
GOF Mutant p53 in Cancers: A Therapeutic Challenge



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
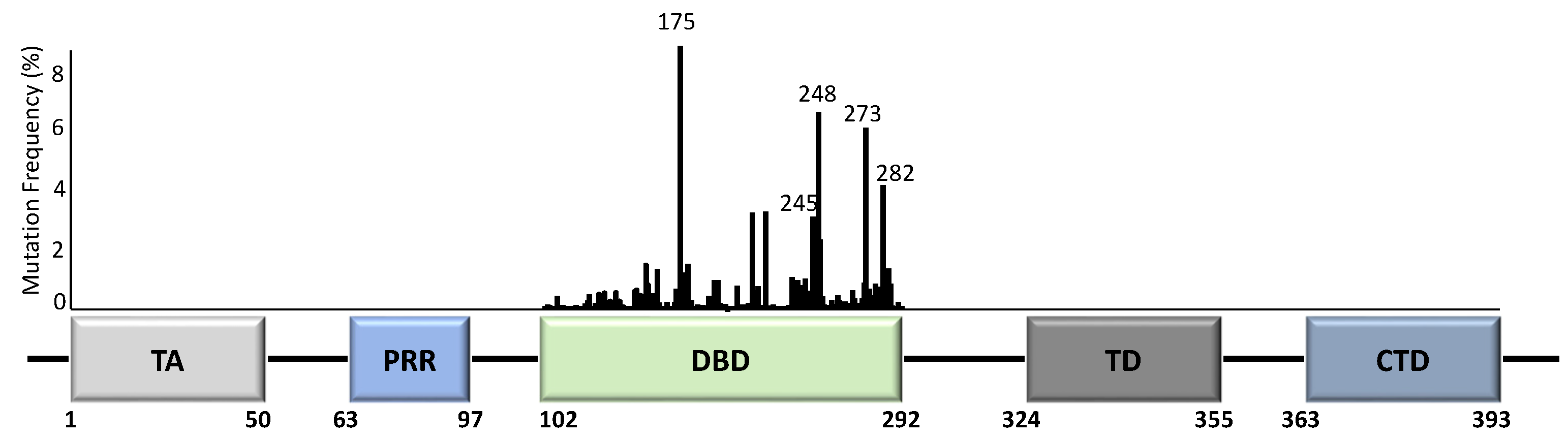
1. p53 and Mutations in Cancers
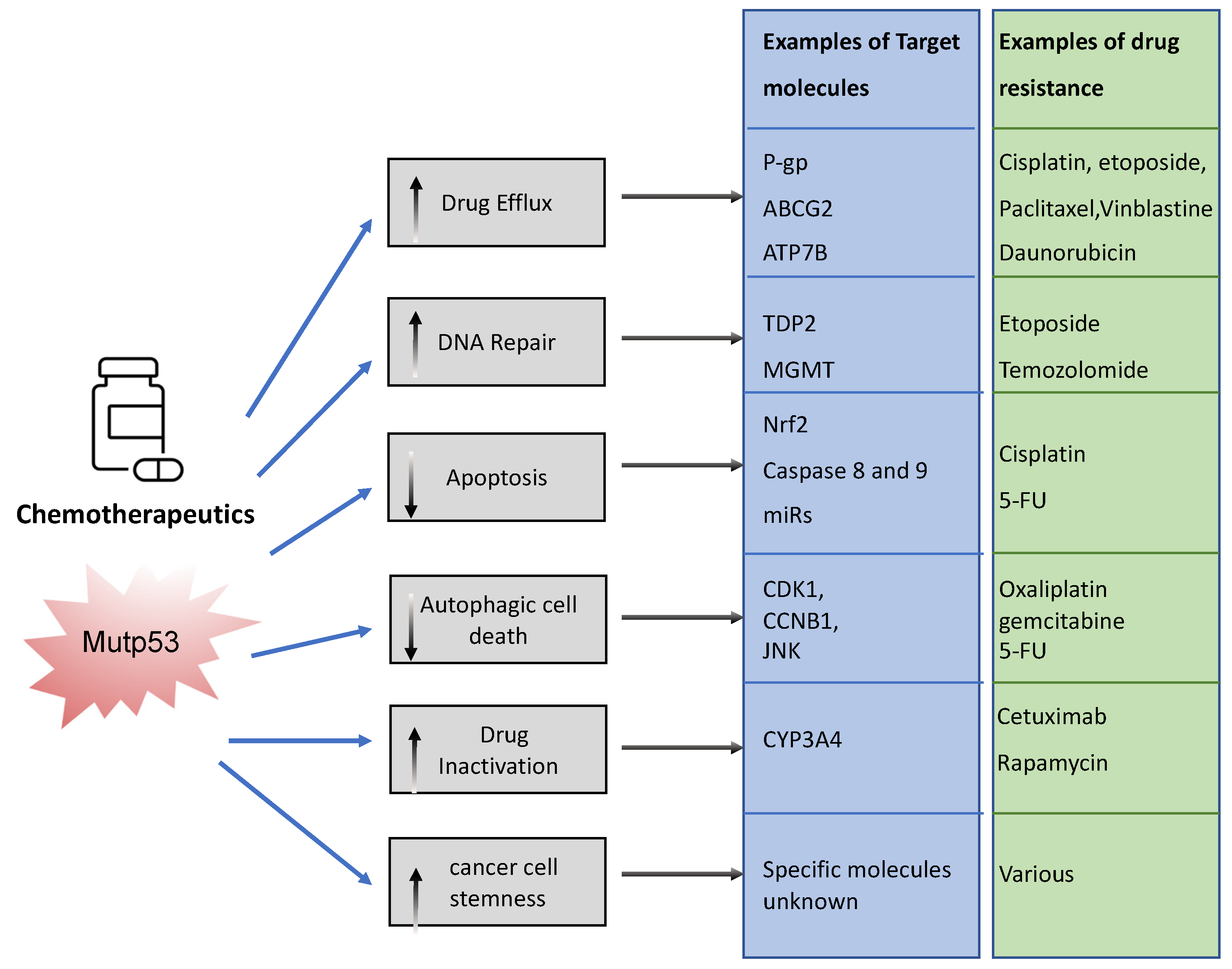
2. Mutant p53 and Chemoresistance
3. Strategies That Exploit Mutant p53 Expression
3.1. Mutant p53-Specific Targeted Therapy
3.1.1. Reactivating Mutant p53 to Behave like a WT Molecule
3.1.2. Induction of Mutp53 Degradation
3.1.3. Disruption of Mutp53 Function
3.2. Mutant p53-Specific Gene Therapy
3.3. Mutant p53 Specific Immunotherapy
3.4. Mutant p53 Specific Combination Therapy
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senturk, E.; Manfredi, J.J. p53 and cell cycle effects after DNA damage. Methods Mol. Biol. 2013, 962, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa, J.N.M.; Verdun, R.E.; Emerson, B.M. p53 Functions through Stress- and Promoter-Specific Recruitment of Transcription Initiation Components before and after DNA Damage. Mol. Cell 2003, 12, 1015–1027. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, U.M.; Petrenko, O. The MDM2-p53 Interaction. Mol. Cancer Res. 2003, 1, 1001. [Google Scholar]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Olivier, M.; Langerød, A.; Carrieri, P.; Bergh, J.; Klaar, S.; Eyfjord, J.; Theillet, C.; Rodriguez, C.; Lidereau, R.; Bièche, I.; et al. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin. Cancer Res. 2006, 12, 1157–1167. [Google Scholar] [CrossRef] [Green Version]
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000, 6, 2969–2972. [Google Scholar] [PubMed]
- Aubele, M.; Werner, M.; Höfler, H. Genetic alterations in presumptive precursor lesions of breast carcinomas. Anal. Cell. Pathol. 2002, 24, 69–76. [Google Scholar] [CrossRef]
- Iakova, P.; Timchenko, L.; Timchenko, N.A. Intracellular signaling and hepatocellular carcinoma. Semin. Cancer Biol. 2011, 21, 28–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozaki, M.; Tada, M.; Kobayashi, H.; Zhang, C.L.; Sawamura, Y.; Abe, H.; Ishii, N.; Van Meir, E.G. Roles of the functional loss of p53 and other genes in astrocytoma tumorigenesis and progression. Neuro. Oncol. 1999, 1, 124–137. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Paraskeva, C.; Markowitz, S.; Willson, J.K.; Hamilton, S.; Vogelstein, B. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. 1990, 50, 7717–7722. [Google Scholar]
- Teoh, P.J.; Chung, T.H.; Sebastian, S.; Choo, S.N.; Yan, J.; Ng, S.B.; Fonseca, R.; Chng, W.J. p53 haploinsufficiency and functional abnormalities in multiple myeloma. Leukemia 2014, 28, 2066–2074. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell. Rep. 2019, 28, 1370–1384.e1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soussi, T.; Wiman, K.G. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X. P53: A Target and a Biomarker of Cancer Therapy? Recent Adv. Cancer Res. Ther. 2012, 12, 197–213. [Google Scholar] [CrossRef]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iggo, R.; Gatter, K.; Bartek, J.; Lane, D.; Harris, A.L. Increased expression of mutant forms of p53 oncogene in primary lung cancer. Lancet 1990, 335, 675–679. [Google Scholar] [CrossRef]
- Gannon, J.V.; Greaves, R.; Iggo, R.; Lane, D.P. Activating mutations in p53 produce a common conformational effect. A monoclonal antibody specific for the mutant form. Embo J. 1990, 9, 1595–1602. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Phatak, V.; von Grabowiecki, Y.; Janus, J.; Officer, L.; Behan, C.; Aschauer, L.; Pinon, L.; Mackay, H.; Zanivan, S.; Norman, J.C.; et al. Mutant p53 promotes RCP-dependent chemoresistance coinciding with increased delivery of P-glycoprotein to the plasma membrane. Cell Death Dis. 2021, 12, 207. [Google Scholar] [CrossRef] [PubMed]
- Mackay, H.L.; Moore, D.; Hall, C.; Birkbak, N.J.; Jamal-Hanjani, M.; Karim, S.A.; Phatak, V.M.; Pinon, L.; Morton, J.P.; Swanton, C.; et al. Genomic instability in mutant p53 cancer cells upon entotic engulfment. Nat. Commun. 2018, 9, 3070. [Google Scholar] [CrossRef] [Green Version]
- von Grabowiecki, Y.; Phatak, V.; Aschauer, L.; Muller, P.A.J. Rab11-FIP1/RCP Functions as a Major Signalling Hub in the Oncogenic Roles of Mutant p53 in Cancer. Front. Oncol. 2021, 11, 804107. [Google Scholar] [CrossRef]
