
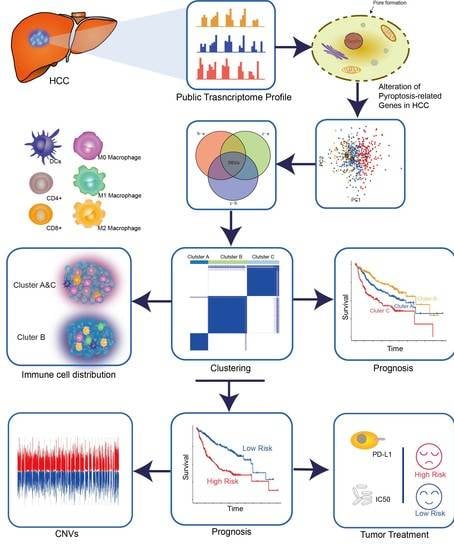
Novel Risk Classification Based on Pyroptosis-Related Genes Defines Immune Microenvironment and Pharmaceutical Landscape for Hepatocellular Carcinoma

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Data Availability and Preprocessing
2.2. Unsupervised Clustering Method of Proptosis-Related Subtypes
2.3. Gene Set Variation Analysis (GSVA), Enrichment and Visualization, and Single-Sample Gene Set Enrichment Analysis (ssGSEA)
2.4. Estimation of Immune Infiltration and Stromal Score
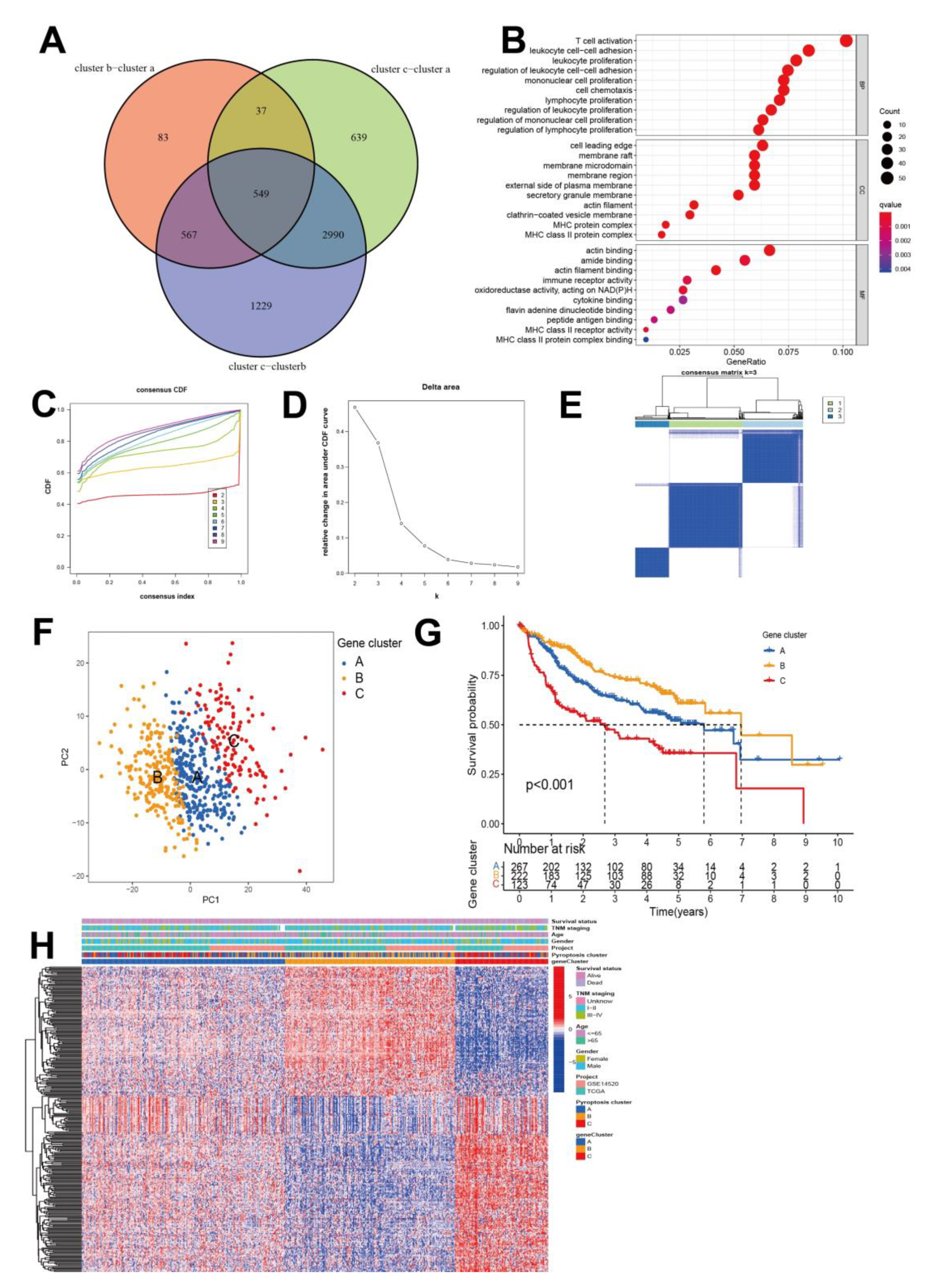
2.5. Generation of DEGs between Pyroptosis Distinct Clusters
2.6. Construction of the Pyroptosis Gene Signature
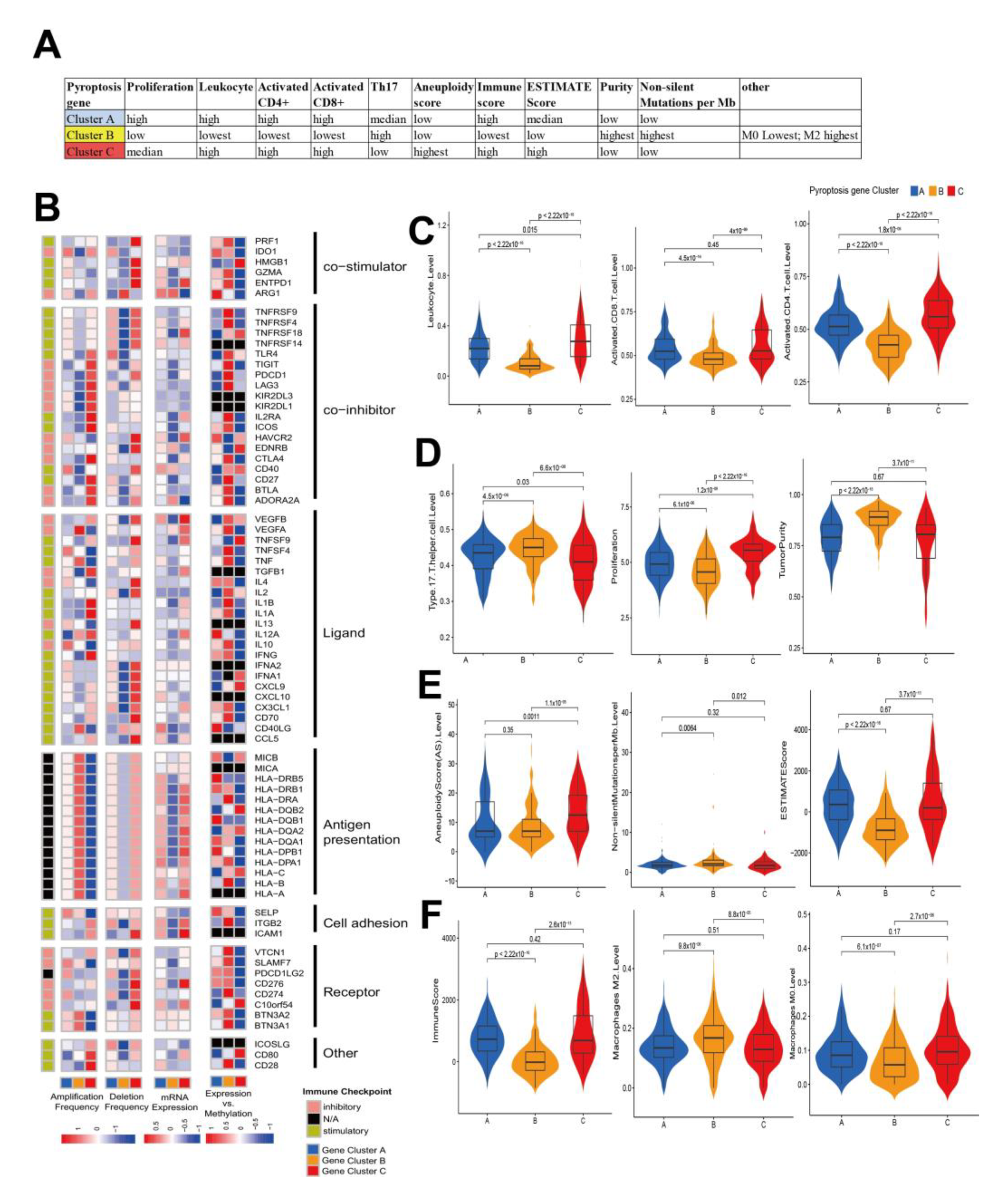
2.7. The Relationship between Immunomodulators (IMs) and Gene Clusters
2.8. The Immunophenoscore (IPS) Analysis
2.9. Statistical Analysis
3. Results
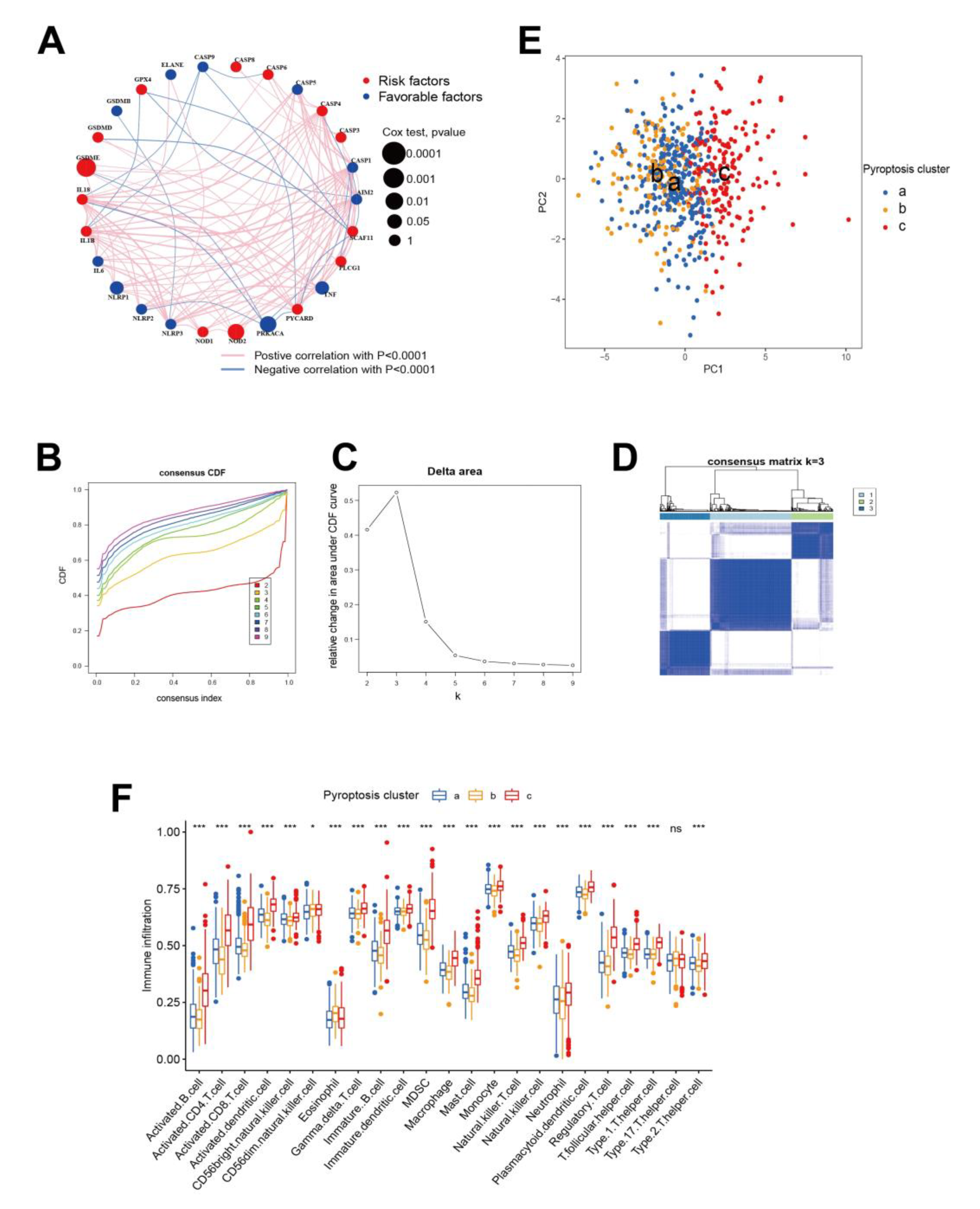
3.1. Expression and Copy Number Variation (CNV) of Pyroptosis Related Genes in HCC
3.2. Pyroptosis Patterns Mediated by 26 Regulators
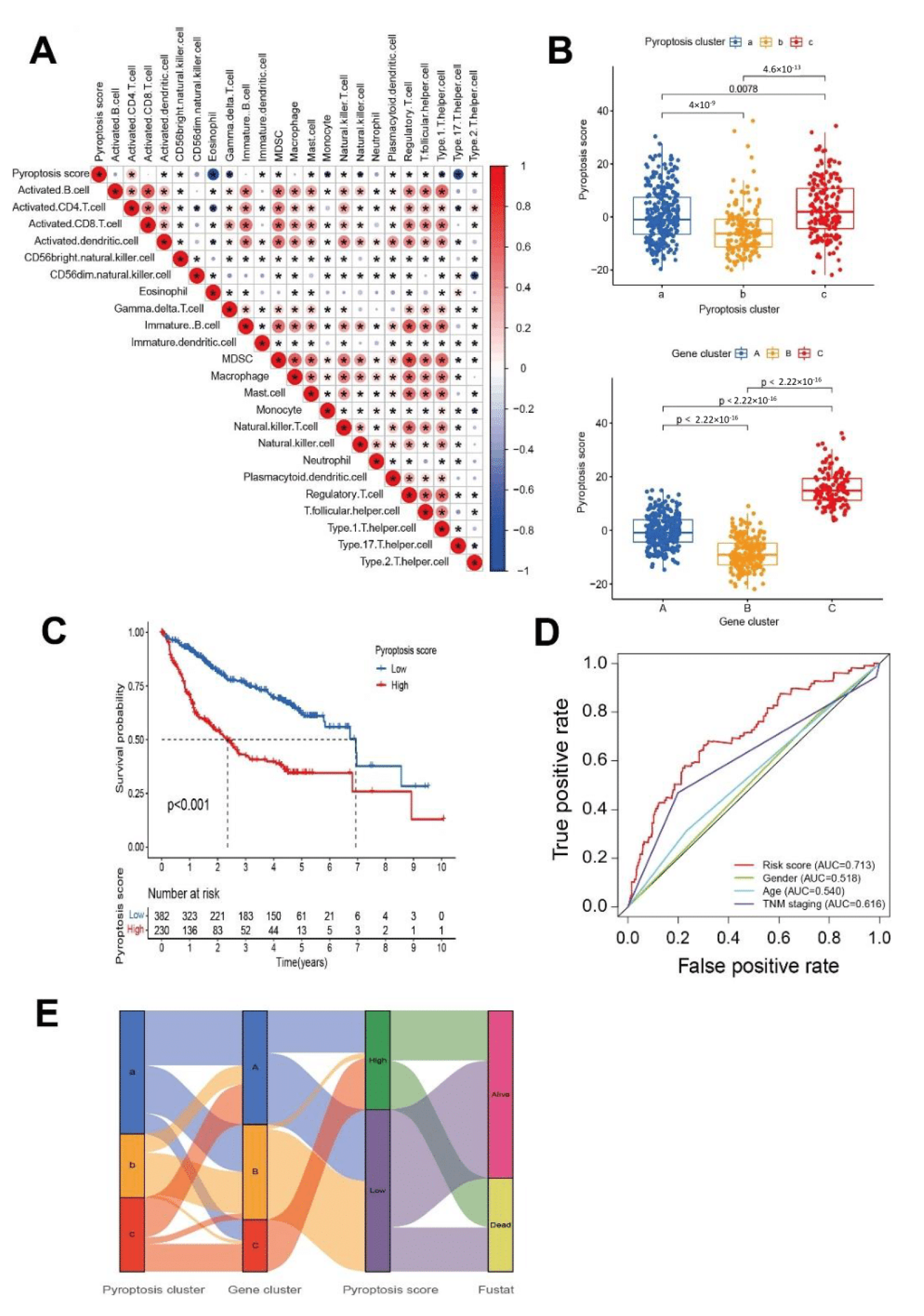
3.3. Characterization of Pyroptosis Gene Subtypes
3.4. Generation of Pyroptosis Gene Signatures and Correlations with Clinical Parameters
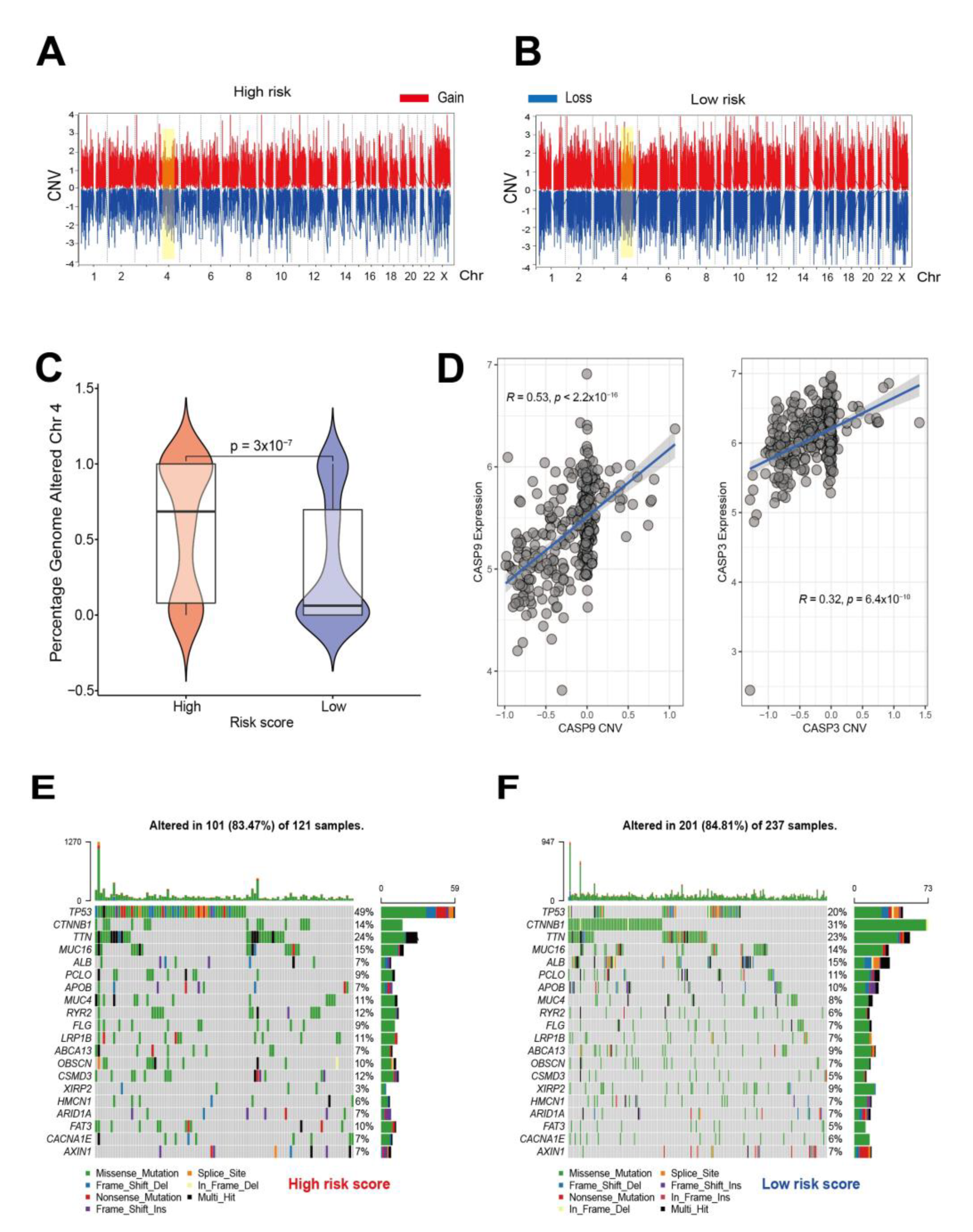
3.5. Genomic Characteristics of Pyroptosis Signatures
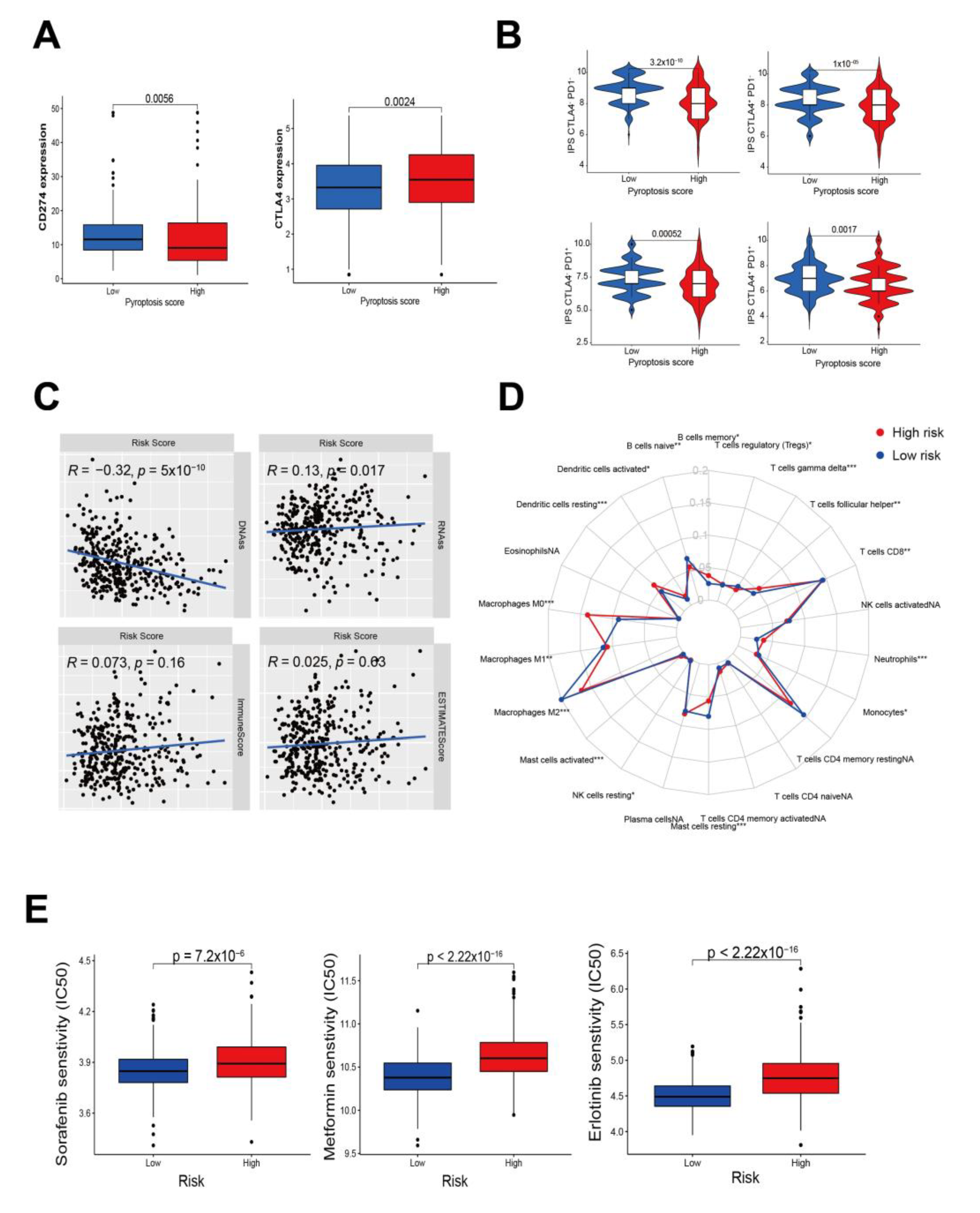
3.6. Predictive Response of Pyroptosis Risk Score to Immunotherapy and Systemic Therapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Saborowski, A. Current strategies for the treatment of intermediate and advanced hepatocellular carcinoma. Cancer Treat Rev. 2020, 82, 101946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruf, B.; Heinrich, B.; Greten, T.F. Immunobiology and immunotherapy of HCC: Spotlight on innate and innate-like immune cells. Cell. Mol. Immunol. 