
STAT3 Enhances Sensitivity of Glioblastoma to Drug-Induced Autophagy-Dependent Cell Death


 , , , ,
, , , , 
Abstract
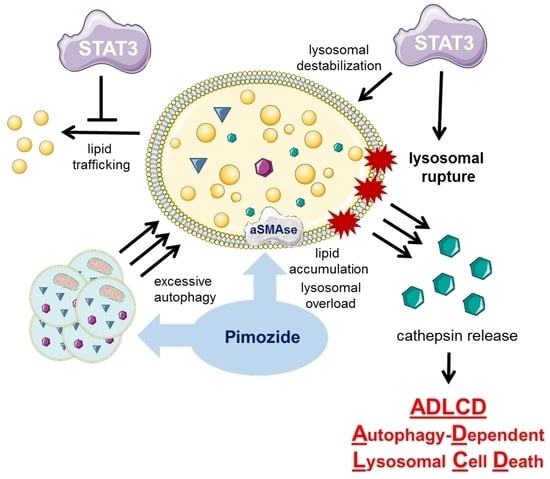
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Knockout of STAT3 Attenuates Autophagy-Dependent Cell Death
2.2. Knockout of STAT3 Confers Sensitivity to Lysosomal Membrane Permeabilization
3. Discussion
4. Materials and Methods
4.1. Cells and Cell Culture
4.2. Compounds, Antibodies and Taqman-Probes
4.3. SDS-PAGE and Western Blot
4.4. Cell-Based Assays
4.5. Flow Cytometry-Based Assays
4.6. Immunofluorescence Microscopy
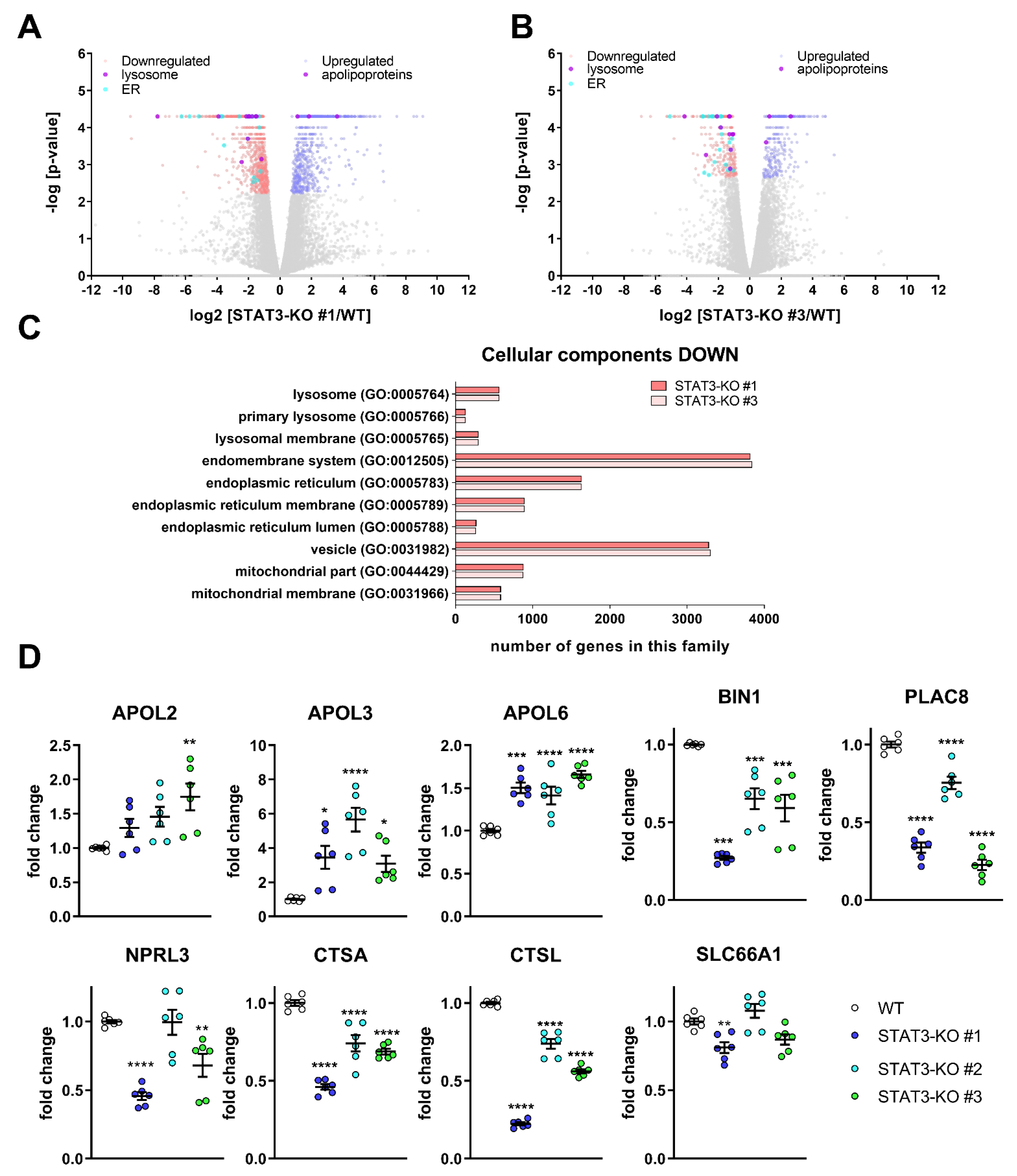
4.7. Whole Transcriptome Analyses
4.8. Taqman-Based Gene Expression Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, K.P.; Yu, J. Status quo—standard-of-care medical and radiation therapy for glioblastoma. Cancer J. 2012, 18, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Olar, A.; Aldape, K.D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 2014, 232, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef]
- Hossain, A.; Gumin, J.; Gao, F.; Figueroa, J.; Shinojima, N.; Takezaki, T.; Priebe, W.; Villarreal, D.; Kang, S.G.; Joyce, C.; et al. Mesenchymal stem cells isolated from human gliomas increase proliferation and maintain stemness of glioma stem cells through the IL-6/gp130/STAT3 pathway. Stem Cells 2015, 33, 2400–2415. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Galoczova, M.; Coates, P.; Vojtesek, B. STAT3, stem cells, cancer stem cells and p63. Cell. Mol. Biol. Lett. 2018, 23, 12. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Patel, M.; Ruzevick, J.; Jackson, C.M.; Lim, M. STAT3 Activation in glioblastoma: Biochemical and therapeutic implications. Cancers 2014, 6, 376–395. [Google Scholar] [CrossRef] [Green Version]
- Brantley, E.C.; Benveniste, E.N. Signal transducer and activator of transcription-3: A molecular hub for signaling pathways in gliomas. Mol. Cancer Res. 2008, 6, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Luwor, R.B.; Baradaran, B.; Taylor, L.E.; Iaria, J.; Nheu, T.V.; Amiry, N.; Hovens, C.M.; Wang, B.; Kaye, A.H.; Zhu, H.J. Targeting Stat3 and Smad7 to restore TGF-beta cytostatic regulation of tumor cells in vitro and in vivo. Oncogene 2013, 32, 2433–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermarck, J.; Kahari, V.M. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 1999, 13, 781–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendt, M.K.; Balanis, N.; Carlin, C.R.; Schiemann, W.P. STAT3 and epithelial-mesenchymal transitions in carcinomas. JAK-STAT 2014, 3, e28975. [Google Scholar] [CrossRef] [Green Version]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Esposito, C.L.; Nuzzo, S.; Catuogno, S.; Romano, S.; de Nigris, F.; de Franciscis, V. STAT3 gene silencing by aptamer-siRNA chimera as selective therapeutic for glioblastoma. Mol. Ther. Nucleic