
Construction of a Prognostic and Early Diagnosis Model for LUAD Based on Necroptosis Gene Signature and Exploration of Immunotherapy Potential
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
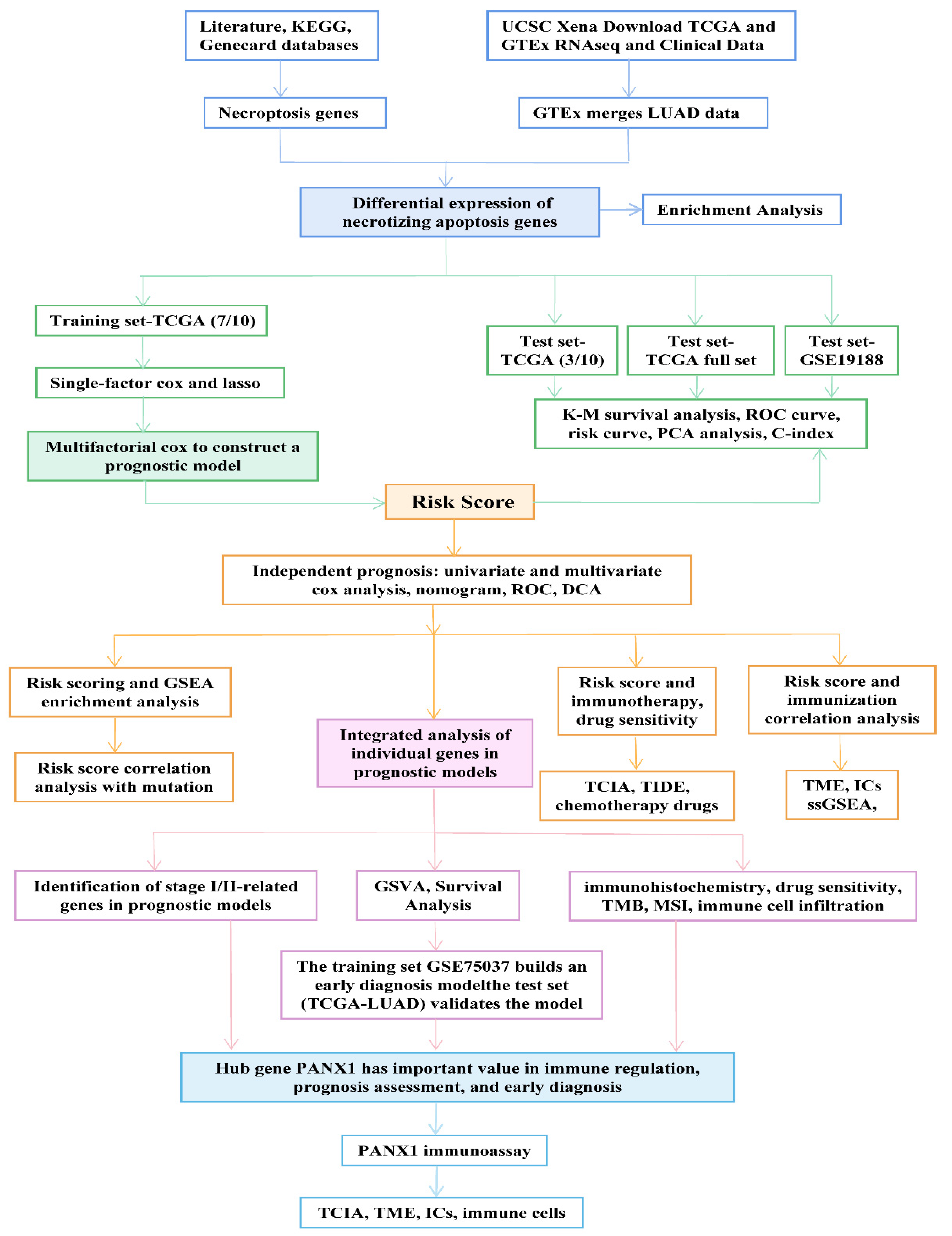