- Kennedy, M.C.; Lowe, S.W. Mutant p53: It’s not all one and the same. Cell Death Differ. 2022, 29, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Stein, Y.; Aloni-Grinstein, R.; Rotter, V. Mutant p53 oncogenicity: Dominant-negative or gain-of-function? Carcinogenesis 2020, 41, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Ortiz, E.; de la Cruz-López, K.G.; Becerril-Rico, J.; Sarabia-Sánchez, M.A.; Ortiz-Sánchez, E.; García-Carrancá, A. Mutant p53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front. Cell Dev. Biol. 2020, 8, 607670. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, L.; Singh, A.; Kaur, P.; Ethayathulla, A.S. Comprehensive omics studies of p53 mutants in human cancer. Brief Funct. Genom. 2022, elac015. [Google Scholar] [CrossRef]
- Friedlander, P.; Haupt, Y.; Prives, C.; Oren, M. A mutant p53 that discriminates between p53-responsive genes cannot induce apoptosis. Mol. Cell. Biol. 1996, 16, 4961–4971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.S.; Ramos, H.; Inga, A.; Sousa, E.; Saraiva, L. Structural and Drug Targeting Insights on Mutant p53. Cancers 2021, 13, 3344. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Li, L.; Guan, X.; Xiong, L.; Miao, X. Mutant p53 Gain of Function and Chemoresistance: The Role of Mutant p53 in Response to Clinical Chemotherapy. Chemotherapy 2017, 62, 43–53. [Google Scholar] [CrossRef]
- Bergh, J.; Norberg, T.; Sjögren, S.; Lindgren, A.; Holmberg, L. Complete sequencing of the p53 gene provides prognostic information in breast cancer patients, particularly in relation to adjuvant systemic therapy and radiotherapy. Nat. Med. 1995, 1, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Righetti, S.C.; Della Torre, G.; Pilotti, S.; Ménard, S.; Ottone, F.; Colnaghi, M.I.; Pierotti, M.A.; Lavarino, C.; Cornarotti, M.; Oriana, S.; et al. A comparative study of p53 gene mutations, protein accumulation, and response to cisplatin-based chemotherapy in advanced ovarian carcinoma. Cancer Res. 1996, 56, 689–693. [Google Scholar]
- Horio, Y.; Takahashi, T.; Kuroishi, T.; Hibi, K.; Suyama, M.; Niimi, T.; Shimokata, K.; Yamakawa, K.; Nakamura, Y.; Ueda, R.; et al. Prognostic significance of p53 mutations and 3p deletions in primary resected non-small cell lung cancer. Cancer Res. 1993, 53, 1–4. [Google Scholar]
- Wattel, E.; Preudhomme, C.; Hecquet, B.; Vanrumbeke, M.; Quesnel, B.; Dervite, I.; Morel, P.; Fenaux, P. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood 1994, 84, 3148–3157. [Google Scholar] [CrossRef] [Green Version]
- Chin, K.V.; Ueda, K.; Pastan, I.; Gottesman, M.M. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science 1992, 255, 459–462. [Google Scholar] [CrossRef]
- Cascorbi, I. P-glycoprotein: Tissue distribution, substrates, and functional consequences of genetic variations. Handb. Exp. Pharmacol. 2011, 2011, 261–283. [Google Scholar] [CrossRef]
- Sampath, J.; Sun, D.; Kidd, V.J.; Grenet, J.; Gandhi, A.; Shapiro, L.H.; Wang, Q.; Zambetti, G.P.; Schuetz, J.D. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J. Biol. Chem. 2001, 276, 39359–39367. [Google Scholar] [CrossRef] [Green Version]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef]
- Alam, S.K.; Yadav, V.K.; Bajaj, S.; Datta, A.; Dutta, S.K.; Bhattacharyya, M.; Bhattacharya, S.; Debnath, S.; Roy, S.; Boardman, L.A.; et al. DNA damage-induced ephrin-B2 reverse signaling promotes chemoresistance and drives EMT in colorectal carcinoma harboring mutant p53. Cell Death Differ. 2016, 23, 707–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Wang, J.; Hu, Y.; Qian, J.; Xu, B.; Chen, H.; Zou, W.; Fang, J.Y. Unequal prognostic potentials of p53 gain-of-function mutations in human cancers associate with drug-metabolizing activity. Cell Death Dis. 2014, 5, e1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, P.M.; Varanasi, L.; Fan, S.; Li, C.; Kubacka, I.; Newman, V.; Chauhan, K.; Daniels, S.R.; Boccetta, M.; Garrett, M.R.; et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 2012, 26, 830–845. [Google Scholar] [CrossRef] [PubMed]