Acids 2018, 10, 398–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiprianova, I.; Remy, J.; Milosch, N.; Mohrenz, I.V.; Seifert, V.; Aigner, A.; Kogel, D. Sorafenib sensitizes glioma cells to the BH3 mimetic ABT-737 by targeting MCL1 in a STAT3-dependent manner. Neoplasia 2015, 17, 564–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priester, M.; Copanaki, E.; Vafaizadeh, V.; Hensel, S.; Bernreuther, C.; Glatzel, M.; Seifert, V.; Groner, B.; Kogel, D.; Weissenberger, J. STAT3 silencing inhibits glioma single cell infiltration and tumor growth. Neuro-Oncology 2013, 15, 840–852. [Google Scholar] [CrossRef] [Green Version]
- Senft, C.; Priester, M.; Polacin, M.; Schroder, K.; Seifert, V.; Kogel, D.; Weissenberger, J. Inhibition of the JAK-2/STAT3 signaling pathway impedes the migratory and invasive potential of human glioblastoma cells. J. Neuro-Oncol. 2011, 101, 393–403. [Google Scholar] [CrossRef]
- Sherry, M.M.; Reeves, A.; Wu, J.L.K.; Cochran, B.H. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Kaisho, T.; Yoshida, N.; Takeda, J.; Kishimoto, T.; Akira, S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: Generation and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 1998, 161, 4652–4660. [Google Scholar] [CrossRef] [Green Version]
- Linder, B.; Weirauch, U.; Ewe, A.; Uhmann, A.; Seifert, V.; Mittelbronn, M.; Harter, P.N.; Aigner, A.; Kogel, D. Therapeutic targeting of Stat3 using lipopolyplex nanoparticle-formulated siRNA in a syngeneic orthotopic mouse glioma model. Cancers 2019, 11, 333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Munoz, A.R.; Chan, D.; Ghosh, R.; Kumar, A.P. STAT3 down regulates LC3 to inhibit autophagy and pancreatic cancer cell growth. Oncotarget 2014, 5, 2529–2541. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Hu, L.; Cogdell, D.; Lu, L.; Gao, C.; Tian, W.; Zhang, Z.; Kang, Y.; Fleming, J.B.; Zhang, W. MIR506 induces autophagy-related cell death in pancreatic cancer cells by targeting the STAT3 pathway. Autophagy 2017, 13, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.E.; Lee, H.G.; Cho, I.H.; Chung, D.H.; Yoon, S.H.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.W.; Ye, S.K.; et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005, 19, 1296–1298. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.E.; Kim, H.S.; Lee, C.S.; Shin, Y.J.; Kim, Y.N.; Kang, G.H.; Kim, T.Y.; Juhnn, Y.S.; Kim, S.J.; Park, J.W.; et al. STAT3 inhibits the degradation of HIF-1alpha by pVHL-mediated ubiquitination. Exp. Mol. Med. 2008, 40, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Pratt, J.; Annabi, B. Induction of autophagy biomarker BNIP3 requires a JAK2/STAT3 and MT1-MMP signaling interplay in Concanavalin-A-activated U87 glioblastoma cells. Cell. Signal. 2014, 26, 917–924. [Google Scholar] [CrossRef]
