
Aberrant Methylation of SLIT2 Gene in Plasma Cell-Free DNA of Non-Small Cell Lung Cancer Patients

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population and Samples
2.2. Analysis of SLITs Methylation
2.3. Feature Selection and Model Building for Lung Cancer Prediction
2.4. Analysis of SLIT2 Methylation in Plasma
2.5. Statistical Analysis
3. Results
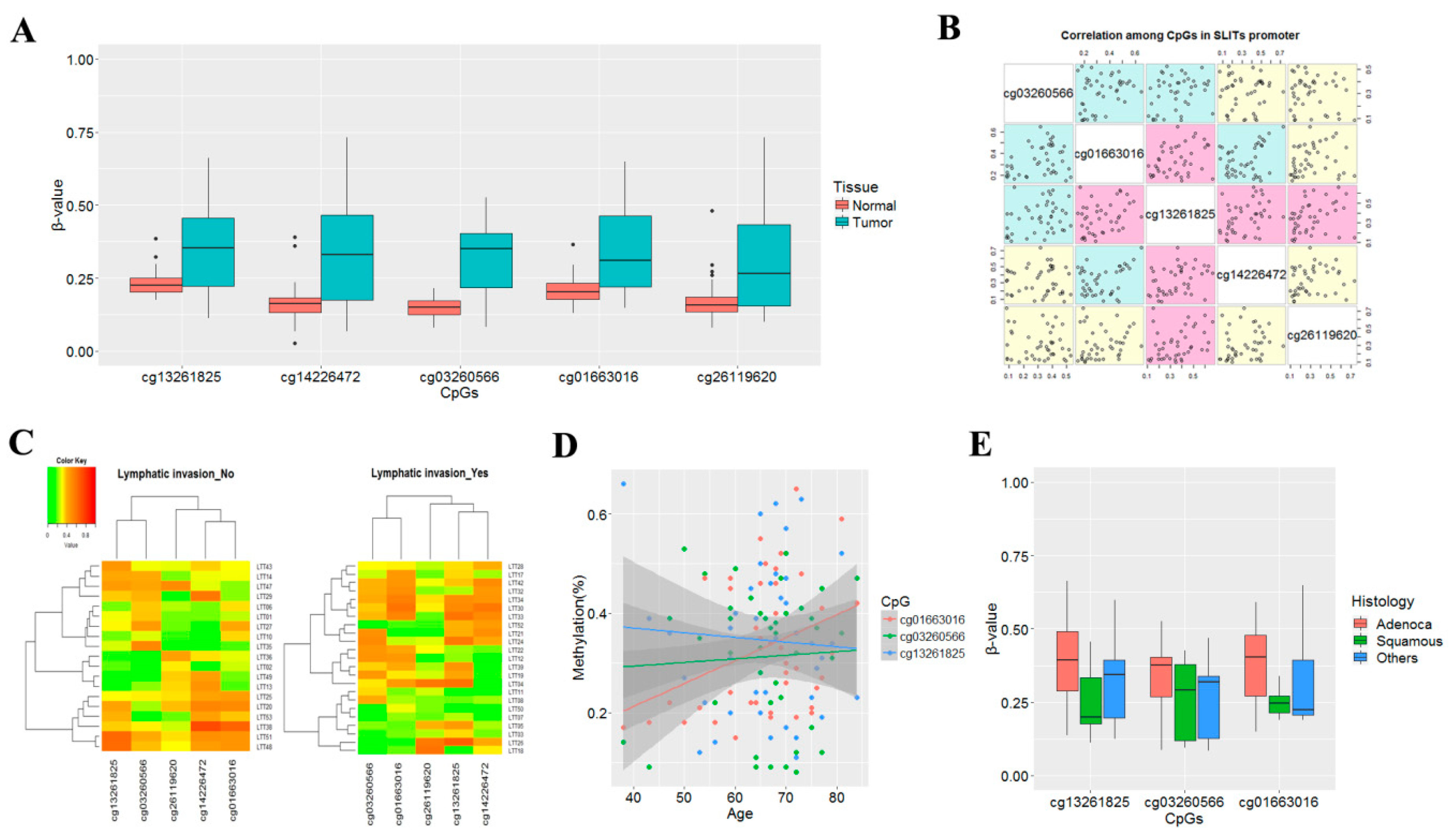
3.1. Aberrant Methylation of SLIT Genes in Primary Non-Small Cell Lung Cancer
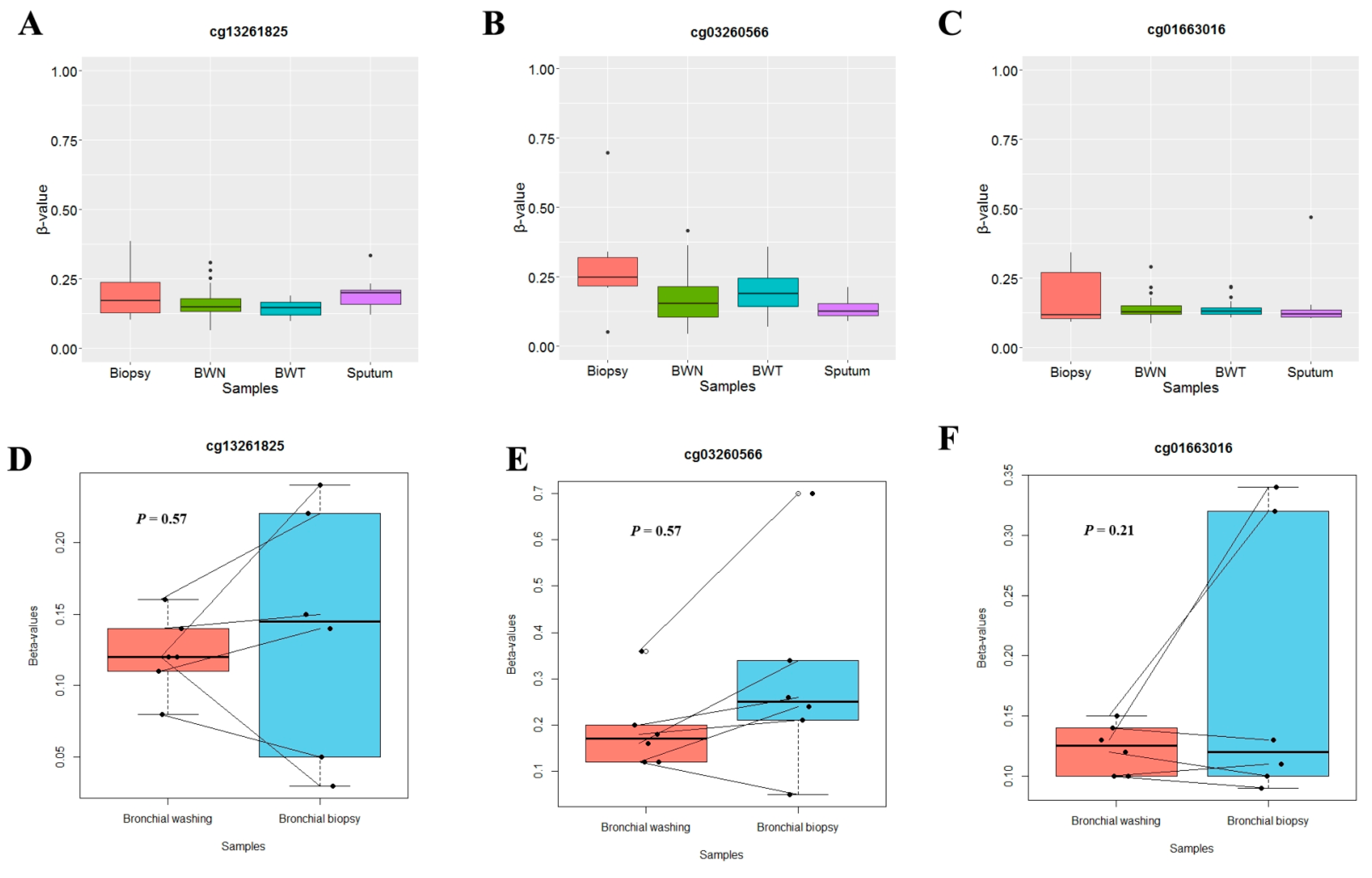
3.2. Aberrant Methylation of SLIT Genes in Bronchial Washing, Biopsy, and Sputum Samples
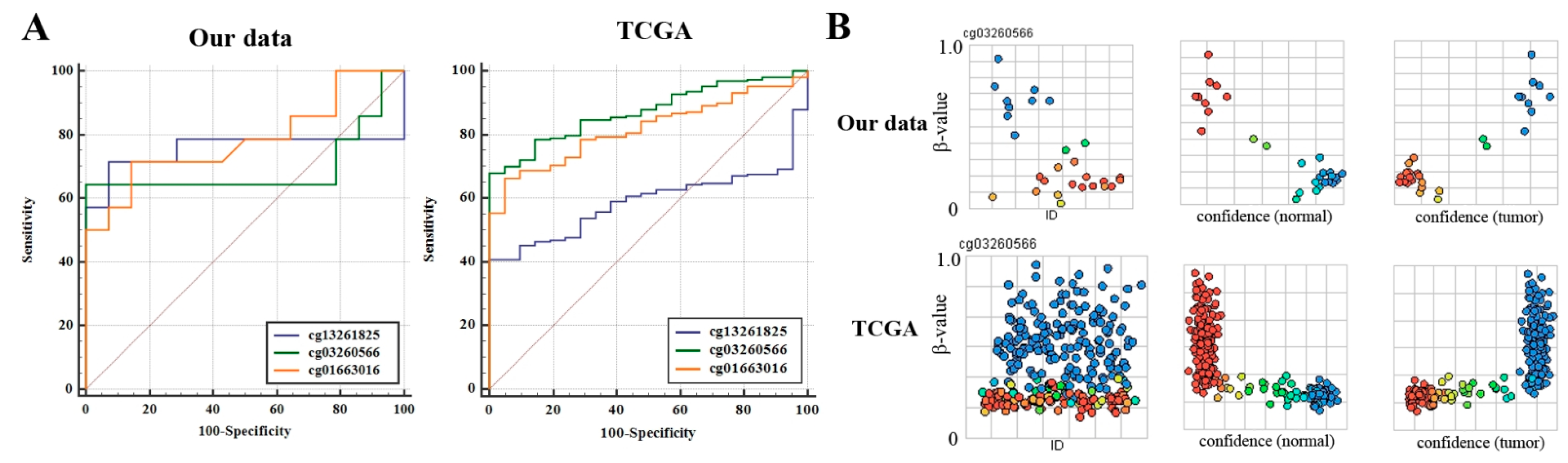
3.3. Prediction of NSCLC Using Methylation Levels of SLIT Genes in Lung Tumor Tissues
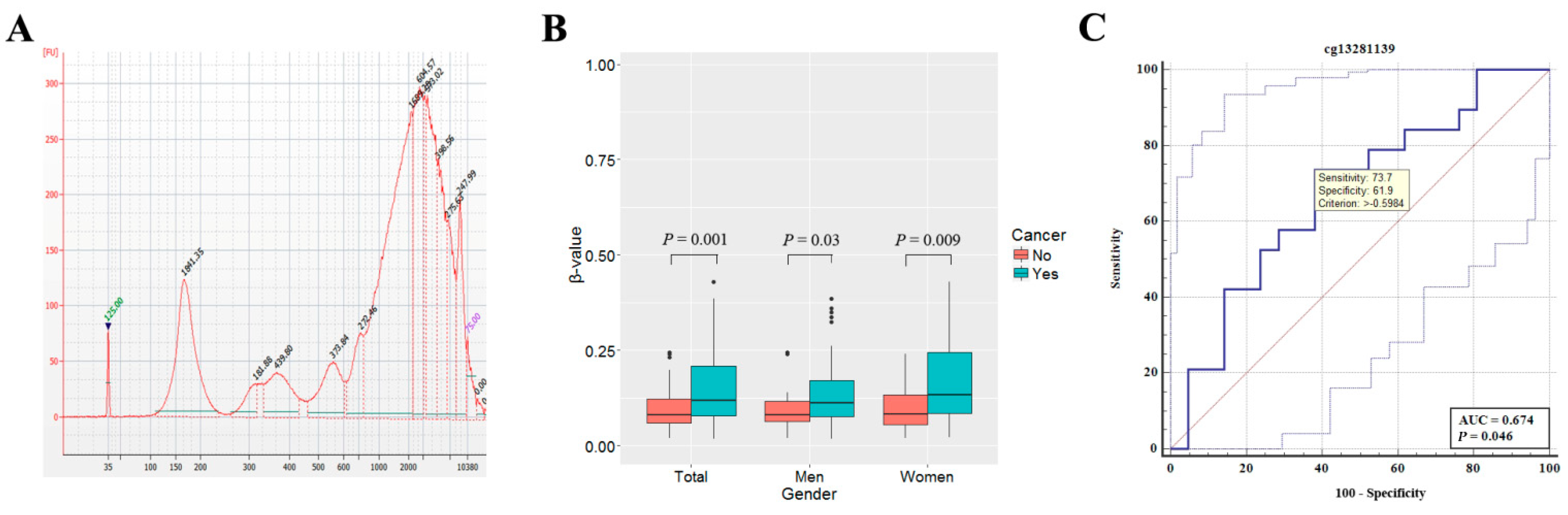
3.4. Aberrant Methylation of SLIT2 in Plasma Cell-Free DNA of Patients with NSCLC
3.5. SLIT2 Hypermethylation in Plasma Cell-Free DNA Is Associated with Poor Recurrence-Free Survival of NSCLC Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.; Jun, T.; Nie, Y.; Hao, J.; Fan, D. The Role of the Slit/Robo Signaling Pathway. J. Cancer 2019, 10, 2694–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Liang, G.; Xiao, Y.; Qin, T.; Chen, X.; Wu, E.; Ma, Q.; Wang, Z. Targeting the SLIT/ROBO pathway in tumor progression: Molecular mechanisms and therapeutic perspectives. Ther. Adv. Med. Oncol. 2019, 11, 1758835919855238. [Google Scholar] [CrossRef]
- Marlow, R.; Strickland, P.; Lee, J.S.; Wu, X.; PeBenito, M.; Binnewies, M.; Hinck, L. SLITs suppress tumor growth in vivo by silencing Sdf1/Cxcr4 within breast epithelium. Cancer Res. 2008, 68, 7819–7827. [Google Scholar] [CrossRef] [Green Version]