- Ledesma, F.C.; El Khamisy, S.F.; Zuma, M.C.; Osborn, K.; Caldecott, K.W. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 2009, 461, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, J.X.; Liu, Y.H.; You, C.; Mao, Q. Mutant TP53 enhances the resistance of glioblastoma cells to temozolomide by up-regulating O(6)-methylguanine DNA-methyltransferase. Neurol. Sci. 2013, 34, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Tung, M.C.; Lin, P.L.; Wang, Y.C.; He, T.Y.; Lee, M.C.; Yeh, S.D.; Chen, C.Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chee, J.L.; Saidin, S.; Lane, D.P.; Leong, S.M.; Noll, J.E.; Neilsen, P.M.; Phua, Y.T.; Gabra, H.; Lim, T.M. Wild-type and mutant p53 mediate cisplatin resistance through interaction and inhibition of active caspase-9. Cell Cycle 2013, 12, 278–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrhardt, H.; Häcker, S.; Wittmann, S.; Maurer, M.; Borkhardt, A.; Toloczko, A.; Debatin, K.M.; Fulda, S.; Jeremias, I. Cytotoxic drug-induced, p53-mediated upregulation of caspase-8 in tumor cells. Oncogene 2008, 27, 783–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, H.; Schroering, A.; Ding, H.F. p53 mediates DNA damaging drug-induced apoptosis through a caspase-9-dependent pathway in SH-SY5Y neuroblastoma cells. Mol. Cancer Ther. 2002, 1, 679–686. [Google Scholar]
- Donzelli, S.; Fontemaggi, G.; Fazi, F.; Di Agostino, S.; Padula, F.; Biagioni, F.; Muti, P.; Strano, S.; Blandino, G. MicroRNA-128-2 targets the transcriptional repressor E2F5 enhancing mutant p53 gain of function. Cell Death Differ. 2012, 19, 1038–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morselli, E.; Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Kepp, O.; Criollo, A.; Vicencio, J.M.; Soussi, T.; Kroemer, G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008, 7, 3056–3061. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef]
- Martins, C.P.; Brown-Swigart, L.; Evan, G.I. Modeling the Therapeutic Efficacy of p53 Restoration in Tumors. Cell 2006, 127, 1323–1334. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Chen, L.; Li, C.; Lu, W.; Chen, J. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J. Biol. Chem. 2001, 276, 40583–40590. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [Green Version]
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.V.; Feinstein, E.; et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 6302–6307. [Google Scholar] [CrossRef] [Green Version]
- Bártek, J.; Bártková, J.; Vojtĕsek, B.; Stasková, Z.; Lukás, J.; Rejthar, A.; Kovarík, J.; Midgley, C.A.; Gannon, J.V.; Lane, D.P. Aberrant expression of the p53 oncoprotein is a common feature of a wide spectrum of human malignancies. Oncogene 1991, 6, 1699–1703. [Google Scholar] [PubMed]
- Klimovich, B.; Merle, N.; Neumann, M.; Elmshäuser, S.; Nist, A.; Mernberger, M.; Kazdal, D.; Stenzinger, A.; Timofeev, O.; Stiewe, T. p53 partial loss-of-function mutations sensitize to chemotherapy. Oncogene 2022, 41, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Foster, B.A.; Coffey, H.A.; Morin, M.J.; Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef] [Green Version]
- Rippin, T.M.; Bykov, V.J.N.; Freund, S.M.V.; Selivanova, G.; Wiman, K.G.; Fersht, A.R. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 2002, 21, 2119–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykov, V.J.N.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K.G. Reactivation of Mutant p53 and Induction of Apoptosis in Human Tumor Cells by Maleimide Analogs*. J. Biol. Chem. 2005, 280, 30384–30391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Zache, N.; Lambert, J.M.; Wiman, K.G.; Bykov, V.J. PRIMA-1MET inhibits growth of mouse tumors carrying mutant p53. Cell Oncol. 2008, 30, 411–418. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Wiman, K.G. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 2014, 588, 2622–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through p53-dependent and -independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Synnott, N.C.; O’Connell, D.; Crown, J.; Duffy, M.J. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res. Treat. 2020, 179, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Salim, K.Y.; Maleki Vareki, S.; Danter, W.R.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef]