- Pensa, S.; Lloyd-Lewis, B.; Sargeant, T.J.; Resemann, H.K.; Kahn, C.R.; Watson, C.J. Signal transducer and activator of transcription 3 and the phosphatidylinositol 3-kinase regulatory subunits p55alpha and p50alpha regulate autophagy in vivo. FEBS J. 2014, 281, 4557–4567. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Niso-Santano, M.; Adjemian, S.; Takehara, T.; Malik, S.A.; Minoux, H.; Souquere, S.; Marino, G.; Lachkar, S.; Senovilla, L.; et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol. Cell 2012, 48, 667–680. [Google Scholar] [CrossRef] [Green Version]
- Szczepanek, K.; Chen, Q.; Larner, A.C.; Lesnefsky, E.J. Cytoprotection by the modulation of mitochondrial electron transport chain: The emerging role of mitochondrial STAT3. Mitochondrion 2012, 12, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Szczepanek, K.; Lesnefsky, E.J.; Larner, A.C. Multi-tasking: Nuclear transcription factors with novel roles in the mitochondria. Trends Cell Biol. 2012, 22, 429–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Sargeant, T.J.; Lloyd-Lewis, B.; Resemann, H.K.; Ramos-Montoya, A.; Skepper, J.; Watson, C.J. Stat3 controls cell death during mammary gland involution by regulating uptake of milk fat globules and lysosomal membrane permeabilization. Nat. Cell Biol. 2014, 16, 1057–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuzaler, P.A.; Staniszewska, A.D.; Li, W.; Omidvar, N.; Kedjouar, B.; Turkson, J.; Poli, V.; Flavell, R.A.; Clarkson, R.W.; Watson, C.J. Stat3 controls lysosomal-mediated cell death in vivo. Nat. Cell Biol. 2011, 13, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sun, B.; Gao, Y.; Niu, H.; Yuan, H.; Lou, H. STAT3 contributes to lysosomal-mediated cell death in a novel derivative of riccardin D-treated breast cancer cells in association with TFEB. Biochem. Pharmacol. 2018, 150, 267–279. [Google Scholar] [CrossRef]
- de Duve, C. Lysosomes revisited. Eur. J. Biochem. 1983, 137, 391–397. [Google Scholar] [CrossRef]
- Serrano-Puebla, A.; Boya, P. Lysosomal membrane permeabilization in cell death: New evidence and implications for health and disease. Ann. N. Y. Acad. Sci. 2016, 1371, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Henkel, L.; Linder, B.; Zielke, S.; Tascher, G.; Trautmann, S.; Geisslinger, G.; Münch, C.; Fulda, S.; Tegeder, I.; et al. Autophagy activation, lipotoxicity and lysosomal membrane permeabilization synergize to promote pimozide- and loperamide-induced glioma cell death. Autophagy 2020, 17, 3424–3443. [Google Scholar] [CrossRef] [PubMed]
