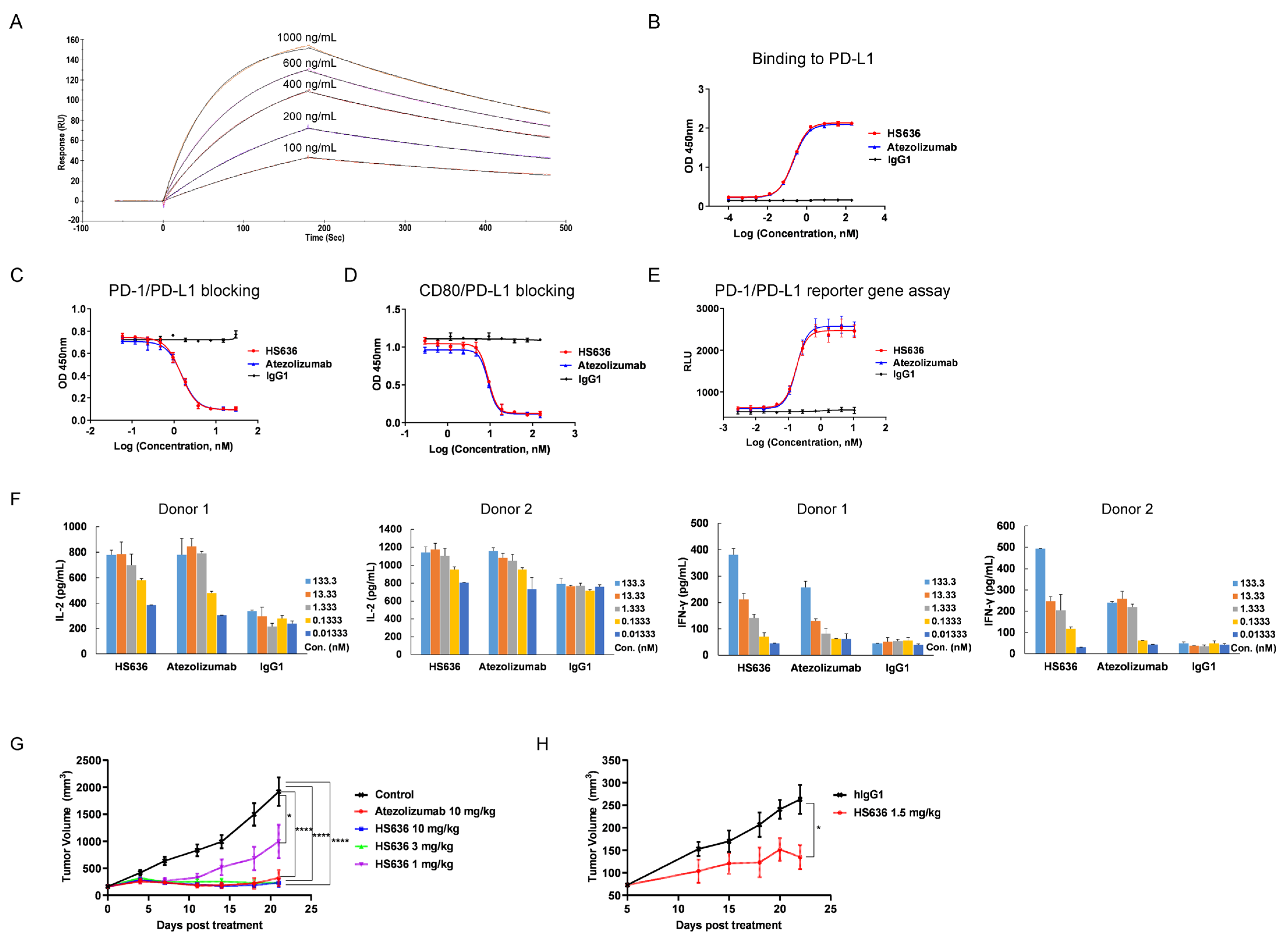
3.1. Screen and Biological Activity Evaluation of An Anti-PD-L1 Antibody
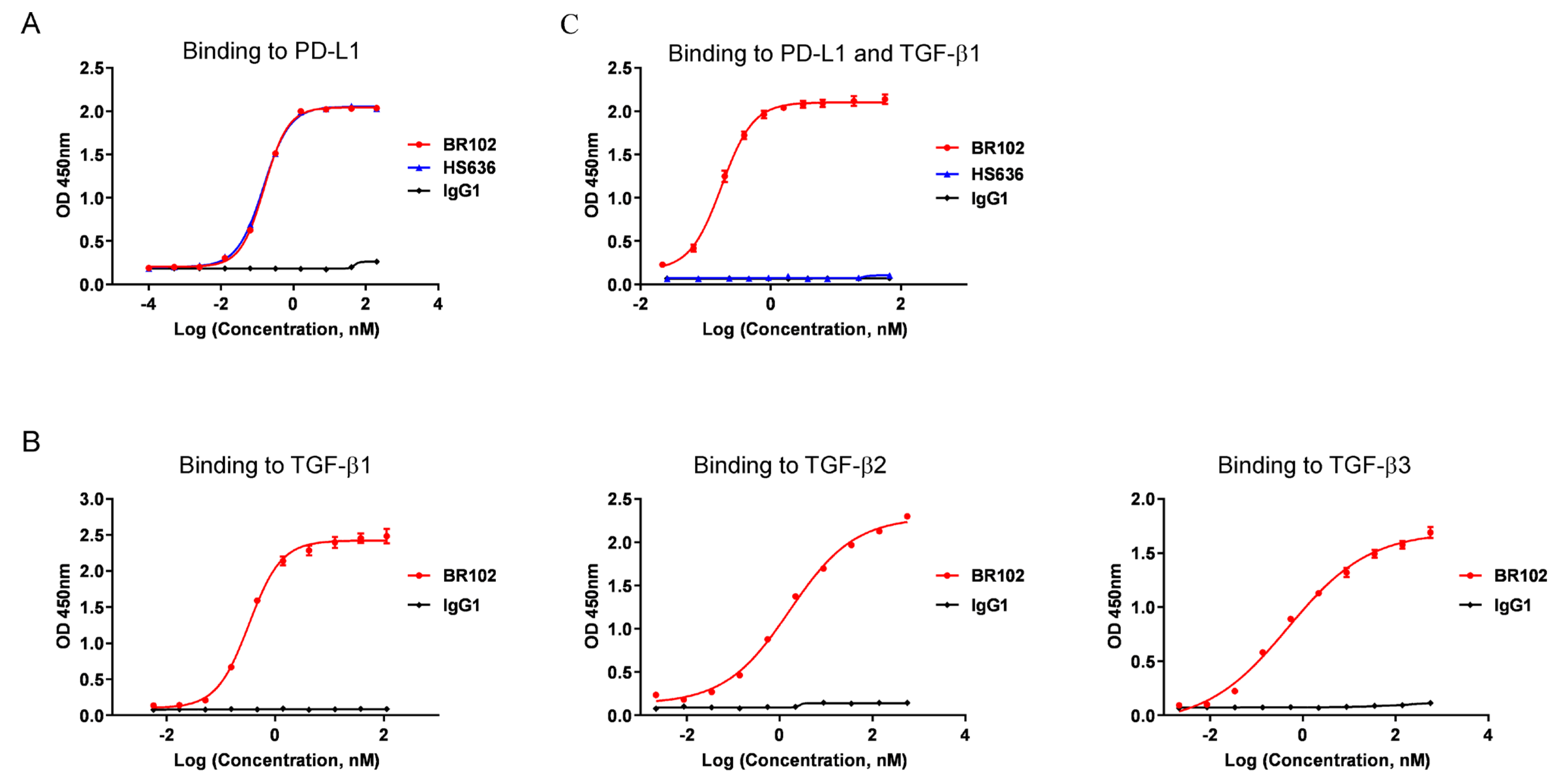
3.2. BR102 Could Simultaneously Recognize TGF-β and PD-L1
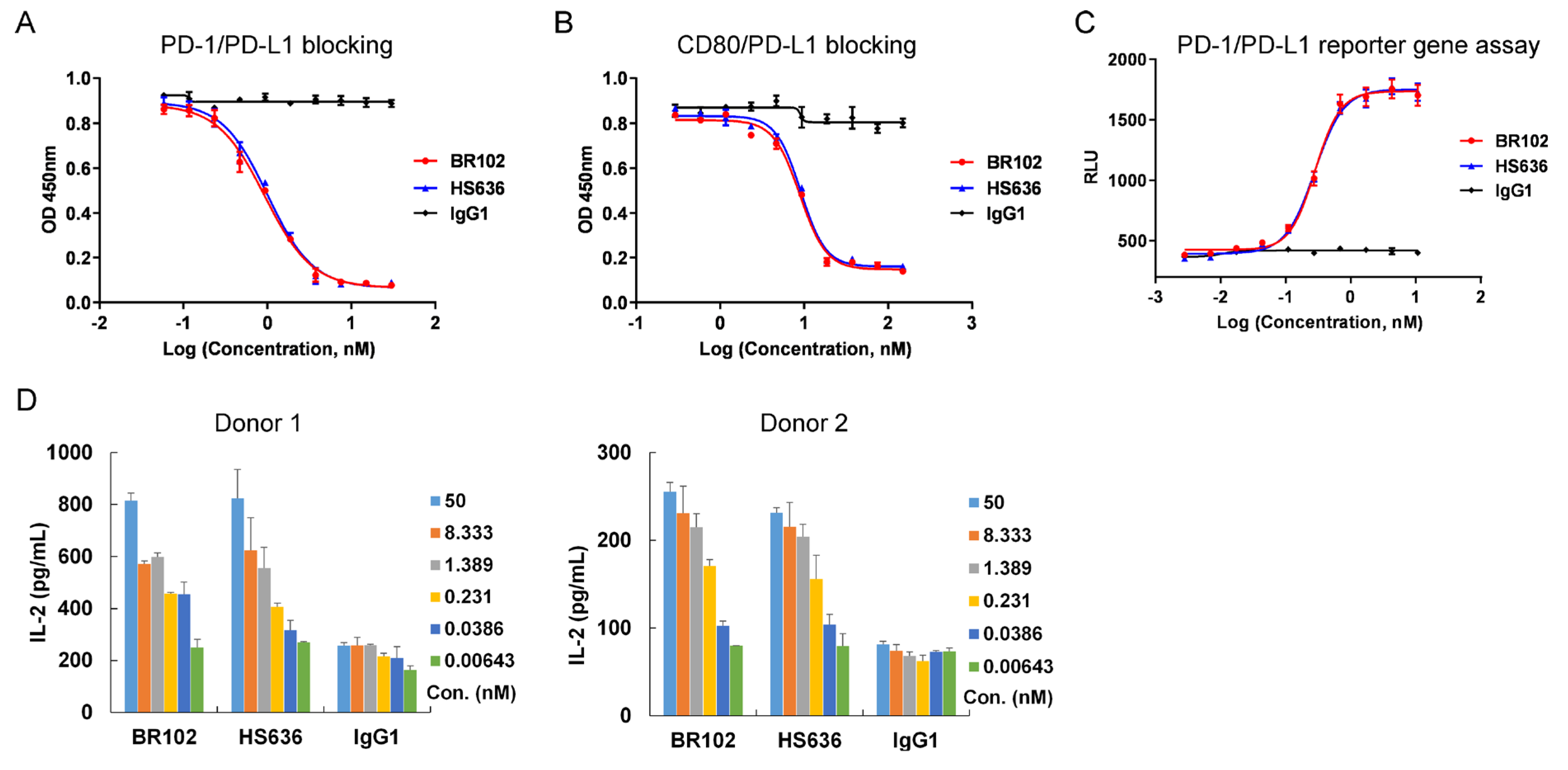
3.3. BR102 Disrupts PD-L1-Mediated Signal and Induces Activation of T Cell
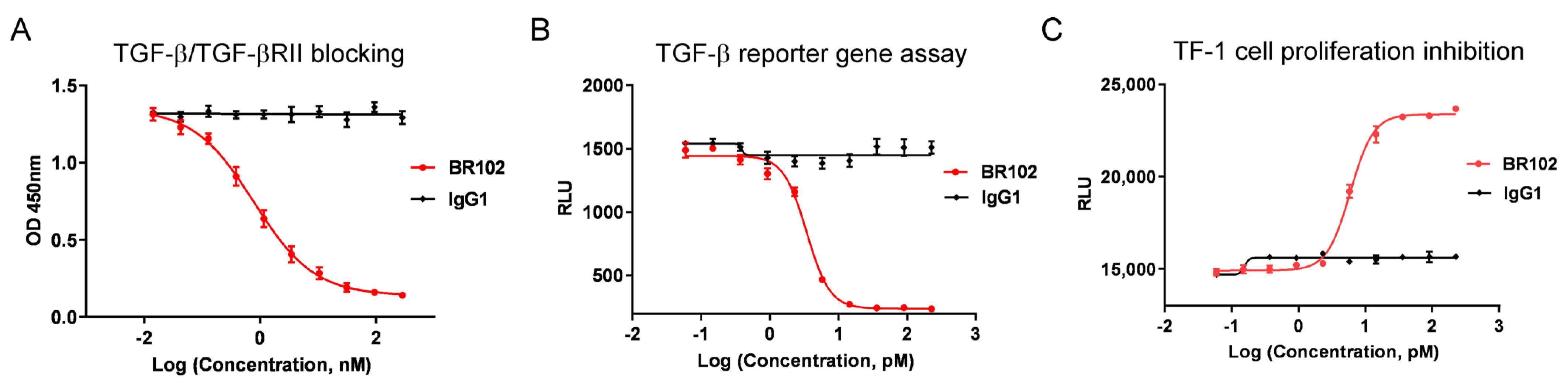
3.4. BR102 Inhibits TGF-β Signal In Vitro
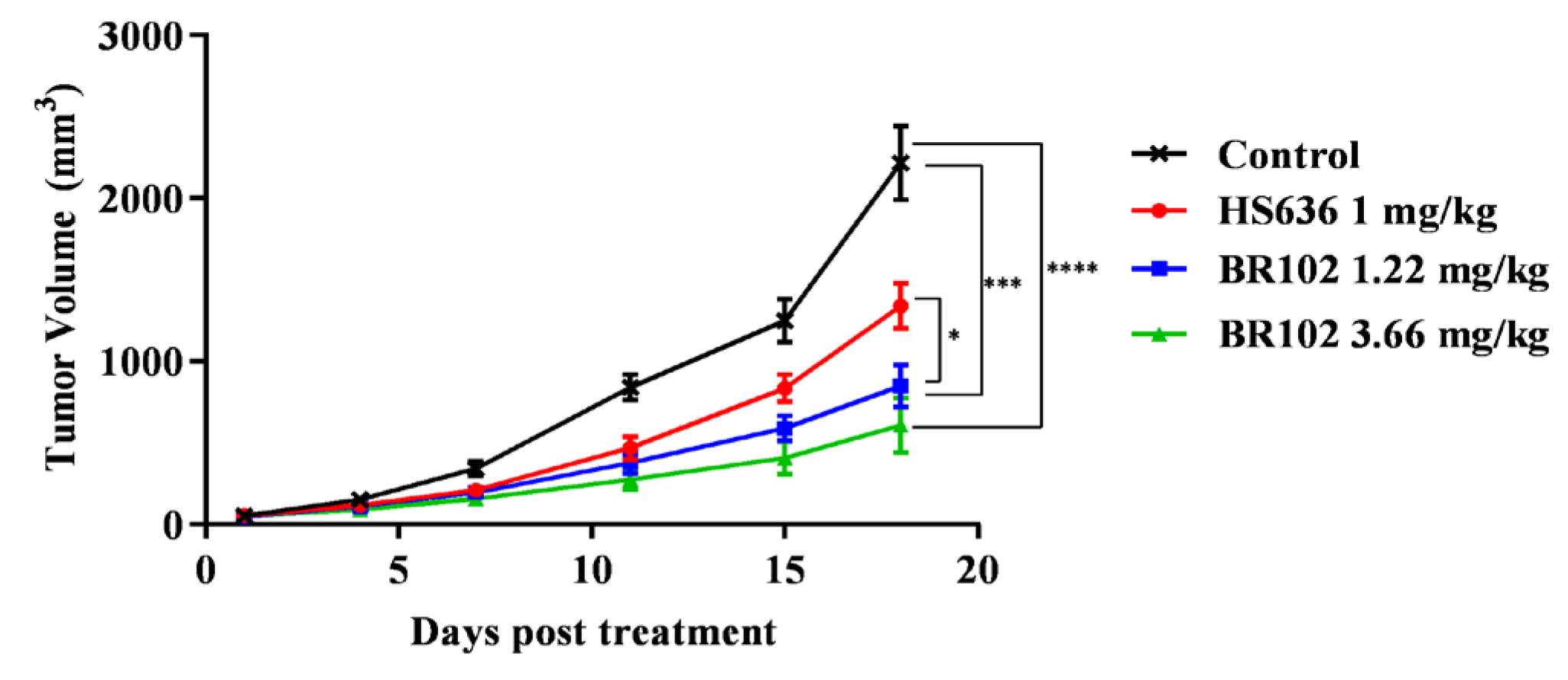
3.5. BR102 Treatment Inhibits Tumor Growth In Vivo
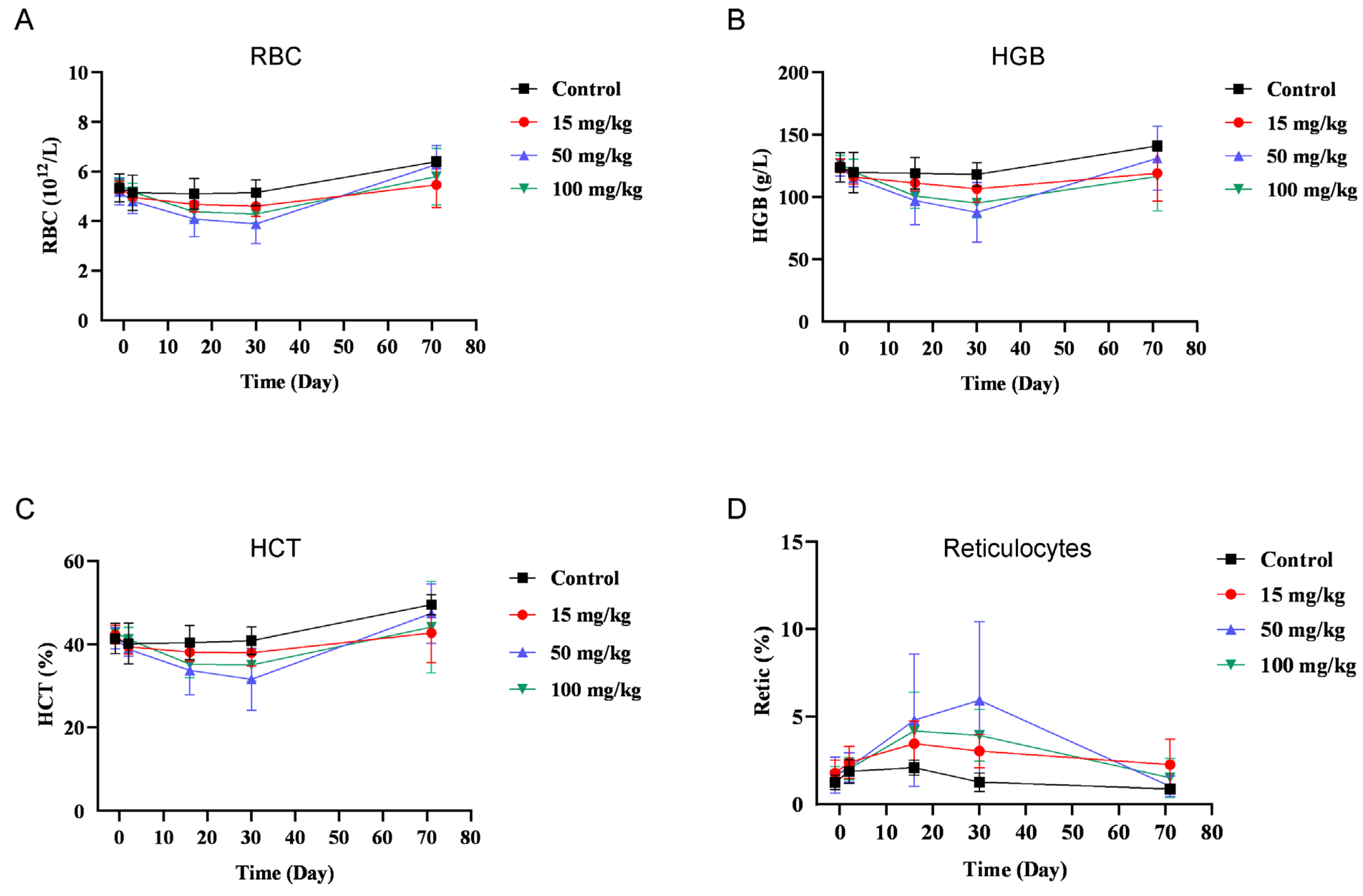
3.6. BR102 Shows A Favorable Safety Profile In Vivo
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Hamid, O.; Daud, A.; Wolchok, J.D.; Joshua, A.M.; Hwu, W.J.; Weber, J.S.; Gangadhar, T.C.; Joseph, R.W.; et al. Durable Complete Response After Discontinuation of Pembrolizumab in Patients With Metastatic Melanoma. J. Clin. Oncol. 2018, 36, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121–126. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef]
- Derynck, R.; Muthusamy, B.P.; Saeteurn, K.Y. Signaling pathway cooperation in TGF-beta-induced epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2014, 31, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-beta-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Lin, R.L.; Zhao, L.J. Mechanistic basis and clinical relevance of the role of