- Yuasa-Kawada, J.; Kinoshita-Kawada, M.; Rao, Y.; Wu, J.Y. Deubiquitinating enzyme USP33/VDU1 is required for Slit signaling in inhibiting breast cancer cell migration. Proc. Natl. Acad. Sci. USA 2009, 106, 14530–14535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, P.-H.; Hwang-Verslues, W.W.; Chang, Y.-C.; Chen, C.-C.; Hsiao, M.; Jeng, Y.-M.; Chang, K.-J.; Lee, E.Y.-H.; Shew, J.-Y.; Lee, W.-H. Activation of Robo1 signaling of breast cancer cells by Slit2 from stromal fibroblast restrains tumorigenesis via blocking PI3K/Akt/β-catenin pathway. Cancer Res. 2012, 72, 4652–4661. [Google Scholar] [CrossRef] [Green Version]
- Dallol, A.; Morton, D.; Maher, E.R.; Latif, F. SLIT2 axon guidance molecule is frequently inactivated in colorectal cancer and suppresses growth of colorectal carcinoma cells. Cancer Res. 2003, 63, 1054–1058. [Google Scholar] [PubMed]
- Huang, Z.; Wen, P.; Kong, R.; Cheng, H.; Zhang, B.; Quan, C.; Bian, Z.; Chen, M.; Zhang, Z.; Chen, X.; et al. USP33 mediates Slit-Robo signaling in inhibiting colorectal cancer cell migration. Int. J. Cancer 2015, 136, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Yang, Z.; Liu, W.; Liu, B.; Xu, Z.; Zhang, Z. Knockdown of Slit2 promotes growth and motility in gastric cancer cells via activation of AKT/β-catenin. Oncol. Rep. 2014, 31, 812–818. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Wang, L.; Xu, Z.; Kong, R.; Wang, F.; Yin, K.; Xu, J.; Li, B.; He, Z.; Wang, L.; et al. Reduced USP33 expression in gastric cancer decreases inhibitory effects of Slit2-Robo1 signalling on cell migration and EMT. Cell Prolif. 2019, 52, e12606. [Google Scholar] [CrossRef] [Green Version]