- Aits, S.; Jaattela, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Sintes, R.; Ledesma, M.D.; Boya, P. Lysosomal cell death mechanisms in aging. Ageing Res. Rev. 2016, 32, 150–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 2016, 32, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Kallunki, T.; Olsen, O.D.; Jaattela, M. Cancer-associated lysosomal changes: Friends or foes? Oncogene 2013, 32, 1995–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piao, S.; Amaravadi, R.K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 2016, 1371, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Senft, C.; Polacin, M.; Priester, M.; Seifert, V.; Kogel, D.; Weissenberger, J. The nontoxic natural compound Curcumin exerts anti-proliferative, anti-migratory, and anti-invasive properties against malignant gliomas. BMC Cancer 2010, 10, 491. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Li, G.; Deng, L.; Liu, Q.; Dai, J.; Shen, J.; Zhang, J. IL-6 augments the invasiveness of U87MG human glioblastoma multiforme cells via up-regulation of MMP-2 and fascin-1. Oncol. Rep. 2010, 23, 1553–1559. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’Angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging 2010, 2, 823–842. [Google Scholar] [CrossRef] [Green Version]
- Shchors, K.; Massaras, A.; Hanahan, D. Dual targeting of the autophagic regulatory circuitry in gliomas with repurposed drugs elicits cell-lethal autophagy and therapeutic benefit. Cancer Cell 2015, 28, 456–471. [Google Scholar] [CrossRef] [Green Version]
- Zielke, S.; Meyer, N.; Mari, M.; Abou-El-Ardat, K.; Reggiori, F.; van Wijk, S.J.L.; Kogel, D.; Fulda, S. Loperamide, pimozide, and STF-62247 trigger autophagy-dependent cell death in glioblastoma cells. Cell Death Dis. 2018, 9, 994. [Google Scholar] [CrossRef] [PubMed]
- Stringer, B.W.; Day, B.W.; D’Souza, R.C.J.; Jamieson, P.R.; Ensbey, K.S.; Bruce, Z.C.; Lim, Y.C.; Goasdoue, K.; Offenhauser, C.; Akgul, S.; et al. A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci. Rep. 2019, 9, 4902. [Google Scholar] [CrossRef] [Green Version]
- Campos, B.; Gal, Z.; Baader, A.; Schneider, T.; Sliwinski, C.; Gassel, K.; Bageritz, J.; Grabe, N.; von Deimling, A.; Beckhove, P.; et al. Aberrant self-renewal and quiescence contribute to the aggressiveness of glioblastoma. J. Pathol. 2014, 234, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Campos, B.; Wan, F.; Farhadi, M.; Ernst, A.; Zeppernick, F.; Tagscherer, K.E.; Ahmadi, R.; Lohr, J.; Dictus, C.; Gdynia, G.; et al. Differentiation therapy exerts antitumor effects on stem-like glioma cells. Clin. Cancer Res. 2010, 16, 2715–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaizuka, T.; Morishita, H.; Hama, Y.; Tsukamoto, S.; Matsui, T.; Toyota, Y.; Kodama, A.; Ishihara, T.; Mizushima, T.; Mizushima, N. An autophagic flux probe that releases an internal control. Mol. Cell 2016, 64, 835–849. [Google Scholar] [CrossRef] [Green Version]