- Chen, S.; Wu, J.L.; Liang, Y.; Tang, Y.G.; Song, H.X.; Wu, L.L.; Xing, Y.F.; Yan, N.; Li, Y.T.; Wang, Z.Y.; et al. Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell 2021, 39, 225–239.e228. [Google Scholar] [CrossRef]
- Decitabine, Cytarabine and Arsenic Trioxide for Acute Myeloid Leukemia with p53 Mutations. Available online: https://ClinicalTrials.gov/show/NCT03381781 (accessed on 1 January 2022).
- Demma, M.; Maxwell, E.; Ramos, R.; Liang, L.; Li, C.; Hesk, D.; Rossman, R.; Mallams, A.; Doll, R.; Liu, M.; et al. SCH529074, a small molecule activator of mutant p53, which binds p53 DNA binding domain (DBD), restores growth-suppressive function to mutant p53 and interrupts HDM2-mediated ubiquitination of wild type p53. J. Biol. Chem. 2010, 285, 10198–10212. [Google Scholar] [CrossRef] [Green Version]
- Friedler, A.; Hansson, L.O.; Veprintsev, D.B.; Freund, S.M.; Rippin, T.M.; Nikolova, P.V.; Proctor, M.R.; Rüdiger, S.; Fersht, A.R. A peptide that binds and stabilizes p53 core domain: Chaperone strategy for rescue of oncogenic mutants. Proc. Natl. Acad. Sci. USA 2002, 99, 937–942. [Google Scholar] [CrossRef] [Green Version]
- Tal, P.; Eizenberger, S.; Cohen, E.; Goldfinger, N.; Pietrokovski, S.; Oren, M.; Rotter, V. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget 2016, 7, 11817–11837. [Google Scholar] [CrossRef] [Green Version]
- Gogna, R.; Madan, E.; Kuppusamy, P.; Pati, U. Chaperoning of mutant p53 protein by wild-type p53 protein causes hypoxic tumor regression. J. Biol. Chem. 2012, 287, 2907–2914. [Google Scholar] [CrossRef] [Green Version]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Floquet, C.; Deforges, J.; Rousset, J.P.; Bidou, L. Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res. 2011, 39, 3350–3362. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Feng, J.; Song, W.; Wang, J.; Tsai, B.; Zhang, Y.; Scaringe, W.A.; Hill, K.A.; Margaritis, P.; High, K.A.; et al. A mouse model for nonsense mutation bypass therapy shows a dramatic multiday response to geneticin. Proc. Natl. Acad. Sci. USA 2007, 104, 15394–15399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GANNET53: Ganetespib in Metastatic, p53-Mutant, Platinum-Resistant Ovarian Cancer. 2013. Available online: https://ClinicalTrials.gov/show/NCT02012192 (accessed on 1 January 2022).
- Mutant p53-Based Personalized Trial Using Decitabine and Arsenic Trioxide on AML/MDS. 2019. Available online: https://ClinicalTrials.gov/show/NCT03855371 (accessed on 1 January 2022).
- Vorinostat in Treating Patients with Metastatic or Unresectable Melanoma. 2015. Available online: https://ClinicalTrials.gov/show/NCT00121225 (accessed on 1 September 2022).
- Vorinostat and Alvocidib in Treating Patients with Advanced Solid Tumors. 2006. Available online: https://ClinicalTrials.gov/show/NCT00324480 (accessed on 1 September 2022).
- MLN9708 and Vorinostat in Patients with Advanced p53 Mutant Malignancies. 2014. Available online: https://ClinicalTrials.gov/show/NCT02042989 (accessed on 1 September 2022).
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrell, F.K.; Kerr, E.M.; Gao, M.; Thorpe, H.; Doherty, G.J.; Cridge, J.; Shorthouse, D.; Speed, A.; Samarajiwa, S.; Hall, B.A.; et al. Lung tumors with distinct p53 mutations respond similarly to p53 targeted therapy but exhibit genotype-specific statin sensitivity. Genes Dev. 2017, 31, 1339–1353. [Google Scholar] [CrossRef]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Zhu, H.-B.; Yang, K.; Xie, Y.-Q.; Lin, Y.-W.; Mao, Q.-Q.; Xie, L.-P. Silencing of mutant p53 by siRNA induces cell cycle arrest and apoptosis in human bladder cancer cells. World J. Surg. Oncol. 2013, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V.; Toretsky, J.; Neckers, L. Geldanamycin selectively destabilizes and conformationally alters mutated p53. Oncogene 1995, 11, 933–939. [Google Scholar]
- A Phase 3 Study of Ganetespib in Combination with Docetaxel versus Docetaxel alone in Patients with Advanced NSCLC. Available online: https://clinicaltrials.gov/ct2/show/NCT01798485 (accessed on 24 September 2022).