- Stella, M.C.; Trusolino, L.; Comoglio, P.M. The Slit/Robo system suppresses hepatocyte growth factor-dependent invasion and morphogenesis. Mol. Biol. Cell 2009, 20, 642–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Ning, Z.; Wang, A.; Chen, D.; Liu, X.; Xia, T.; Tekcham, D.S.; Wang, W.; Li, T.; Liu, X.; et al. USP10 suppresses tumor progression by inhibiting mTOR activation in hepatocellular carcinoma. Cancer Lett. 2018, 436, 139–148. [Google Scholar] [CrossRef]
- Tseng, R.C.; Lee, S.H.; Hsu, H.S.; Chen, B.H.; Tsai, W.C.; Tzao, C.; Wang, Y.C. SLIT2 attenuation during lung cancer progression deregulates beta-catenin and E-cadherin and associates with poor prognosis. Cancer Res. 2010, 70, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Kong, R.; Yi, F.; Wen, P.; Liu, J.; Chen, X.; Ren, J.; Li, X.; Shang, Y.; Nie, Y.; Wu, K.; et al. Myo9b is a key player in SLIT/ROBO-mediated lung tumor suppression. J. Clin. Investif. 2015, 125, 4407–4420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Li, T.; Zhao, Y.; Huang, L.; Sun, H.; Wu, H.; Jiang, X. USP10 inhibits lung cancer cell growth and invasion by upregulating PTEN. Mol. Cell Biochem. 2018, 441, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, S.; Bao, H.; Mu, S.; Zhang, B.; Ma, H.; Ma, S. MicroRNA-365 promotes lung carcinogenesis by downregulating the USP33/SLIT2/ROBO1 signalling pathway. Cancer Cell Int. 2018, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.J.; Lim, S.; You, M.-H.; Park, Y.; Song, D.E.; Sim, S.; Kim, T.Y.; Shong, Y.K.; Kim, W.B. The role of Slit2 as a tumor suppressor in thyroid cancer. Mol. Cell Endocrinol. 2019, 483, 87–96. [Google Scholar] [CrossRef]