2. Materials and Methods
2.1. Summary of Necrotizing Apoptosis Genes
2.2. Open Data Sets and Pre-Processing
2.3. Identification of Differentially Expressed Genes in NRGs and the Related Enrichment Analysis
2.4. Construction and Validation of Prognostic Models
2.5. Relationship between Risk Score and Independent Prognosis
2.6. Nomogram Construction and Verification
2.7. Relationships between Risk Score, Gene Set Enrichment Analysis (GSEA), and Mutations
2.8. Risk Score and Immunization Correlation Analysis
2.9. Relationships between Risk Scores, Immunotherapy, and Drug Sensitivity
2.10. Relationship between Risk Score and GSVA
2.11. DENRGs Associated with Early Diagnosis of LUAD Was Used to Construct a Diagnostic Model
2.12. Online Database
3. Results
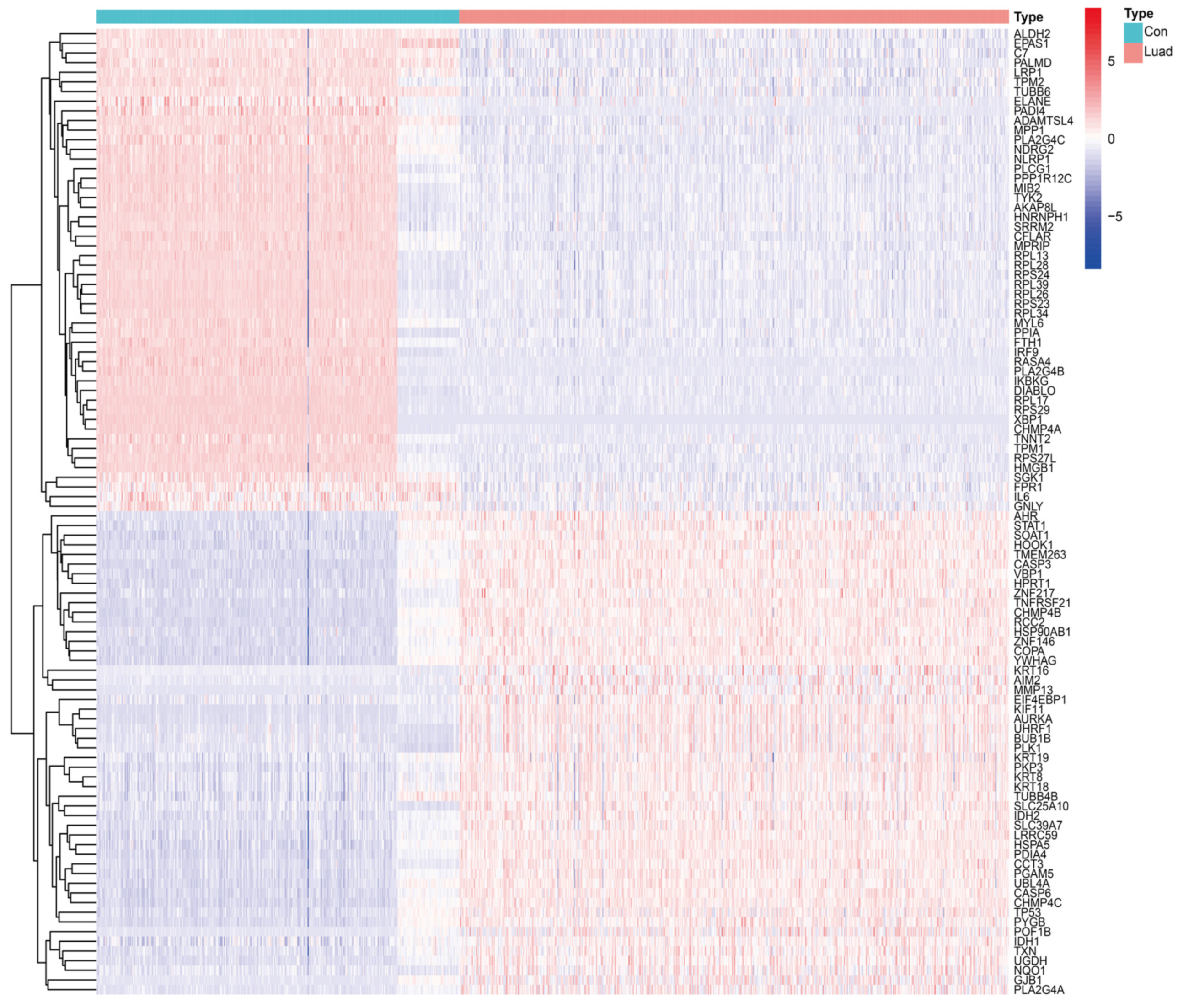
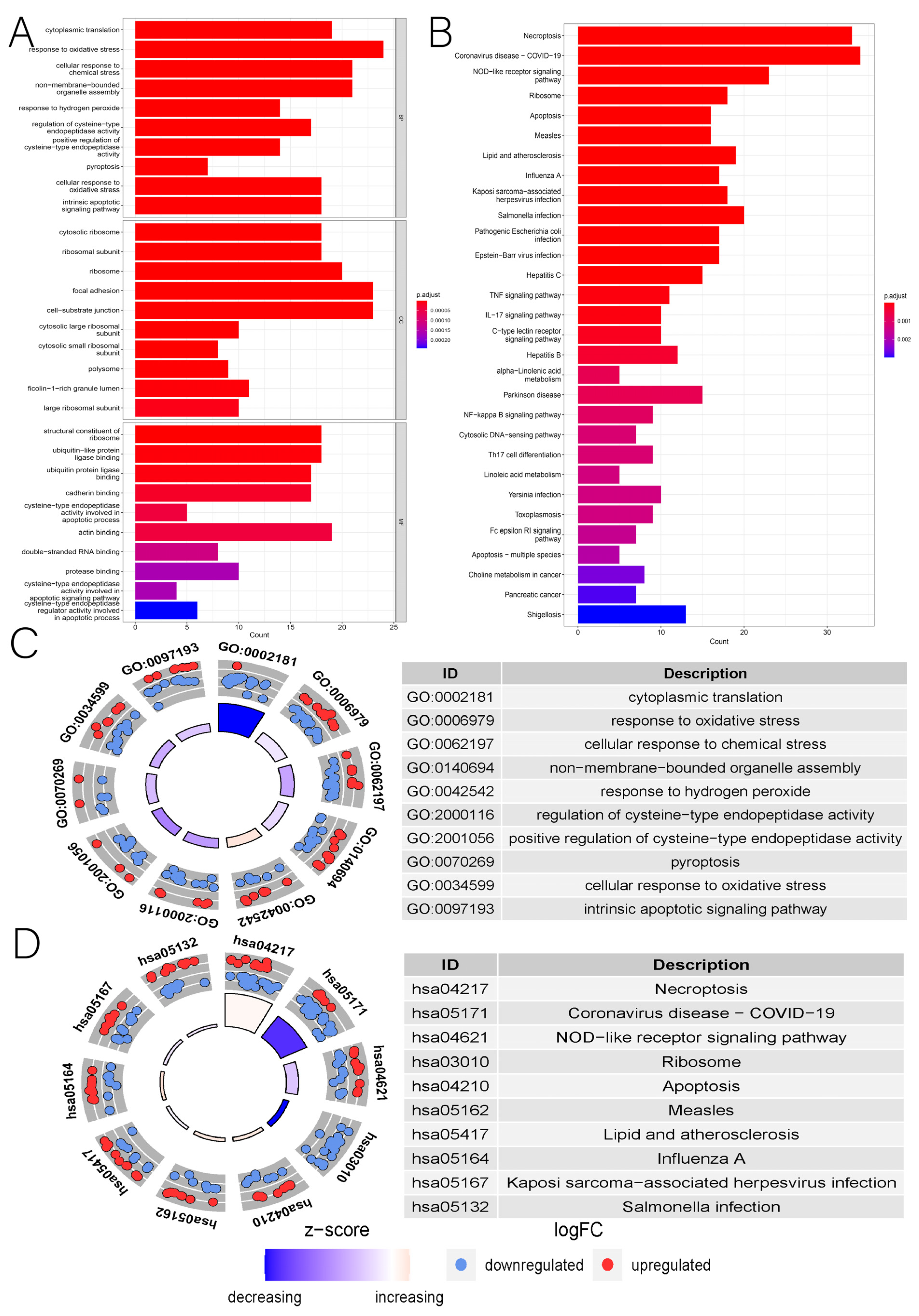
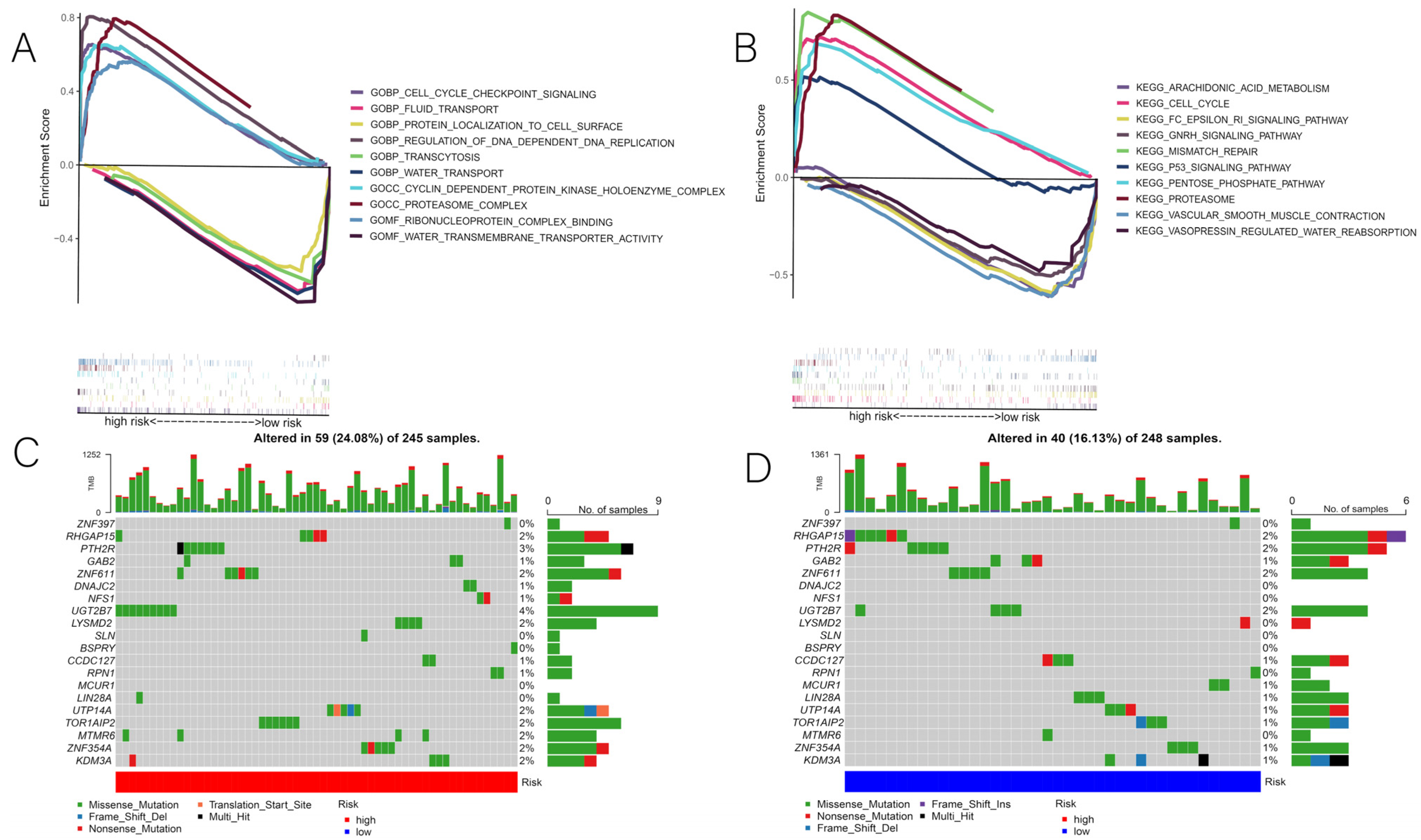
3.1. Identification of DENRGs and Enrichment Analysis Related to DENRGs