- Duchateau, P.N.; Pullinger, C.R.; Orellana, R.E.; Kunitake, S.T.; Naya-Vigne, J.; O’Connor, P.M.; Malloy, M.J.; Kane, J.P. Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas: Identification, cloning, characterization, and plasma distribution of apolipoprotein L. J. Biol. Chem. 1997, 272, 25576–25582. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Meyer, N.; Zielke, S.; Michaelis, J.B.; Linder, B.; Warnsmann, V.; Rakel, S.; Osiewacz, H.D.; Fulda, S.; Mittelbronn, M.; Munch, C.; et al. AT 101 induces early mitochondrial dysfunction and HMOX1 (heme oxygenase 1) to trigger mitophagic cell death in glioma cells. Autophagy 2018, 14, 1693–1709. [Google Scholar] [CrossRef]
- Pan, K.; Liang, X.T.; Zhang, H.K.; Zhao, J.J.; Wang, D.D.; Li, J.J.; Lian, Q.; Chang, A.E.; Li, Q.; Xia, J.C. Characterization of bridging integrator 1 (BIN1) as a potential tumor suppressor and prognostic marker in hepatocellular carcinoma. Mol. Med. 2012, 18, 507–518. [Google Scholar] [CrossRef]
- Kinsey, C.; Balakrishnan, V.; O’Dell, M.R.; Huang, J.L.; Newman, L.; Whitney-Miller, C.L.; Hezel, A.F.; Land, H. Plac8 links oncogenic mutations to regulation of autophagy and is critical to pancreatic cancer progression. Cell Rep. 2014, 7, 1143–1155. [Google Scholar] [CrossRef] [Green Version]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-Oncology 2017, 19, 139–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Kristiana, I.; Sharpe, L.J.; Catts, V.S.; Lutze-Mann, L.H.; Brown, A.J. Antipsychotic drugs upregulate lipogenic gene expression by disrupting intracellular trafficking of lipoprotein-derived cholesterol. Pharmacogenomics J. 2010, 10, 396–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, C.; Kirchner, P.; Bug, M.; Grum, D.; Koerver, L.; Schulze, N.; Poehler, R.; Dressler, A.; Fengler, S.; Arhzaouy, K.; et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 2017, 36, 135–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aits, S.; Kricker, J.; Liu, B.; Ellegaard, A.M.; Hamalisto, S.; Tvingsholm, S.; Corcelle-Termeau, E.; Hogh, S.; Farkas, T.; Jonassen, A.H.; et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 2015, 11, 1408–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, D.E.; Lee, C.K. What does Stat3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Lin, J. STAT3 as a therapeutic target for glioblastoma. Anti-Cancer Agents Med. Chem. 2010, 10, 512–519. [Google Scholar] [CrossRef]
- Shi, Y.; Guryanova, O.A.; Zhou, W.; Liu, C.; Huang, Z.; Fang, X.; Wang, X.; Chen, C.; Wu, Q.; He, Z.; et al. Ibrutinib inactivates BMX-STAT3 in glioma stem cells to impair malignant growth and radioresistance. Sci. Transl. Med. 2018, 10, 600. [Google Scholar] [CrossRef] [Green Version]
- Voss, V.; Senft, C.; Lang, V.; Ronellenfitsch, M.W.; Steinbach, J.P.; Seifert, V.; Kogel, D. The pan-Bcl-2 inhibitor (−)-gossypol triggers autophagic cell death in malignant glioma. Mol. Cancer Res. 2010, 8, 1002–1016. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Tiedra, S.; Fabrias, G.; Davila, D.; Salanueva, I.J.; Casas, J.; Montes, L.R.; Anton, Z.; Garcia-Taboada, E.; Salazar-Roa, M.; Lorente, M.; et al. Dihydroceramide accumulation mediates cytotoxic autophagy of cancer cells via autolysosome destabilization. Autophagy 2016, 12, 2213–2229. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernandez-Tiedra, S.; Lorente, M.; Egia, A.; Vazquez, P.; Blazquez, C.; Torres, S.; Garcia, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Invest. 2009, 119, 1359–1372. [Google Scholar] [CrossRef] [Green Version]