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef]
- Blagosklonny, M.V.; Trostel, S.; Kayastha, G.; Demidenko, Z.N.; Vassilev, L.T.; Romanova, L.Y.; Bates, S.; Fojo, T. Depletion of Mutant p53 and Cytotoxicity of Histone Deacetylase Inhibitors. Cancer Res. 2005, 65, 7386–7392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foggetti, G.; Ottaggio, L.; Russo, D.; Mazzitelli, C.; Monti, P.; Degan, P.; Miele, M.; Fronza, G.; Menichini, P. Autophagy induced by SAHA affects mutant P53 degradation and cancer cell survival. Biosci. Rep. 2019, 39, BSR20181345. [Google Scholar] [CrossRef] [Green Version]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrales, A.; Thoenen, E.; Iwakuma, T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2018, 25, 460–470. [Google Scholar] [CrossRef]
- Chou, C.W.; Lin, C.H.; Hsiao, T.H.; Lo, C.C.; Hsieh, C.Y.; Huang, C.C.; Sher, Y.P. Therapeutic effects of statins against lung adenocarcinoma via p53 mutant-mediated apoptosis. Sci. Rep. 2019, 9, 20403. [Google Scholar] [CrossRef]
- Paranjpe, A.; Srivenugopal, K.S. Degradation of NF-κB, p53 and other regulatory redox-sensitive proteins by thiol-conjugating and -nitrosylating drugs in human tumor cells. Carcinogenesis 2013, 34, 990–1000. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.W.; Kang, H.J.; Kim, H.J.; Kong, Y.; Brown, M.L.; Bae, I. Targeting mutant p53 by a SIRT1 activator YK-3-237 inhibits the proliferation of triple-negative breast cancer cells. Oncotarget 2013, 4, 984–994. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Jung, Y.S.; Zhang, Y.; Chen, X. Arsenic trioxide reactivates proteasome-dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PLoS ONE 2014, 9, e103497. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, L.; El-Deiry, W.S. Small-Molecule NSC59984 Induces Mutant p53 Degradation through a ROS-ERK2-MDM2 Axis in Cancer Cells. Mol. Cancer Res. 2022, 20, 622–636. [Google Scholar] [CrossRef]
- García-Garrido, E.; Cordani, M.; Somoza, Á. Modified Gold Nanoparticles to Overcome the Chemoresistance to Gemcitabine in Mutant p53 Cancer Cells. Pharmaceutics 2021, 13, 2067. [Google Scholar] [CrossRef] [PubMed]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Tsai, K.Y.; Crowley, D.; Sengupta, S.; Yang, A.; McKeon, F.; Jacks, T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002, 416, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Pflaum, J.; Schlosser, S.; Muller, M. p53 Family and Cellular Stress Responses in Cancer. Front. Oncol. 2014, 4, 285. [Google Scholar] [CrossRef] [Green Version]
- Rinne, T.; Brunner, H.G.; van Bokhoven, H. p63-Associated Disorders. Cell Cycle 2007, 6, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Pozzi, S.; Zambelli, F.; Merico, D.; Pavesi, G.; Robert, A.; Maltère, P.; Gidrol, X.; Mantovani, R.; Vigano, M.A. Transcriptional Network of p63 in Human Keratinocytes. PLoS ONE 2009, 4, e5008. [Google Scholar] [CrossRef]
- Koster, M.I.; Dai, D.; Roop, D.R. Conflicting Roles for p63 in Skin Development and Carcinogenesis. Cell Cycle 2007, 6, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Crum, C.P.; McKeon, F.D. p63in Epithelial Survival, Germ Cell Surveillance, and Neoplasia. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 349–371. [Google Scholar] [CrossRef]
- Maeso-Alonso, L.; López-Ferreras, L.; Marques, M.M.; Marin, M.C. p73 as a Tissue Architect. Front. Cell Dev. Biol. 2021, 9, 716957. [Google Scholar] [CrossRef]
- Santos Guasch, G.L.; Beeler, J.S.; Marshall, C.B.; Shaver, T.M.; Sheng, Q.; Johnson, K.N.; Boyd, K.L.; Venters, B.J.; Cook, R.S.; Pietenpol, J.A. p73 Is Required for Ovarian Follicle Development and Regulates a Gene Network Involved in Cell-to-Cell Adhesion. iScience 2018, 8, 236–249. [Google Scholar] [CrossRef] [Green Version]
- Holembowski, L.; Kramer, D.; Riedel, D.; Sordella, R.; Nemajerova, A.; Dobbelstein, M.; Moll, U.M. TAp73 is essential for germ cell adhesion and maturation in testis. J. Cell Biol. 2014, 204, 1173–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, C.B.; Mays, D.J.; Beeler, J.S.; Rosenbluth, J.M.; Boyd, K.L.; Santos Guasch, G.L.; Shaver, T.M.; Tang, L.J.; Liu, Q.; Shyr, Y.; et al. p73 Is Required for Multiciliogenesis and Regulates the Foxj1-Associated Gene Network. Cell Rep. 2016, 14, 2289–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuertes-Alvarez, S.; Maeso-Alonso, L.; Villoch-Fernandez, J.; Wildung, M.; Martin-Lopez, M.; Marshall, C.; Villena-Cortes, A.J.; Diez-Prieto, I.; Pietenpol, J.A.; Tissir, F.; et al. p73 regulates ependymal planar cell polarity by modulating actin and microtubule cytoskeleton. Cell Death Dis. 2018, 9, 1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Como, C.J.; Gaiddon, C.; Prives, C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol. Cell Biol. 1999, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, B.; Prabhu, V.V.; Zhang, S.; van den Heuvel, A.P.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Prodigiosin rescues deficient p53 signaling and antitumor effects via upregulating p73 and disrupting its interaction with mutant p53. Cancer Res. 2014, 74, 1153–1165. [Google Scholar] [CrossRef] [Green Version]