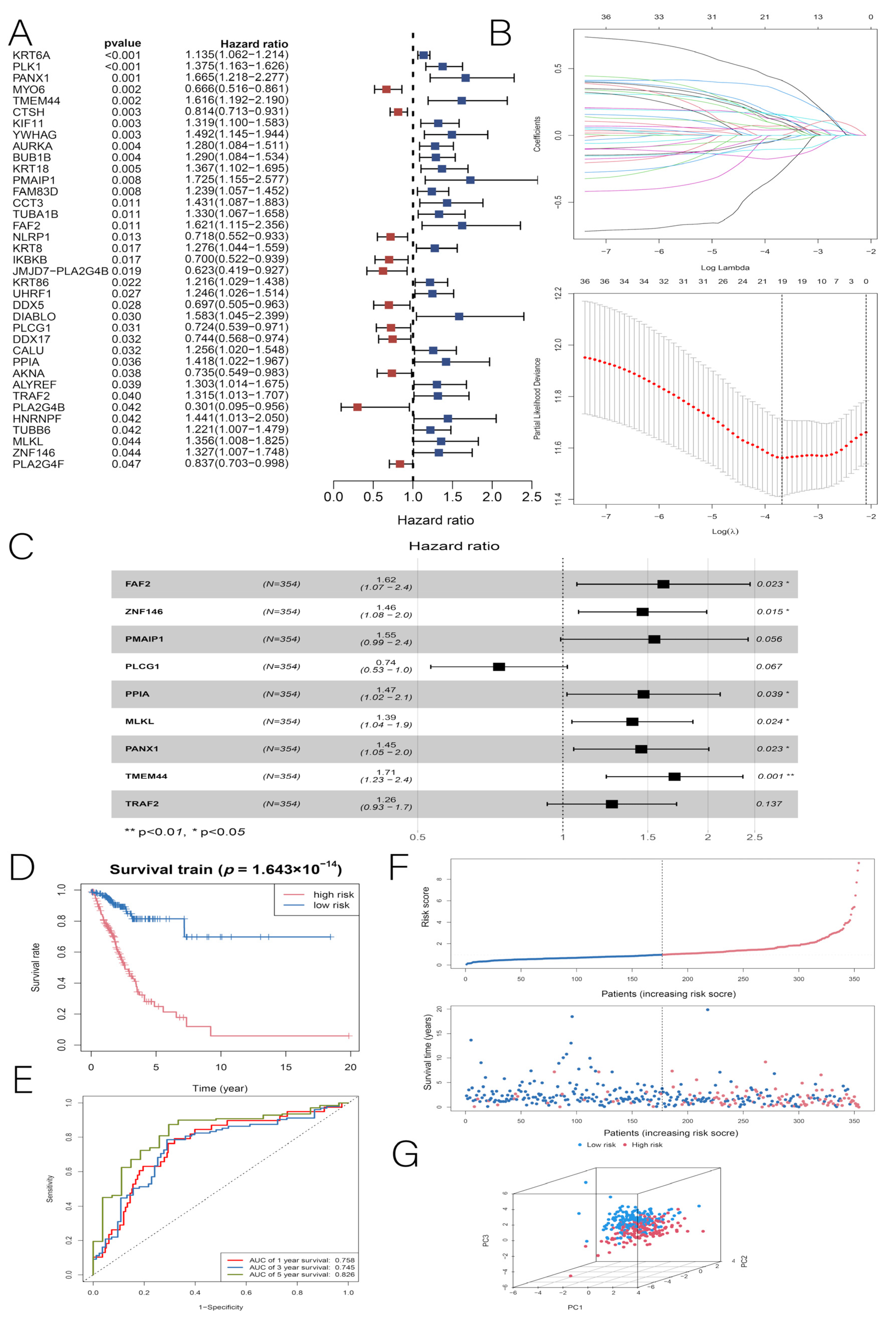
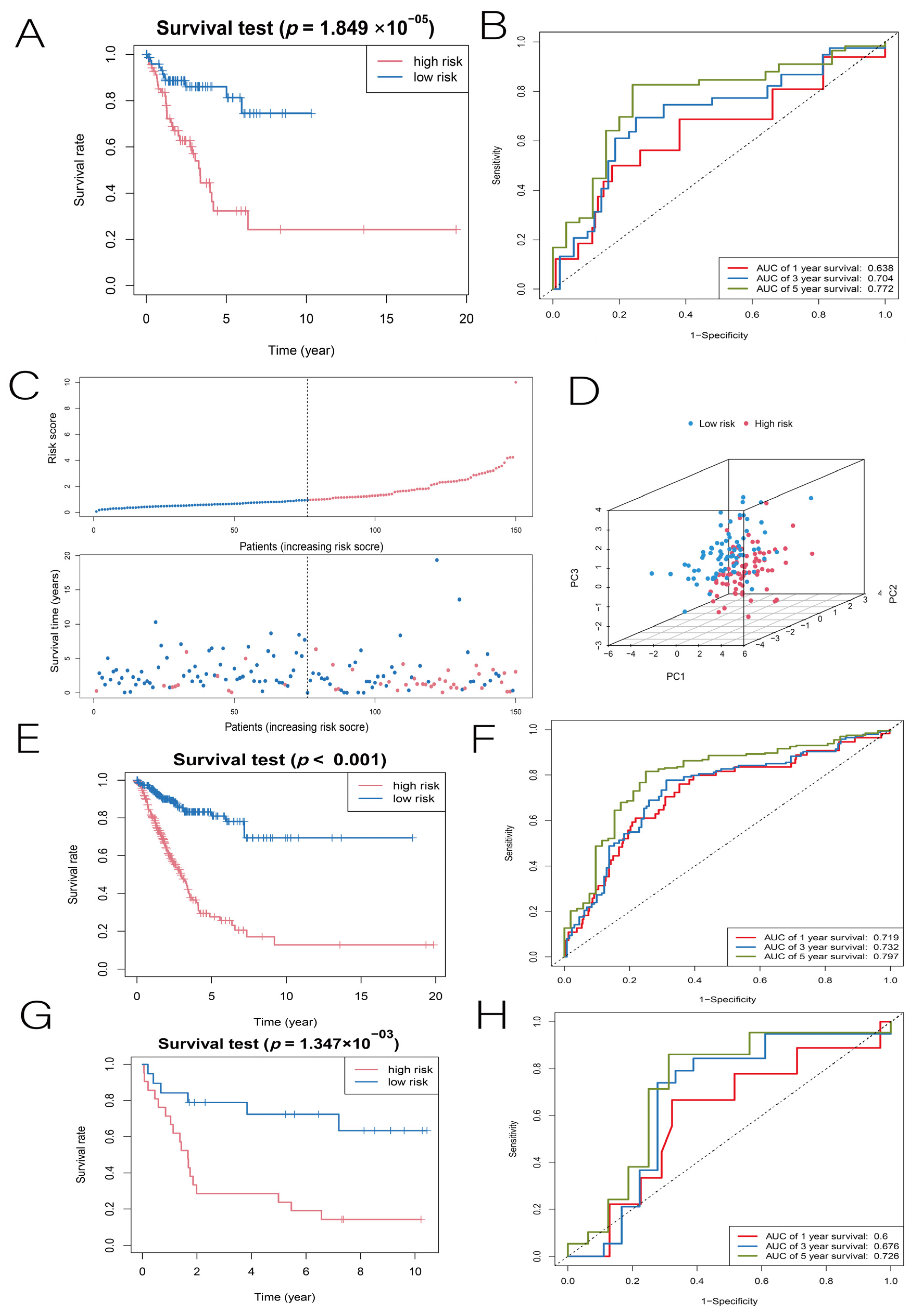
3.2. Construction and Validation of the LUAD Prognostic Model
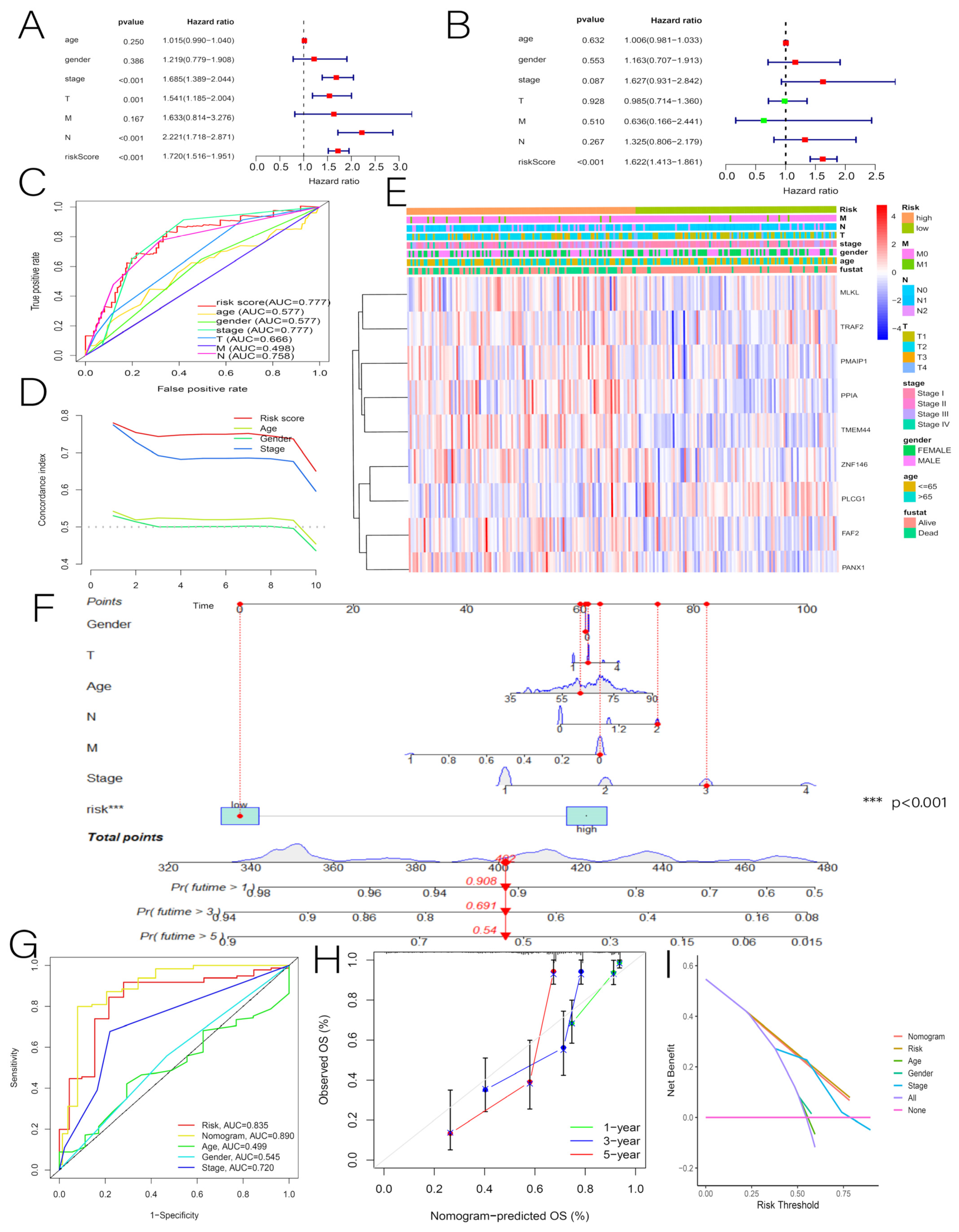
3.3. Independent Predictive Value of Risk Scores and Clinical Characteristics