- Narayan, G.; Goparaju, C.; Arias-Pulido, H.; Kaufmann, A.M.; Schneider, A.; Dürst, M.; Murty, V.V. Promoter hypermethylation-mediated inactivation of multiple Slit-Robo pathway genes in cervical cancer progression. Mol. Cancer 2006, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Beggs, A.D.; Jones, A.; El-Bahrawy, M.; Abulafi, M.; Hodgson, S.V.; Tomlinson, I.P. Whole-genome methylation analysis of benign and malignant colorectal tumours. J. Pathol. 2013, 229, 697–704. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, R.E.; Dallol, A.; Bieche, I.; Krex, D.; Morton, D.; Maher, E.R.; Latif, F. Epigenetic inactivation of SLIT3 and SLIT1 genes in human cancers. Br. J. Cancer 2004, 91, 2071–2078. [Google Scholar] [CrossRef] [Green Version]
- Dallol, A.; Forgacs, E.; Martinez, A.; Sekido, Y.; Walker, R.L.; Kishida, T.; Rabbitts, P.H.; Maher, E.R.; Minna, J.D.; Latif, F. Tumour specific promoter region methylation of the human homologue of the Drosophila Roundabout gene DUTT1 (ROBO1) in human cancers. Oncogene 2002, 21, 3020–3028. [Google Scholar] [CrossRef] [Green Version]
- Dammann, R.; Strunnikova, M.; Schagdarsurengin, U.; Rastetter, M.; Papritz, M.; Hattenhorst, U.E.; Hofmann, H.-S.; Silber, R.-E.; Burdach, S.; Hansen, G. CpG island methylation and expression of tumour-associated genes in lung carcinoma. Eur. J. Cancer 2005, 41, 1223–1236. [Google Scholar] [CrossRef]