transforming growth factor-beta in cancer. Cancer Biol. Med. 2015, 12, 385–393. [Google Scholar] [CrossRef]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical development of therapies targeting TGFbeta: Current knowledge and future perspectives. Ann. Oncol. 2020, 31, 1336–1349. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Dominguez, C.; McCampbell, K.K.; Gulley, J.L.; Schlom, J.; Palena, C. A novel bifunctional anti-PD-L1/TGF-beta Trap fusion protein (M7824) efficiently reverts mesenchymalization of human lung cancer cells. Oncoimmunology 2017, 6, e1349589. [Google Scholar] [CrossRef] [Green Version]
- Park, B.V.; Freeman, Z.T.; Ghasemzadeh, A.; Chattergoon, M.A.; Rutebemberwa, A.; Steigner, J.; Winter, M.E.; Huynh, T.V.; Sebald, S.M.; Lee, S.J.; et al. TGFbeta1-Mediated SMAD3 Enhances PD-1 Expression on Antigen-Specific T Cells in Cancer. Cancer Discov. 2016, 6, 1366–1381. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [Green Version]
- de Streel, G.; Bertrand, C.; Chalon, N.; Lienart, S.; Bricard, O.; Lecomte, S.; Devreux, J.; Gaignage, M.; De Boeck, G.; Marien, L.; et al. Selective inhibition of TGF-beta1 produced by GARP-expressing Tregs overcomes resistance to PD-1/PD-L1 blockade in cancer. Nat. Commun. 2020, 11, 4545. [Google Scholar] [CrossRef]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernandez, V.M.; et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Sci. Transl. Med. 2018, 10, eaan5488. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.Z.; Li, C.; Chen, G.; Yuan, J.J.; Zhou, Y.Q.; Jiao, J.Y.; Nie, L.; Qi, J.; Yang, Y.; Chen, S.Q.; et al. Optimization strategies for expression of a novel bifunctional anti-PD-L1/TGFBR2-ECD fusion protein. Protein. Expr. Purif. 2022, 189, 105973. [Google Scholar] [CrossRef]
- Mitra, M.S.; Lancaster, K.; Adedeji, A.O.; Palanisamy, G.S.; Dave, R.A.; Zhong, F.; Holdren, M.S.; Turley, S.J.; Liang, W.C.; Wu, Y.; et al. A Potent Pan-TGFbeta Neutralizing Monoclonal Antibody Elicits Cardiovascular Toxicity in Mice and Cynomolgus Monkeys. Toxicol. Sci. 2020, 175, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Anderton, M.J.; Mellor, H.R.; Bell, A.; Sadler, C.; Pass, M.; Powell, S.; Steele, S.J.; Roberts, R.R.; Heier, A. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol. Pathol. 2011, 39, 916–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.M.; Chen, D.S. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massague, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Viel, S.; Marcais, A.; Guimaraes, F.S.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef]
- Motz, G.T.; Coukos, G. Deciphering and reversing tumor immune suppression. Immunity 2013, 39, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.; Gara, S.K.; Achyut, B.R.; Li, Z.; Yan, H.H.; Day, C.P.; Weiss, J.M.; Trinchieri, G.; Morris, J.C.; Yang, L. TGF-beta signaling in myeloid cells is required for tumor metastasis. Cancer Discov. 2013, 3, 936–951. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFbeta biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef]
- Yi, M.; Zhang, J.; Li, A.; Niu, M.; Yan, Y.; Jiao, Y.; Luo, S.; Zhou, P.; Wu, K. The construction, expression, and enhanced anti-tumor activity of YM101: A bispecific antibody simultaneously targeting TGF-beta and PD-L1. J. Hematol. Oncol. 2021, 14, 27. [Google Scholar] [CrossRef]
- Strauss, J.; Heery, C.R.; Schlom, J.; Madan, R.A.; Cao, L.; Kang, Z.; Lamping, E.; Marte, J.L.; Donahue, R.N.; Grenga, I.; et al. Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD-L1 and TGFbeta, in Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 1287–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khasraw, M.; Weller, M.; Lorente, D.; Kolibaba, K.; Lee, C.K.; Gedye, C.; La Fuente, M.I.; Vicente, D.; Reardon, D.A.; Gan, H.K.; et al. Bintrafusp alfa (M7824), a bifunctional fusion protein targeting TGF-beta and PD-L1: Results from a phase I expansion cohort in patients with recurrent glioblastoma. Neurooncol. Adv. 2021, 3, vdab058. [Google Scholar] [CrossRef] [PubMed]