3.4. Correlation of Risk Scores with GSEA and Mutations
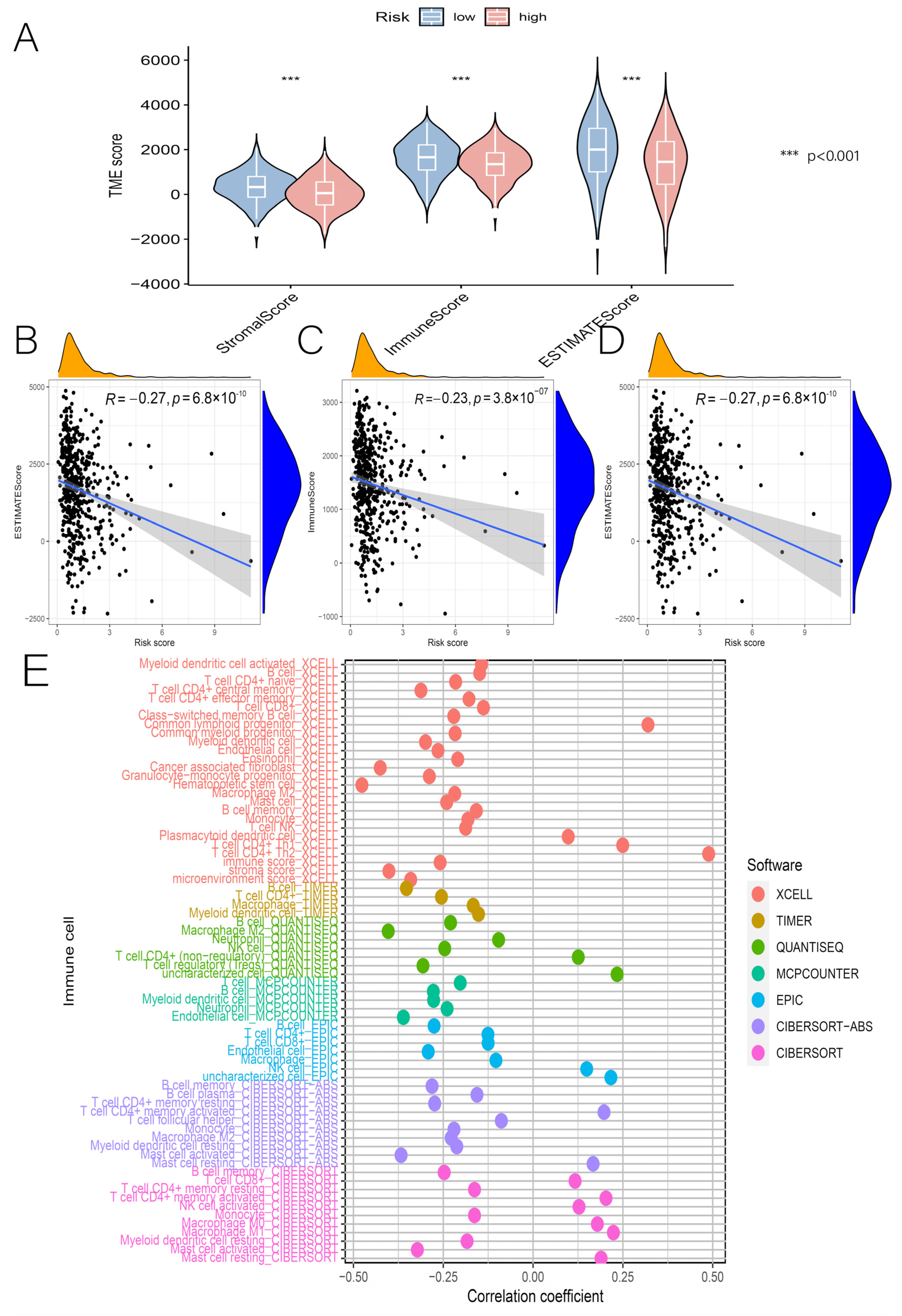
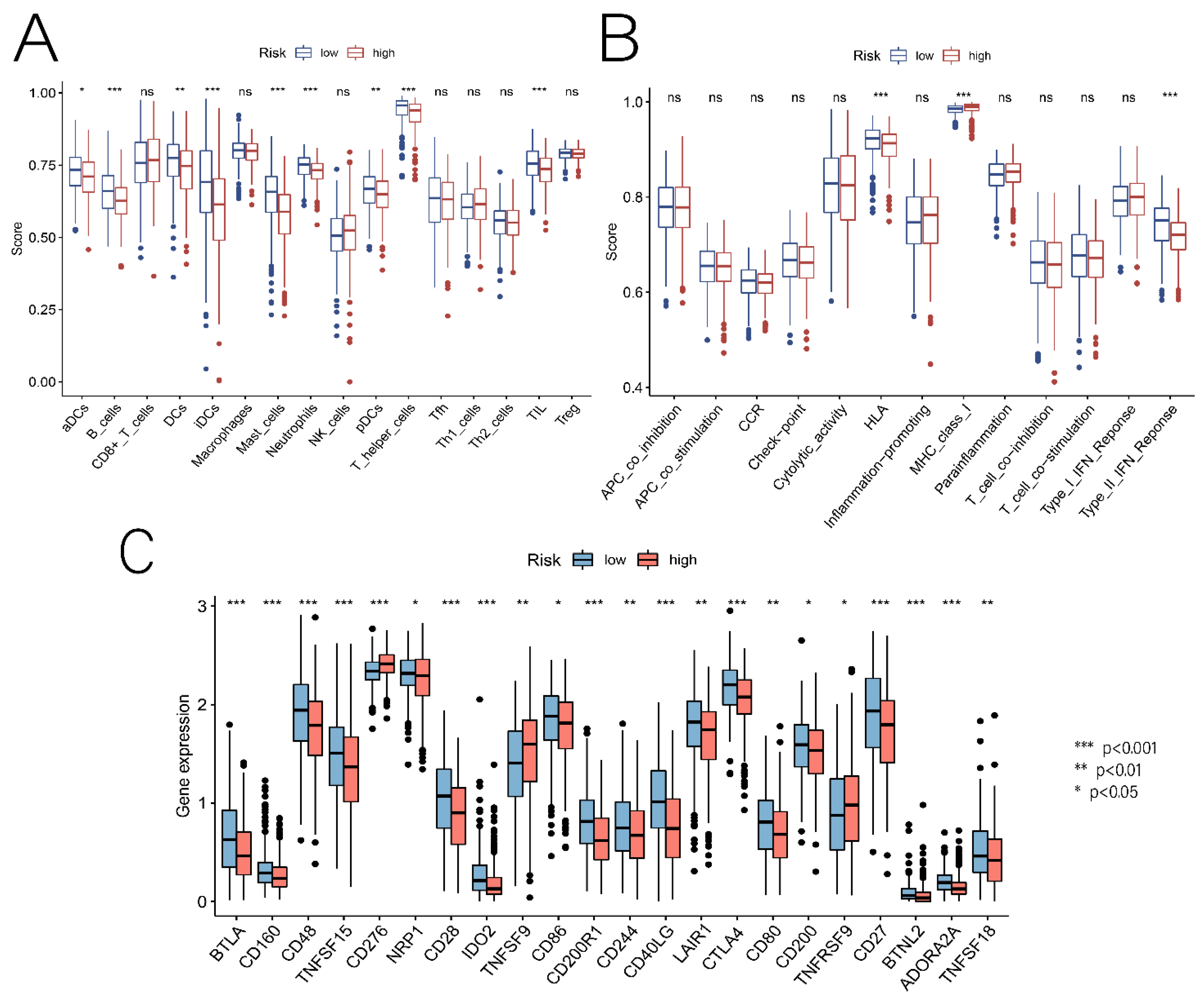
3.5. Relationship between Risk Scores and TME, Immune Cells, and ICs