- Kornhuber, J.; Tripal, P.; Reichel, M.; Muhle, C.; Rhein, C.; Muehlbacher, M.; Groemer, T.W.; Gulbins, E. Functional inhibitors of acid sphingomyelinase (FIASMAs): A novel pharmacological group of drugs with broad clinical applications. Cell. Physiol. Biochem. 2010, 26, 9–20. [Google Scholar] [CrossRef]
- Petersen, N.H.; Olsen, O.D.; Groth-Pedersen, L.; Ellegaard, A.M.; Bilgin, M.; Redmer, S.; Ostenfeld, M.S.; Ulanet, D.; Dovmark, T.H.; Lønborg, A.; et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell 2013, 24, 379–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabande-Rodriguez, E.; Boya, P.; Labrador, V.; Dotti, C.G.; Ledesma, M.D. High sphingomyelin levels induce lysosomal damage and autophagy dysfunction in Niemann Pick disease type A. Cell Death Differ. 2014, 21, 864–875. [Google Scholar] [CrossRef]
- Groth-Pedersen, L.; Jäättelä, M. Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer Lett. 2013, 332, 265–274. [Google Scholar] [CrossRef]
- Liu, B.; Palmfeldt, J.; Lin, L.; Colaco, A.; Clemmensen, K.K.B.; Huang, J.; Xu, F.; Liu, X.; Maeda, K.; Luo, Y.; et al. STAT3 associates with vacuolar H(+)-ATPase and regulates cytosolic and lysosomal pH. Cell Res. 2018, 28, 996–1012. [Google Scholar] [CrossRef] [Green Version]
- Wiklund, E.D.; Catts, V.S.; Catts, S.V.; Ng, T.F.; Whitaker, N.J.; Brown, A.J.; Lutze-Mann, L.H. Cytotoxic effects of antipsychotic drugs implicate cholesterol homeostasis as a novel chemotherapeutic target. Int. J. Cancer 2010, 126, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Gowda, R.; Noory, M.A.; Robertson, G.P. Modulating cancer cell survival by targeting intracellular cholesterol transport. Br. J. Cancer 2017, 117, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Villa, G.R.; Hulce, J.J.; Zanca, C.; Bi, J.; Ikegami, S.; Cahill, G.L.; Gu, Y.; Lum, K.M.; Masui, K.; Yang, H.; et al. An LXR-cholesterol axis creates a metabolic co-dependency for brain cancers. Cancer Cell 2016, 30, 683–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetschko, H.; Voss, V.; Horn, S.; Seifert, V.; Prehn, J.H.; Kogel, D. Pharmacological inhibition of Bcl-2 family members reactivates TRAIL-induced apoptosis in malignant glioma. J. Neuro-Oncol. 2008, 86, 265–272. [Google Scholar] [CrossRef]
- Smilowitz, H.M.; Weissenberger, J.; Weis, J.; Brown, J.D.; O’Neill, R.J.; Laissue, J.A. Orthotopic transplantation of v-src-expressing glioma cell lines into immunocompetent mice: Establishment of a new transplantable in vivo model for malignant glioma. J. Neurosurg. 2007, 106, 652–659. [Google Scholar] [CrossRef] [Green Version]