2021, 18, 112–127. [Google Scholar] [CrossRef]
- Cheng, A.L.; Hsu, C.; Chan, S.L.; Choo, S.P.; Kudo, M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J. Hepatol. 2020, 72, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Hilmi, M.; Neuzillet, C.; Calderaro, J.; Lafdil, F.; Pawlotsky, J.M.; Rousseau, B. Angiogenesis and immune checkpoint inhibitors as therapies for hepatocellular carcinoma: Current knowledge and future research directions. J. Immunother. Cancer 2019, 7, 333. [Google Scholar] [CrossRef]
- Dhir, M.; Melin, A.A.; Douaiher, J.; Lin, C.; Zhen, W.K.; Hussain, S.M.; Geschwind, J.F.; Doyle, M.B.; Abou-Alfa, G.K.; Are, C. A Review and Update of Treatment Options and Controversies in the Management of Hepatocellular Carcinoma. Ann. Surg. 2016, 263, 1112–1125. [Google Scholar] [CrossRef]
- de Martel, C.; Maucort-Boulch, D.; Plummer, M.; Franceschi, S. World-wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma. Hepatology 2015, 62, 1190–1200. [Google Scholar] [CrossRef]
- Enomoto, H.; Nakamura, H.; Liu, W.; Nishiguchi, S. Hepatoma-Derived Growth Factor: Its Possible Involvement in the Progression of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2015, 16, 14086–14097. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Villanueva, A.; Lachenmayer, A.; Finn, R.S. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat. Rev. Clin. Oncol. 2015, 12, 408–424. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef]