- Kwon, Y.-J.; Lee, S.J.; Koh, J.S.; Kim, S.H.; Lee, H.W.; Kang, M.C.; Bae, J.B.; Kim, Y.-J.; Park, J.H. Genome-wide analysis of DNA methylation and the gene expression change in lung cancer. J. Thorac. Oncol. 2012, 7, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Yu, X.-F.; Ouyang, N.; Luo, Q.-L.; Zhao, S.-Y.; Guan, X.-F.; Chen, T.; Li, J.-X. Multi-platform analysis of methylation-regulated genes in human lung adenocarcinoma. J. Toxicol. Environ. Health A 2019, 82, 37–45. [Google Scholar] [CrossRef]
- Um, S.W.; Kim, H.K.; Kim, Y.; Lee, B.B.; Kim, D.; Han, J.; Kim, D.H. Bronchial biopsy specimen as a surrogate for DNA methylation analysis in inoperable lung cancer. Clin. Epigenet. 2017, 9, 131. [Google Scholar] [CrossRef]
- Kim, J.S.; Han, J.; Shim, Y.M.; Park, J.; Kim, D.H. Aberrant methylation of H-cadherin (CDH13) promoter is associated with tumor progression in primary nonsmall cell lung carcinoma. Cancer 2005, 104, 1825–1833. [Google Scholar]
- Edge, S.B.; Byrd, D.R.; Compton, C.C.; Fritz, A.G.; Greene, F.L.; Troth, A. American Joint Committee on Cancer. In AJCC Cancer Staging Manual, 7th ed.; Springer: New York, NY, USA, 2010; pp. 253–270. [Google Scholar]
- Samejima, K.; Earnshaw, W.C. Trashing the genome: The role of nucleases during apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.; Baranova, A.; Butler, T.; Spellman, P.; Mileyko, V. Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genom. 2015, 16, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Bin Lee, B.; Kim, D.; Um, S.; Cho, E.Y.; Han, J.; Shim, Y.M.; Kim, D. Clinicopathological significance of RUNX1 in non-small cell lung cancer. J. Clin. Med. 2020, 9, 1694. [Google Scholar] [CrossRef]