- Di Agostino, S.; Cortese, G.; Monti, O.; Dell’Orso, S.; Sacchi, A.; Eisenstein, M.; Citro, G.; Strano, S.; Blandino, G. The disruption of the protein complex mutantp53/p73 increases selectively the response of tumor cells to anticancer drugs. Cell Cycle 2008, 7, 3440–3447. [Google Scholar] [CrossRef]
- Gomes, S.; Raimundo, L.; Soares, J.; Loureiro, J.B.; Leão, M.; Ramos, H.; Monteiro, M.N.; Lemos, A.; Moreira, J.; Pinto, M.; et al. New inhibitor of the TAp73 interaction with MDM2 and mutant p53 with promising antitumor activity against neuroblastoma. Cancer Lett. 2019, 446, 90–102. [Google Scholar] [CrossRef]
- Zhang, G.; Li, L.; Bi, J.; Wu, Y.; Li, E. Targeting DNA and mutant p53 by a naphthalimide derivative, NA20, exhibits selective inhibition in gastric tumorigenesis by blocking mutant p53-EGFR signaling pathway. Eur. J. Pharmacol. 2020, 887, 173584. [Google Scholar] [CrossRef]
- Yang, M.; Schell, M.J.; Loboda, A.; Nebozhyn, M.; Li, J.; Teer, J.K.; Pledger, W.J.; Yeatman, T.J. Repurposing EGFR Inhibitor Utility in Colorectal Cancer in Mutant APC and TP53 Subpopulations. Cancer Epidemiol. Biomarkers Prev. 2019, 28, 1141–1152. [Google Scholar] [CrossRef] [Green Version]
- Rokudai, S. High-Throughput RNA Interference Screen Targeting Synthetic-Lethal Gain-of-Function of Oncogenic Mutant TP53 in Triple-Negative Breast Cancer. Methods Mol. Biol. 2020, 2108, 297–303. [Google Scholar] [CrossRef]
- Moser, R.; Gurley, K.E.; Nikolova, O.; Qin, G.; Joshi, R.; Mendez, E.; Shmulevich, I.; Ashley, A.; Grandori, C.; Kemp, C.J. Synthetic lethal kinases in Ras/p53 mutant squamous cell carcinoma. Oncogene 2022, 41, 3355–3369. [Google Scholar] [CrossRef] [PubMed]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, M.R.; Siau, J.W.; Kannan, S.; Nguyen, M.N.; Ouaray, Z.; Kwoh, C.K.; Lane, D.P.; Ghadessy, F.; Verma, C.S. Simulations of mutant p53 DNA binding domains reveal a novel druggable pocket. Nucleic Acids Res. 2019, 47, 1637–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, L.L.; Dell, J.; Maxwell, E.; Armstrong, L.; Maneval, D.; Catino, J.J. Efficacy of p53 adenovirus-mediated gene therapy against human breast cancer xenografts. Cancer Gene Ther. 1997, 4, 129–138. [Google Scholar] [PubMed]
- Harris, M.P.; Sutjipto, S.; Wills, K.N.; Hancock, W.; Cornell, D.; Johnson, D.E.; Gregory, R.J.; Shepard, H.M.; Maneval, D.C. Adenovirus-mediated p53 gene transfer inhibits growth of human tumor cells expressing mutant p53 protein. Cancer Gene Ther. 1996, 3, 121–130. [Google Scholar]
- Lane, D.P.; Cheok, C.F.; Lain, S. p53-based cancer therapy. Cold Spring Harb. Perspect. Biol. 2010, 2, a001222. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Cai, D.W.; Georges, R.N.; Mukhopadhyay, T.; Grimm, E.A.; Roth, J.A. Therapeutic effect of a retroviral wild-type p53 expression vector in an orthotopic lung cancer model. J. Natl. Cancer Inst. 1994, 86, 1458–1462. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Zhang, W.; Li, L.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; Zhang, M.J.; Chandler, L.A.; Lin, H.; et al. The First Approved Gene Therapy Product for Cancer Ad-p53 (Gendicine): 12 Years in the Clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef] [Green Version]
- van Beusechem, V.W.; van den Doel, P.B.; Grill, J.; Pinedo, H.M.; Gerritsen, W.R. Conditionally replicative adenovirus expressing p53 exhibits enhanced oncolytic potency. Cancer Res. 2002, 62, 6165–6171. [Google Scholar]
- O’Shea, C.C.; Soria, C.; Bagus, B.; McCormick, F. Heat shock phenocopies E1B-55K late functions and selectively sensitizes refractory tumor cells to ONYX-015 oncolytic viral therapy. Cancer Cell 2005, 8, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Su, C.; Cao, H.; Li, K.; Chen, J.; Jiang, L.; Zhang, Q.; Wu, X.; Jia, X.; Liu, Y.; et al. A novel triple-regulated oncolytic adenovirus carrying p53 gene exerts potent antitumor efficacy on common human solid cancers. Mol. Cancer Ther. 2008, 7, 1598–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Fang, H. Clinical trials with oncolytic adenovirus in China. Curr. Cancer Drug. Targets 2007, 7, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Pirollo, K.F.; Tang, W.H.; Rait, A.; Chang, E.H. Transferrin-liposome-mediated systemic p53 gene therapy in combination with radiation results in regression of human head and neck cancer xenografts. Hum. Gene Ther. 1999, 10, 2941–2952. [Google Scholar] [CrossRef]
- Senzer, N.; Nemunaitis, J.; Nemunaitis, D.; Bedell, C.; Edelman, G.; Barve, M.; Nunan, R.; Pirollo, K.F.; Rait, A.; Chang, E.H. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol. Ther. 2013, 21, 1096–1103. [Google Scholar] [CrossRef] [Green Version]
- Pirollo, K.F.; Nemunaitis, J.; Leung, P.K.; Nunan, R.; Adams, J.; Chang, E.H. Safety and Efficacy in Advanced Solid Tumors of a Targeted Nanocomplex Carrying the p53 Gene Used in Combination with Docetaxel: A Phase 1b Study. Mol.Ther. 2016, 24, 1697–1706. [Google Scholar] [CrossRef] [Green Version]