- Rosazza, T.; Warner, J.; Sollberger, G. NET formation—mechanisms and how they relate to other cell death pathways. FEBS J. 2020, 288, 3334–3350. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.F.; Kuo, Y.T.; Chen, T.Y.; Chien, C.T. Quercetin-Rich Guava (Psidium guajava) Juice in Combination with Trehalose Reduces Autophagy, Apoptosis and Pyroptosis Formation in the Kidney and Pancreas of Type II Diabetic Rats. Molecules 2016, 21, 334. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Zhu, R.; Zhu, J.; Zhao, R.; Li, M. E2-Induced Activation of the NLRP3 Inflammasome Triggers Pyroptosis and Inhibits Autophagy in HCC Cells. Oncol. Res. 2019, 27, 827–834. [Google Scholar] [CrossRef]
- Ummanni, R.; Lehnigk, U.; Zimmermann, U.; Woenckhaus, C.; Walthe, R.; Giebel, J. Immunohistochemical expression of caspase-1 and -9, uncleaved caspase-3 and -6, cleaved caspase-3 and -6 as well as Bcl-2 in benign epithelium and cancer of the prostate. Exp. Ther. Med. 2010, 1, 47–52. [Google Scholar] [CrossRef]
- Hu, B.; Elinav, E.; Huber, S.; Booth, C.J.; Strowig, T.; Jin, C.; Eisenbarth, S.C.; Flavell, R.A. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl. Acad. Sci. USA 2010, 107, 21635–21640. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Chu, Q.; Jiang, Y.; Zhang, W.; Xu, C.; Du, W.; Tuguzbaeva, G.; Qin, Y.; Li, A.; Zhang, L.; Sun, G.; et al. Pyroptosis is involved in the pathogenesis of human hepatocellular carcinoma. Oncotarget 2016, 7, 84658–84665. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Lan, T.; Li, M.; Liu, J.; Wu, X.; Shen, S.; Chen, W.; Peng, B. Six Metabolism Related mRNAs Predict the Prognosis of Patients with Hepatocellular Carcinoma. Front. Mol. Biosci. 2021, 8, 621232. [Google Scholar] [CrossRef]