- Remy, J.; Linder, B.; Weirauch, U.; Konovalova, J.; Marschalek, R.; Aigner, A.; Kogel, D. Inhibition of PIM1 blocks the autophagic flux to sensitize glioblastoma cells to ABT-737-induced apoptosis. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 175–189. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Ilic, N.; Shrestha, Y.; Zou, L.; Kamburov, A.; Zhu, C.; Yang, X.; Lubonja, R.; Tran, N.; Nguyen, C.; et al. Systematic functional interrogation of rare cancer variants identifies oncogenic alleles. Cancer Discov. 2016, 6, 714–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campeau, E.; Ruhl, V.E.; Rodier, F.; Smith, C.L.; Rahmberg, B.L.; Fuss, J.O.; Campisi, J.; Yaswen, P.; Cooper, P.K.; Kaufman, P.D. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 2009, 4, e6529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrain, C.; Creagh, E.M.; Martin, S.J. Apoptosis-associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. EMBO J. 2001, 20, 6627–6636. [Google Scholar] [CrossRef] [Green Version]
- Linder, B.; Wehle, A.; Hehlgans, S.; Bonn, F.; Dikic, I.; Rodel, F.; Seifert, V.; Kogel, D. Arsenic trioxide and (−)-gossypol synergistically target glioma stem-like cells via inhibition of hedgehog and notch signaling. Cancers 2019, 11, 350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinci, M.; Box, C.; Eccles, S.A. Three-dimensional (3D) tumor spheroid invasion assay. J. Vis. Exp. 2015, 99. [Google Scholar] [CrossRef] [Green Version]
- Stokowy, T.; Eszlinger, M.; Swierniak, M.; Fujarewicz, K.; Jarzab, B.; Paschke, R.; Krohn, K. Analysis options for high-throughput sequencing in miRNA expression profiling. BMC Res. Notes 2014, 7, 144. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Remy, J.; Linder, B.; Weirauch, U.; Day, B.W.; Stringer, B.W.; Herold-Mende, C.; Aigner, A.; Krohn, K.; Kögel, D. STAT3 Enhances Sensitivity of Glioblastoma to Drug-Induced Autophagy-Dependent Cell Death. Cancers 2022, 14, 339. https://doi.org/10.3390/cancers14020339
Remy J, Linder B, Weirauch U, Day BW, Stringer BW, Herold-Mende C, Aigner A, Krohn K, Kögel D. STAT3 Enhances Sensitivity of Glioblastoma to Drug-Induced Autophagy-Dependent Cell Death. Cancers. 2022; 14(2):339. https://doi.org/10.3390/cancers14020339
Chicago/Turabian StyleRemy, Janina, Benedikt Linder, Ulrike Weirauch, Bryan W. Day, Brett W. Stringer, Christel Herold-Mende, Achim Aigner, Knut Krohn, and Donat Kögel. 2022. "STAT3 Enhances Sensitivity of Glioblastoma to Drug-Induced Autophagy-Dependent Cell Death" Cancers 14, no. 2: 339. https://doi.org/10.3390/cancers14020339
APA StyleRemy, J., Linder, B., Weirauch, U., Day, B. W., Stringer, B. W., Herold-Mende, C., Aigner, A., Krohn, K., & Kögel, D. (2022). STAT3 Enhances Sensitivity of Glioblastoma to Drug-Induced Autophagy-Dependent Cell Death. Cancers, 14(2), 339. https://doi.org/10.3390/cancers14020339