- Gueorguieva, I.; Cleverly, A.L.; Stauber, A.; Sada Pillay, N.; Rodon, J.A.; Miles, C.P.; Yingling, J.M.; Lahn, M.M. Defining a therapeutic window for the novel TGF-beta inhibitor LY2157299 monohydrate based on a pharmacokinetic/pharmacodynamic model. Br. J. Clin. Pharm. 2014, 77, 796–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, R.J.; Maldonado, G.; Azaro, A.; Fernandez, M.S.; Romero, F.L.; Sepulveda-Sanchez, J.M.; Corretti, M.; Carducci, M.; Dolan, M.; Gueorguieva, I.; et al. Cardiac Safety of TGF-beta Receptor I Kinase Inhibitor LY2157299 Monohydrate in Cancer Patients in a First-in-Human Dose Study. Cardiovasc. Toxicol. 2015, 15, 309–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodon, J.; Carducci, M.A.; Sepulveda-Sanchez, J.M.; Azaro, A.; Calvo, E.; Seoane, J.; Brana, I.; Sicart, E.; Gueorguieva, I.; Cleverly, A.L.; et al. First-in-human dose study of the novel transforming growth factor-beta receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin. Cancer Res. 2015, 21, 553–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacouture, M.E.; Morris, J.C.; Lawrence, D.P.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Berzofsky, J.A.; Hsu, F.J.; Guitart, J. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor beta by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol. Immunother. 2015, 64, 437–446. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Berlin, J.D.; Cosaert, J.; Kauh, J.; Chan, E.; Piha-Paul, S.A.; Amaya, A.; Tang, S.; Driscoll, K.; Kimbung, R.; et al. A phase 1 study of anti-TGFbeta receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 673–680. [Google Scholar] [CrossRef] [Green Version]
- de Miranda, N.F.; van Dinther, M.; van den Akker, B.E.; van Wezel, T.; ten Dijke, P.; Morreau, H. Transforming Growth Factor beta Signaling in Colorectal Cancer Cells with Microsatellite Instability Despite Biallelic Mutations in TGFBR2. Gastroenterology 2015, 148, 1427–1437.e8. [Google Scholar] [CrossRef] [Green Version]
- Yakicier, M.C.; Irmak, M.B.; Romano, A.; Kew, M.; Ozturk, M. Smad2 and Smad4 gene mutations in hepatocellular carcinoma. Oncogene 1999, 18, 4879–4883. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.J.; Datta, A.; Littlefield, C.; Kalra, A.; Chapron, C.; Wawersik, S.; Dagbay, K.B.; Brueckner, C.T.; Nikiforov, A.; Danehy, F.T., Jr.; et al. Selective inhibition of TGFbeta1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci. Transl. Med. 2020, 12, eaay8456. [Google Scholar] [CrossRef]
- Rodon, L.; Gonzalez-Junca, A.; Inda Mdel, M.; Sala-Hojman, A.; Martinez-Saez, E.; Seoane, J. Active CREB1 promotes a malignant TGFbeta2 autocrine loop in glioblastoma. Cancer Discov. 2014, 4, 1230–1241. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Yan, M.; Zhang, J.; Wang, X.; Shen, Z.; Lv, Z.; Li, Z.; Wei, W.; Chen, W. TGFbeta3-mediated induction of Periostin facilitates head and neck cancer growth and is associated with metastasis. Sci. Rep. 2016, 6, 20587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, A.; Lahn, M.M.; Williams, K.E.; Cleverly, A.L.; Pitou, C.; Kadam, S.K.; Farmen, M.W.; Desaiah, D.; Raju, R.; Conkling, P.; et al. A phase I dose-escalation study to a predefined dose of a transforming growth factor-beta1 monoclonal antibody (TbetaM1) in patients with metastatic cancer. Int. J. Oncol. 2014, 45, 2221–2231. [Google Scholar] [CrossRef] [Green Version]
- De Sousa, E.M.F.; Wang, X.; Jansen, M.; Fessler, E.; Trinh, A.; de Rooij, L.P.; de Jong, J.H.; de Boer, O.J.; van Leersum, R.; Bijlsma, M.F.; et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat. Med. 2013, 19, 614–618. [Google Scholar] [CrossRef]
- Fessler, E.; Drost, J.; van Hooff, S.R.; Linnekamp, J.F.; Wang, X.; Jansen, M.; De Sousa, E.M.F.; Prasetyanti, P.R.; JE, I.J.; Franitza, M.; et al. TGFbeta signaling directs serrated adenomas to the mesenchymal colorectal cancer subtype. EMBO Mol. Med. 2016, 8, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Coulouarn, C.; Factor, V.M.; Thorgeirsson, S.S. Transforming growth factor-beta gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology 2008, 47, 2059–2067. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.-H.; Li, N.; Gao, Z.-Z.; Chen, G.; Nie, L.; Zhou, Y.-Q.; Jiang, M.-Z.; Chen, Y.; Chen, J.; Mei, X.-F.; et al. Development of the Novel Bifunctional Fusion Protein BR102 That Simultaneously Targets PD-L1 and TGF-β for Anticancer Immunotherapy. Cancers 2022, 14, 4964. https://doi.org/10.3390/cancers14194964
Wu Z-H, Li N, Gao Z-Z, Chen G, Nie L, Zhou Y-Q, Jiang M-Z, Chen Y, Chen J, Mei X-F, et al. Development of the Novel Bifunctional Fusion Protein BR102 That Simultaneously Targets PD-L1 and TGF-β for Anticancer Immunotherapy. Cancers. 2022; 14(19):4964. https://doi.org/10.3390/cancers14194964
Chicago/Turabian StyleWu, Zhen-Hua, Na Li, Zhang-Zhao Gao, Gang Chen, Lei Nie, Ya-Qiong Zhou, Mei-Zhu Jiang, Yao Chen, Juan Chen, Xiao-Fen Mei, and et al. 2022. "Development of the Novel Bifunctional Fusion Protein BR102 That Simultaneously Targets PD-L1 and TGF-β for Anticancer Immunotherapy" Cancers 14, no. 19: 4964. https://doi.org/10.3390/cancers14194964
APA StyleWu, Z. -H., Li, N., Gao, Z. -Z., Chen, G., Nie, L., Zhou, Y. -Q., Jiang, M. -Z., Chen, Y., Chen, J., Mei, X. -F., Hu, F., & Wang, H. -B. (2022). Development of the Novel Bifunctional Fusion Protein BR102 That Simultaneously Targets PD-L1 and TGF-β for Anticancer Immunotherapy. Cancers, 14(19), 4964. https://doi.org/10.3390/cancers14194964