- Zhu, J.; Tang, B.; Lv, X.; Meng, M.; Weng, Q.; Zhang, N.; Li, J.; Fan, K.; Zheng, L.; Fang, S.; et al. Identifying Apoptosis-Related Transcriptomic Aberrations and Revealing Clinical Relevance as Diagnostic and Prognostic Biomarker in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 519180. [Google Scholar] [CrossRef]
- Bai, Y.; Qi, W.; Liu, L.; Zhang, J.; Pang, L.; Gan, T.; Wang, P.; Wang, C.; Chen, H. Identification of Seven-Gene Hypoxia Signature for Predicting Overall Survival of Hepatocellular Carcinoma. Front. Genet. 2021, 12, 637418. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Chen, H. Development of a Novel Autophagy-Related Prognostic Signature and Nomogram for Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 591356. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.Y.; Wang, D.S.; Lin, H.C.; Chen, X.X.; Yang, H.; Zheng, Y.; Li, Y.H. A Novel Ferroptosis-related Gene Signature for Overall Survival Prediction in Patients with Hepatocellular Carcinoma. Int. J. Biol. Sci. 2020, 16, 2430–2441. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Kanneganti, T.D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer. 2019, 19, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yin, Q. AIM2 inflammasome activation and regulation: A structural perspective. J. Struct. Biol. 2017, 200, 279–282. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The role of pyroptosis in cancer: Pro-cancer or pro-“host”? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef] [Green Version]