- Tseng, R.-C.; Chang, J.-M.; Chen, J.-H.; Huang, W.-R.; Tang, Y.-A.; Kuo, I.-Y.; Yan, J.-J.; Lai, W.-W.; Wang, Y.-C. Deregulation of SLIT2-mediated Cdc42 activity is associated with esophageal cancer metastasis and poor prognosis. J. Thorac. Oncol. 2015, 10, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Qin, F.; Zhang, H.; Ma, L.; Liu, X.; Dai, K.; Li, W.; Gu, F.; Fu, L.; Ma, Y. Low expression of Slit2 and Robo 1 is associated with poor prognosis and brain-specific metastasis of breast cancer patients. Sci. Rep. 2015, 5, 14430. [Google Scholar] [CrossRef]
- Liu, L.; Li, W.; Geng, S.; Fang, Y.; Sun, Z.; Hu, H.; Liang, Z.; Yan, Z. Slit2 and Robo1 expression as biomarkers for assessing prognosis in brain glioma patients. Surg. Oncol. 2016, 25, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, G.; Talima, S.; Li, L.; Wei, W.; Rudzki, Z.; Allam, R.M.; Simmons, W.; Tao, Q.; Murray, P.G. Low expression and promoter hypermethylation of the tumour suppressor SLIT2, are associated with adverse patient outcomes in diffuse large B cell lymphoma. Pathol. Oncol. Res. 2019, 25, 1223–1231. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Guo, H.; Li, B.; Sui, C.; Zhang, Y.; Xia, X.; Qin, Y.; Ye, L.; Xie, F.; Wang, H.; et al. Effects of Slit3 silencing on the invasive ability of lung carcinoma A549 cells. Oncol. Rep. 2015, 34, 952–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risberg, B.; Tsui, D.W.; Biggs, H.; de Almagro, A.R.-V.M.; Dawson, S.-J.; Hodgkin, C.; Jones, L.; Parkinson, C.; Piskorz, A.; Marass, F.; et al. Effects of collection and procession on plasma circulating cell-free DNA from cancer patients. J. Mol. Diagn. 