- Harford, J.B.; Kim, S.S.; Pirollo, K.F.; Chang, E.H. TP53 Gene Therapy as a Potential Treatment for Patients with COVID-19. Viruses 2022, 14, 739. [Google Scholar] [CrossRef]
- Hann, B.; Balmain, A. Replication of an E1B 55-kilodalton protein-deficient adenovirus (ONYX-015) is restored by gain-of-function rather than loss-of-function p53 mutants. J. Virol. 2003, 77, 11588–11595. [Google Scholar] [CrossRef]
- Royds, J.A.; Hibma, M.; Dix, B.R.; Hananeia, L.; Russell, I.A.; Wiles, A.; Wynford-Thomas, D.; Braithwaite, A.W. p53 promotes adenoviral replication and increases late viral gene expression. Oncogene 2006, 25, 1509–1520. [Google Scholar] [CrossRef] [Green Version]
- Geoerger, B.; Grill, J.; Opolon, P.; Morizet, J.; Aubert, G.; Terrier-Lacombe, M.J.; Bressac De-Paillerets, B.; Barrois, M.; Feunteun, J.; Kirn, D.H.; et al. Oncolytic activity of the E1B-55 kDa-deleted adenovirus ONYX-015 is independent of cellular p53 status in human malignant glioma xenografts. Cancer Res. 2002, 62, 764–772. [Google Scholar]
- Edwards, S.J.; Dix, B.R.; Myers, C.J.; Dobson-Le, D.; Huschtscha, L.; Hibma, M.; Royds, J.; Braithwaite, A.W. Evidence that replication of the antitumor adenovirus ONYX-015 is not controlled by the p53 and p14(ARF) tumor suppressor genes. J. Virol. 2002, 76, 12483–12490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil-Ranedo, J.; Gallego-García, C.; Almendral, J.M. Viral targeting of glioblastoma stem cells with patient-specific genetic and post-translational p53 deregulations. Cell Rep. 2021, 36, 109673. [Google Scholar] [CrossRef] [PubMed]
- Chira, S.; Gulei, D.; Hajitou, A.; Berindan-Neagoe, I. Restoring the p53 ‘Guardian’ Phenotype in p53-Deficient Tumor Cells with CRISPR/Cas9. Trends Biotechnol. 2018, 36, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Mirgayazova, R.; Khadiullina, R.; Chasov, V.; Mingaleeva, R.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Therapeutic Editing of the TP53 Gene: Is CRISPR/Cas9 an Option? Genes 2020, 11, 704. [Google Scholar] [CrossRef]
- Enache, O.M.; Rendo, V.; Abdusamad, M.; Lam, D.; Davison, D.; Pal, S.; Currimjee, N.; Hess, J.; Pantel, S.; Nag, A.; et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet 2020, 52, 662–668. [Google Scholar] [CrossRef]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 2021, 129, 1109–1114. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Couch, M.E.; Ferris, R.L.; Brennan, J.A.; Koch, W.M.; Jaffee, E.M.; Leibowitz, M.S.; Nepom, G.T.; Erlich, H.A.; Sidransky, D. Alteration of cellular and humoral immunity by mutant p53 protein and processed mutant peptide in head and neck cancer. Clin. Cancer Res. 2007, 13, 7199–7206. [Google Scholar] [CrossRef] [Green Version]
- Ito, D.; Visus, C.; Hoffmann, T.K.; Balz, V.; Bier, H.; Appella, E.; Whiteside, T.L.; Ferris, R.L.; DeLeo, A.B. Immunological characterization of missense mutations occurring within cytotoxic T cell-defined p53 epitopes in HLA-A*0201+ squamous cell carcinomas of the head and neck. Int. J. Cancer 2007, 120, 2618–2624. [Google Scholar] [CrossRef]
- Carbone, D.P.; Ciernik, I.F.; Kelley, M.J.; Smith, M.C.; Nadaf, S.; Kavanaugh, D.; Maher, V.E.; Stipanov, M.; Contois, D.; Johnson, B.E.; et al. Immunization with mutant p53- and K-ras-derived peptides in cancer patients: Immune response and clinical outcome. J. Clin. Oncol. 2005, 23, 5099–5107. [Google Scholar] [CrossRef] [Green Version]
- Deniger, D.C.; Pasetto, A.; Robbins, P.F.; Gartner, J.J.; Prickett, T.D.; Paria, B.C.; Malekzadeh, P.; Jia, L.; Yossef, R.; Langhan, M.M.; et al. T-cell Responses to TP53 “Hotspot” Mutations and Unique Neoantigens Expressed by Human Ovarian Cancers. Clin. Cancer Res. 2018, 24, 5562–5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonia, S.J.; Mirza, N.; Fricke, I.; Chiappori, A.; Thompson, P.; Williams, N.; Bepler, G.; Simon, G.; Janssen, W.; Lee, J.-H.; et al. Combination of p53 Cancer Vaccine with Chemotherapy in Patients with Extensive Stage Small Cell Lung Cancer. Clin. Cancer Res. 2006, 12, 878–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayordomo, J.I.; Loftus, D.J.; Sakamoto, H.; De Cesare, C.M.; Appasamy, P.M.; Lotze, M.T.; Storkus, W.J.; Appella, E.; DeLeo, A.B. Therapy of murine tumors with p53 wild-type and mutant sequence peptide-based vaccines. J. Exp. Med. 1996, 183, 1357–1365. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Chada, S.; Stipanov, M.; Nadaf, S.; Ciernik, F.I.; Gabrilovich, D.I.; Carbone, D.P. Dendritic cells transduced with wild-type p53 gene elicit potent anti-tumour immune responses. Clin. Exp. Immunol. 1999, 117, 244–251. [Google Scholar] [CrossRef]
- Nikitina, E.Y.; Clark, J.I.; van Beynen, J.; Chada, S.; Virmani, A.K.; Carbone, D.P.; Gabrilovich, D.I. Dendritic Cells Transduced with Full-Length Wild-Type p53 Generate Antitumor Cytotoxic T Lymphocytes from Peripheral Blood of Cancer Patients1. Clin. Cancer Res. 2001, 7, 127–135. [Google Scholar]
- Vaccine Therapy in Treating Patients with Head and Neck Cancer. 2006. Available online: https://ClinicalTrials.gov/show/NCT00404339 (accessed on 1 January 2022).