- Yabuta, S.; Shidoji, Y. TLR4-mediated pyroptosis in human hepatoma-derived HuH-7 cells induced by a branched-chain polyunsaturated fatty acid, geranylgeranoic acid. Biosci. Rep. 2020, 40, BSR20194118. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Zeng, L.; Zhu, S.; Xie, Y.; Liu, J.; Wen, Q.; Cao, L.; Xie, M.; Ran, Q.; Kroemer, G.; et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 2018, 24, 97–108.e4. [Google Scholar] [CrossRef] [Green Version]
- Linder, A.; Bauernfried, S.; Cheng, Y.; Albanese, M.; Jung, C.; Keppler, O.T.; Hornung, V. CARD8 inflammasome activation triggers pyroptosis in human T cells. EMBO J. 2020, 39, e105071. [Google Scholar] [CrossRef]
- Chen, R.; Zeng, L.; Zhu, S.; Liu, J.; Zeh, H.J.; Kroemer, G.; Wang, H.; Billiar, T.R.; Jiang, J.; Tang, D.; et al. cAMP metabolism controls caspase-11 inflammasome activation and pyroptosis in sepsis. Sci. Adv. 2019, 5, eaav5562. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.Y.; Xie, K.X.; Wang, S.L.; Yuan, L.W. Inflammatory caspase-related pyroptosis: Mechanism, regulation and therapeutic potential for inflammatory bowel disease. Gastroenterol. Rep. 2018, 6, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Sotiriou, C.; Wirapati, P.; Loi, S.; Harris, A.; Fox, S.; Smeds, J.; Nordgren, H.; Farmer, P.; Praz, V.; Haibe-Kains, B.; et al. Gene expression profiling in breast cancer: Understanding the molecular basis of histologic grade to improve prognosis. J. Natl. Cancer Inst. 2006, 98, 262–272. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, G.; Nancy, C.; RStephanie, H. pRRophetic: An R Package for Prediction of Clinical Chemotherapeutic Response from Tumor Gene Expression Levels. PLoS ONE 2014, 9, e107468. [Google Scholar]