2018, 20, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerachian, M.A.; Azghandi, M.; Mozaffari-Jovin, S.; Thierry, A.R. Guidelines for pre-analytical conditions for assessing the methylation of circulating cell-free DNA. Clin. Epigenet. 2021, 13, 193. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Survival | SLIT2 Hypermethylation | HR | 95% CI | p-Value |
|---|---|---|---|---|
| Overall survival | No | 1.00 | - | - |
| Yes | 1.84 | 0.76–5.11 | 0.38 | |
| Recurrence-free survival | No | 1.00 | - | - |
| Yes | 2.19 | 1.21–4.36 | 0.01 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Lee, B.B.; Kim, D.; Um, S.-W.; Han, J.; Shim, Y.M.; Kim, D.-H. Aberrant Methylation of SLIT2 Gene in Plasma Cell-Free DNA of Non-Small Cell Lung Cancer Patients. Cancers 2022, 14, 296. https://doi.org/10.3390/cancers14020296
Kim Y, Lee BB, Kim D, Um S-W, Han J, Shim YM, Kim D-H. Aberrant Methylation of SLIT2 Gene in Plasma Cell-Free DNA of Non-Small Cell Lung Cancer Patients. Cancers. 2022; 14(2):296. https://doi.org/10.3390/cancers14020296
Chicago/Turabian StyleKim, Yujin, Bo Bin Lee, Dongho Kim, Sang-Won Um, Joungho Han, Young Mog Shim, and Duk-Hwan Kim. 2022. "Aberrant Methylation of SLIT2 Gene in Plasma Cell-Free DNA of Non-Small Cell Lung Cancer Patients" Cancers 14, no. 2: 296. https://doi.org/10.3390/cancers14020296
APA StyleKim, Y., Lee, B. B., Kim, D., Um, S. -W., Han, J., Shim, Y. M., & Kim, D. -H. (2022). Aberrant Methylation of SLIT2 Gene in Plasma Cell-Free DNA of Non-Small Cell Lung Cancer Patients. Cancers, 14(2), 296. https://doi.org/10.3390/cancers14020296