- Rahma, O.E.; Ashtar, E.; Czystowska, M.; Szajnik, M.E.; Wieckowski, E.; Bernstein, S.; Herrin, V.E.; Shams, M.A.; Steinberg, S.M.; Merino, M.; et al. A gynecologic oncology group phase II trial of two p53 peptide vaccine approaches: Subcutaneous injection and intravenous pulsed dendritic cells in high recurrence risk ovarian cancer patients. Cancer Immunol. Immunother. CII 2012, 61, 373–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Fan, C.; Zeng, Z.; Young, K.H.; Li, Y. Clinical and Immunological Effects of p53-Targeting Vaccines. Front. Cell Dev. Biol. 2021, 9, 762796. [Google Scholar] [CrossRef]
- Rosenberg, S.A. Phase II Study of Metastatic Cancer That Overexpresses P53 Using Lymphodepleting Conditioning Followed by Infusion of Anti-P53 TCR-Gene Engineered Lymphocytes. 2006. Available online: https://clinicaltrials.gov/ct2/show/NCT00393029 (accessed on 22 April 2022).
- Kuball, J.; Schuler, M.; Antunes Ferreira, E.; Herr, W.; Neumann, M.; Obenauer-Kutner, L.; Westreich, L.; Huber, C.; Wölfel, T.; Theobald, M. Generating p53-specific cytotoxic T lymphocytes by recombinant adenoviral vector-based vaccination in mice, but not man. Gene Ther. 2002, 9, 833–843. [Google Scholar] [CrossRef]
- Sellman, B.H.; Menander, K.B. Safety and Efficacy of p53 Gene Therapy Combined with Immune Checkpoint Inhibitors in Solid Tumors. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03544723 (accessed on 24 April 2022).
- Chada, S.; Wiederhold, D.; Menander, K.B.; Sellman, B.; Talbott, M.; Nemunaitis, J.J.; Ahn, H.M.; Jung, B.-K.; Yun, C.-O.; Sobol, R.E. Tumor suppressor immune gene therapy to reverse immunotherapy resistance. Cancer Gene Ther. 2021, 29, 825–834. [Google Scholar] [CrossRef]
- Hardwick, N.R.; Frankel, P.; Ruel, C.; Kilpatrick, J.; Tsai, W.; Kos, F.; Kaltcheva, T.; Leong, L.; Morgan, R.; Chung, V.; et al. p53-Reactive T Cells Are Associated with Clinical Benefit in Patients with Platinum-Resistant Epithelial Ovarian Cancer After Treatment with a p53 Vaccine and Gemcitabine Chemotherapy. Clin. Cancer Res. 2018, 24, 1315–1325. [Google Scholar] [CrossRef]
- Bykov, V.J.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohell, N.; Alfredsson, J.; Fransson, Å.; Uustalu, M.; Byström, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.N.; Björklund, U.; Wiman, K.G. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda-Walerych, K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 2016, 18, 897–909. [Google Scholar] [CrossRef]
- Krayem, M.; Journe, F.; Wiedig, M.; Morandini, R.; Najem, A.; Salès, F.; van Kempen, L.C.; Sibille, C.; Awada, A.; Marine, J.C.; et al. p53 Reactivation by PRIMA-1(Met) (APR-246) sensitises (V600E/K)BRAF melanoma to vemurafenib. Eur. J. Cancer 2016, 55, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Deben, C.; Lardon, F.; Wouters, A.; Op de Beeck, K.; Van den Bossche, J.; Jacobs, J.; Van Der Steen, N.; Peeters, M.; Rolfo, C.; Deschoolmeester, V.; et al. APR-246 (PRIMA-1(MET)) strongly synergizes with AZD2281 (olaparib) induced PARP inhibition to induce apoptosis in non-small cell lung cancer cell lines. Cancer Lett. 2016, 375, 313–322. [Google Scholar] [CrossRef]
- Kong, L.R.; Ong, R.W.; Tan, T.Z.; Mohamed Salleh, N.A.B.; Thangavelu, M.; Chan, J.V.; Koh, L.Y.J.; Periyasamy, G.; Lau, J.A.; Le, T.B.U.; et al. Targeting codon 158 p53-mutant cancers via the induction of p53 acetylation. Nat. Commun. 2020, 11, 2086. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolma, L.; Muller, P.A.J. GOF Mutant p53 in Cancers: A Therapeutic Challenge. Cancers 2022, 14, 5091. https://doi.org/10.3390/cancers14205091
Dolma L, Muller PAJ. GOF Mutant p53 in Cancers: A Therapeutic Challenge. Cancers. 2022; 14(20):5091. https://doi.org/10.3390/cancers14205091
Chicago/Turabian StyleDolma, Lobsang, and Patricia A. J. Muller. 2022. "GOF Mutant p53 in Cancers: A Therapeutic Challenge" Cancers 14, no. 20: 5091. https://doi.org/10.3390/cancers14205091
APA StyleDolma, L., & Muller, P. A. J. (2022). GOF Mutant p53 in Cancers: A Therapeutic Challenge. Cancers, 14(20), 5091. https://doi.org/10.3390/cancers14205091