- Hazra, A.; Gogtay, N. Biostatistics series module 3: Comparing groups: Numerical variables. Indian J. Dermatol. 2016, 61, 251–260. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Meltzer, P.; Davis, S. RCircos: An R package for Circos 2D track plots. BMC Bioinform. 2013, 14, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Sui, S.; Shang, Y.; Yu, Z.; Han, J.; Zhang, G.; Ntim, M.; Hu, M.; Gong, P.; Chen, H.; et al. The landscape of immune cell infiltration and its clinical implications of pancreatic ductal adenocarcinoma. J. Adv. Res. 2020, 24, 139–148. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; Ding, J.; Wang, C.; Zhou, X.; Gao, W.; Huang, H.; Shao, F.; Liu, Z. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 2020, 579, 421–426. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef]
- Tang, X.; Shu, Z.; Zhang, W.; Cheng, L.; Yu, J.; Zhang, M.; Zheng, S. Clinical significance of the immune cell landscape in hepatocellular carcinoma patients with different degrees of fibrosis. Ann. Transl. Med. 2019, 7, 528. [Google Scholar] [CrossRef]
- Shen, S.; Yan, J.; Zhang, Y.; Dong, Z.; Xing, J.; He, Y. N6-methyladenosine (m6A)-mediated messenger RNA signatures and the tumor immune microenvironment can predict the prognosis of hepatocellular carcinoma. Ann. Transl. Med. 2021, 9, 59. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, B.; Huang, Z.L.; Shi, M.; Yu, X.J.; Zheng, L.; Li, S.; Li, L. Increased circulating Th17 cells after transarterial chemoembolization correlate with improved survival in stage III hepatocellular carcinoma: A prospective study. PLoS ONE 2013, 8, e60444. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Qiu, W.; Wang, B.; Gao, Y.; Tian, Y.; Tian, M.; Chen, Y.; Xu, L.; Yao, T.P.; Li, P.; Yang, P. Targeting Histone Deacetylase 6 Reprograms Interleukin-17-Producing Helper T Cell Pathogenicity and Facilitates Immunotherapies for Hepatocellular Carcinoma. Hepatology 2020, 71, 1967–1987. [Google Scholar] [CrossRef]
- Ye, C.; Li, W.Y.; Zheng, M.H.; Chen, Y.P. T-helper 17 cell: A distinctive cell in liver diseases. Hepatol. Res. 2011, 41, 22–29. [Google Scholar] [CrossRef]
- Feng, R.; Cui, Z.; Liu, Z.; Zhang, Y. Upregulated microRNA-132 in T helper 17 cells activates hepatic stellate cells to promote hepatocellular carcinoma cell migration in vitro. Scand. J. Immunol. 2021, 93, e13007. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, X.; Wang, N.; Wu, J.; Ma, L.; Xie, X.; Zhang, H.; Dang, C.; Kang, H.; Zhang, S.; et al. Correlations between tumor mutation burden and immune infiltrates and their prognostic value in pancreatic cancer by bioinformatic analysis. Life Sci. 2021, 277, 119505. [Google Scholar] [CrossRef]
- Lin, Z.; Xu, Q.; Miao, D.; Yu, F. An Inflammatory Response-Related Gene Signature Can Impact the Immune Status and Predict the Prognosis of Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 644416. [Google Scholar] [CrossRef]
- Araujo, A.N.; Camacho, C.P.; Mendes, T.B.; Lindsey, S.C.; Moraes, L.; Miyazawa, M.; Delcelo, R.; Pellegrino, R.; Mazzotti, D.R.; Maciel, R.M.B.; et al. Comprehensive Assessment of Copy Number Alterations Uncovers Recurrent AIFM3 and DLK1 Copy Gain in Medullary Thyroid Carcinoma. Cancers 2021, 13, 218. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Sanmamed, M.F.; Chen, L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Wang, Y.; Steffani, M.; Stöß, C.; Ankerst, D.; Friess, H.; Hüser, N.; Hartmann, D. Novel Risk Classification Based on Pyroptosis-Related Genes Defines Immune Microenvironment and Pharmaceutical Landscape for Hepatocellular Carcinoma. Cancers 2022, 14, 447. https://doi.org/10.3390/cancers14020447
Wang J, Wang Y, Steffani M, Stöß C, Ankerst D, Friess H, Hüser N, Hartmann D. Novel Risk Classification Based on Pyroptosis-Related Genes Defines Immune Microenvironment and Pharmaceutical Landscape for Hepatocellular Carcinoma. Cancers. 2022; 14(2):447. https://doi.org/10.3390/cancers14020447
Chicago/Turabian StyleWang, Jianye, Ying Wang, Marcella Steffani, Christian Stöß, Donna Ankerst, Helmut Friess, Norbert Hüser, and Daniel Hartmann. 2022. "Novel Risk Classification Based on Pyroptosis-Related Genes Defines Immune Microenvironment and Pharmaceutical Landscape for Hepatocellular Carcinoma" Cancers 14, no. 2: 447. https://doi.org/10.3390/cancers14020447
APA StyleWang, J., Wang, Y., Steffani, M., Stöß, C., Ankerst, D., Friess, H., Hüser, N., & Hartmann, D. (2022). Novel Risk Classification Based on Pyroptosis-Related Genes Defines Immune Microenvironment and Pharmaceutical Landscape for Hepatocellular Carcinoma. Cancers, 14(2), 447. https://doi.org/10.3390/cancers14020447