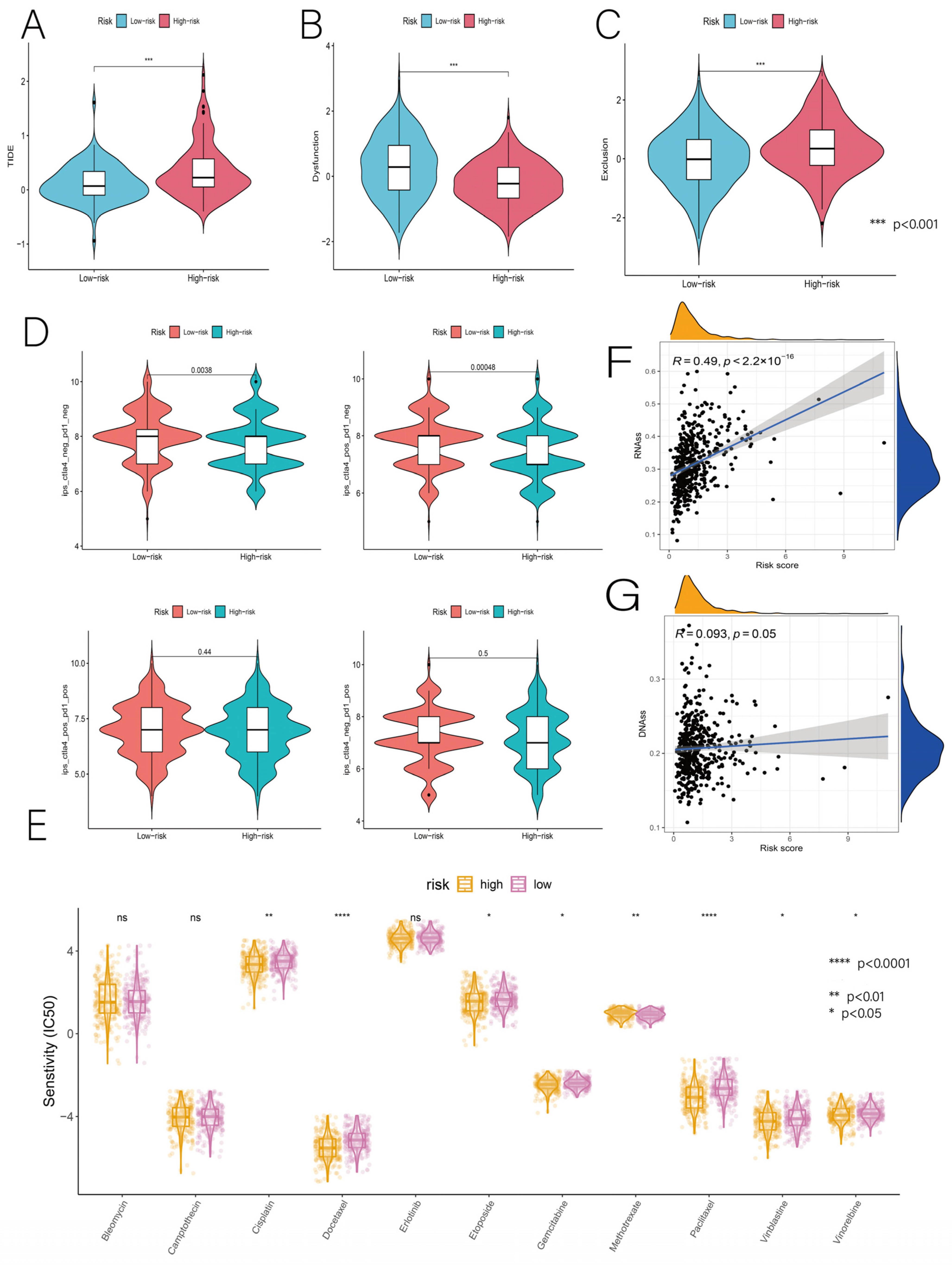
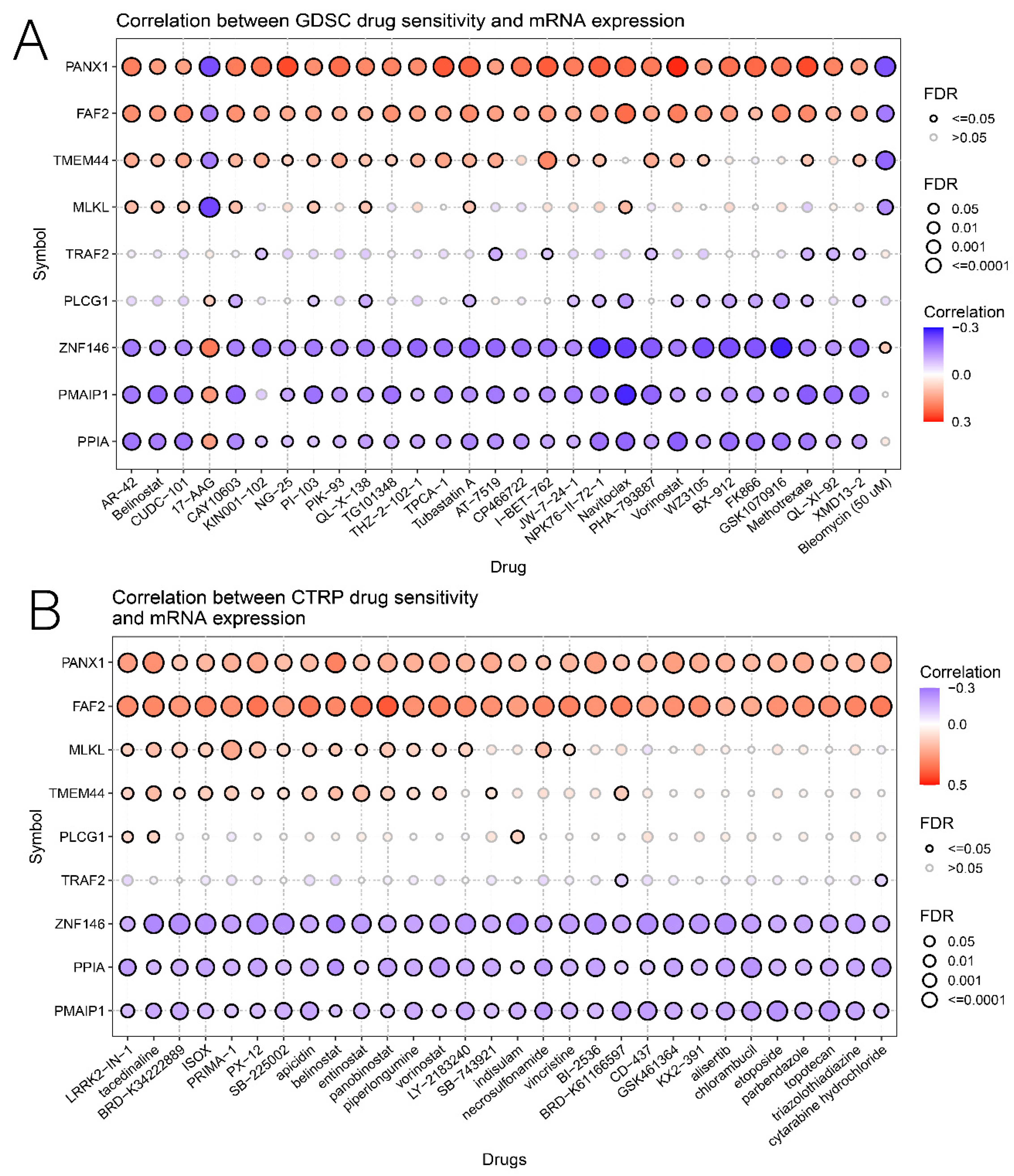
3.6. Relationship of Risk Score with Immunotherapy and Drug Sensitivity
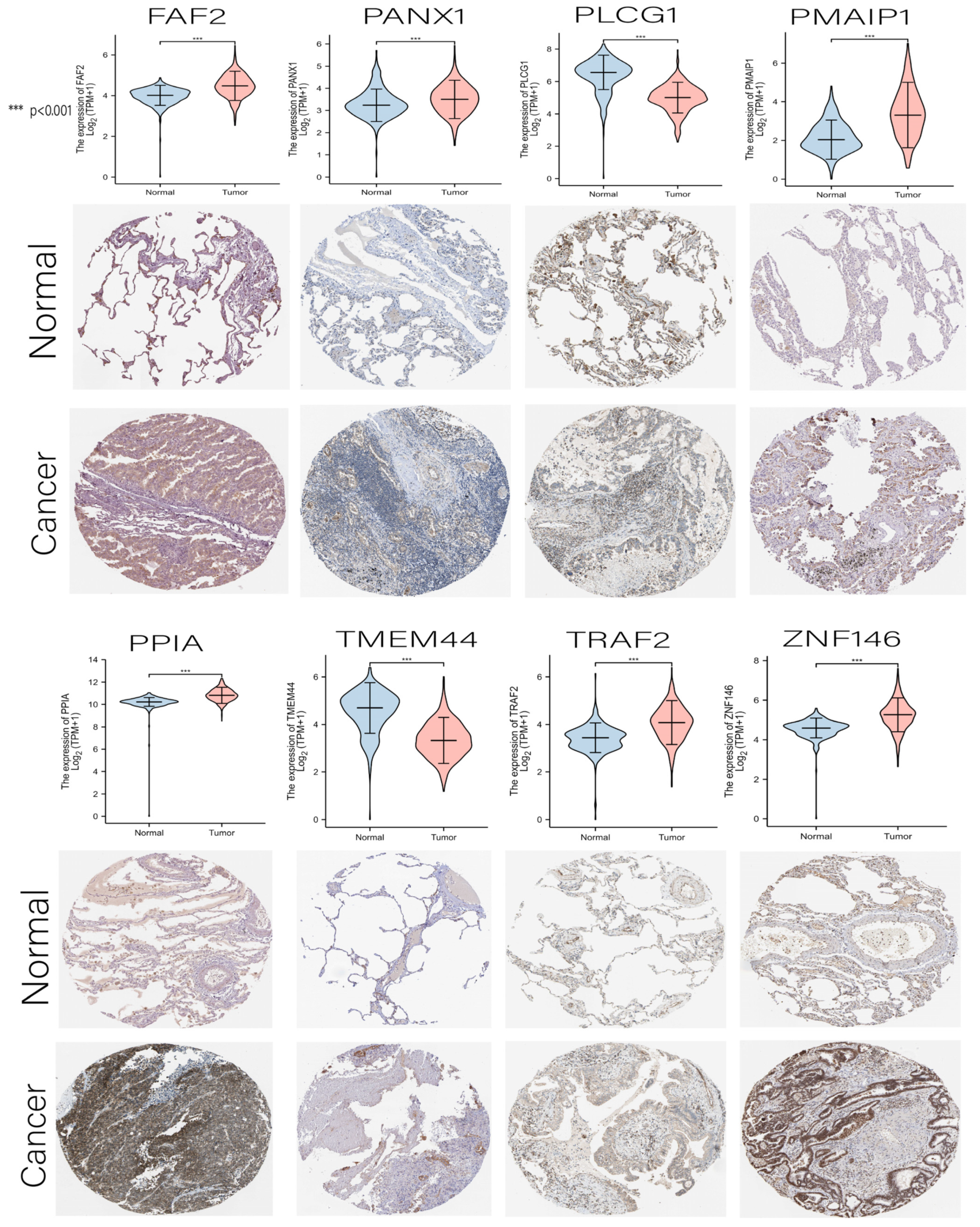
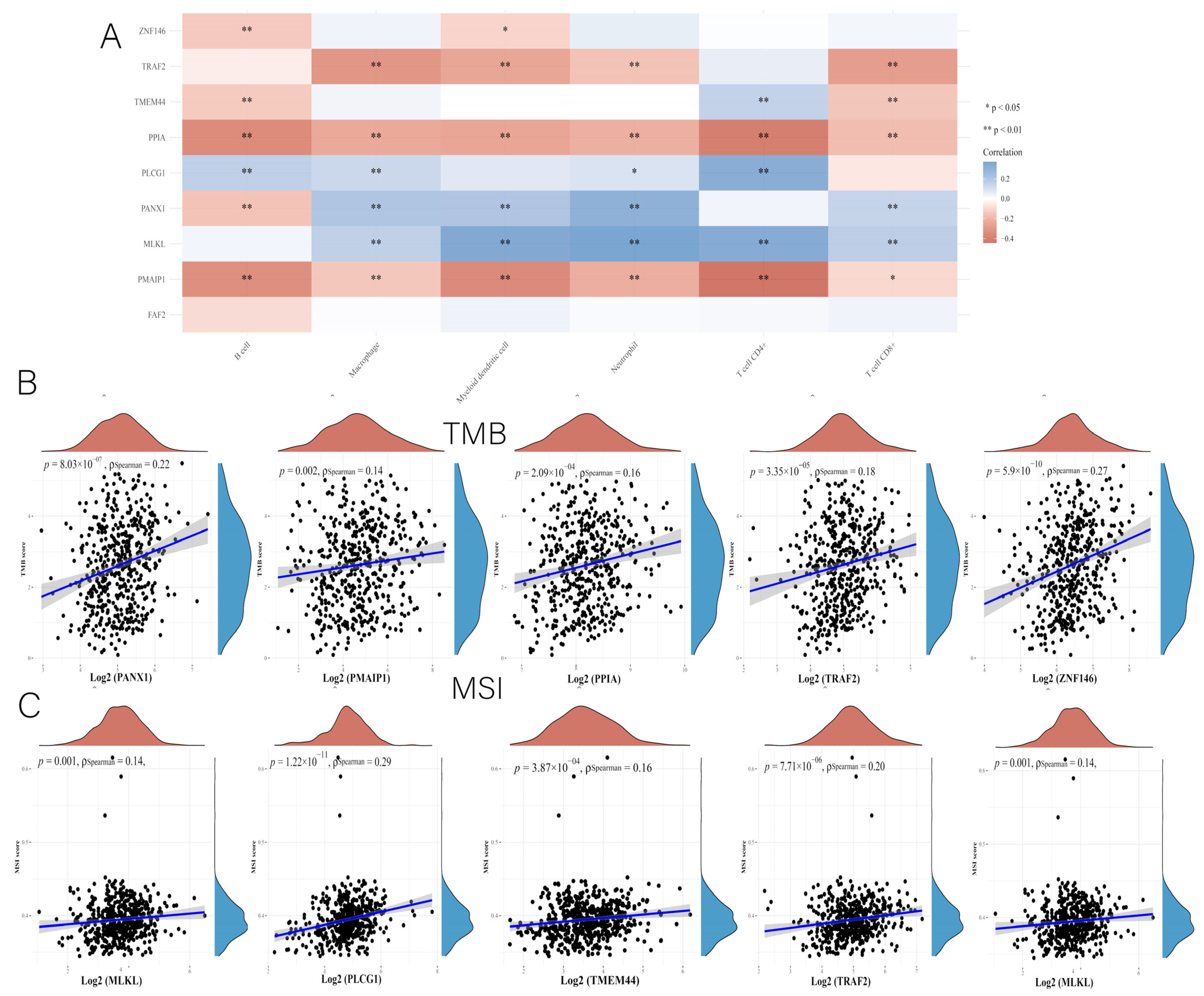
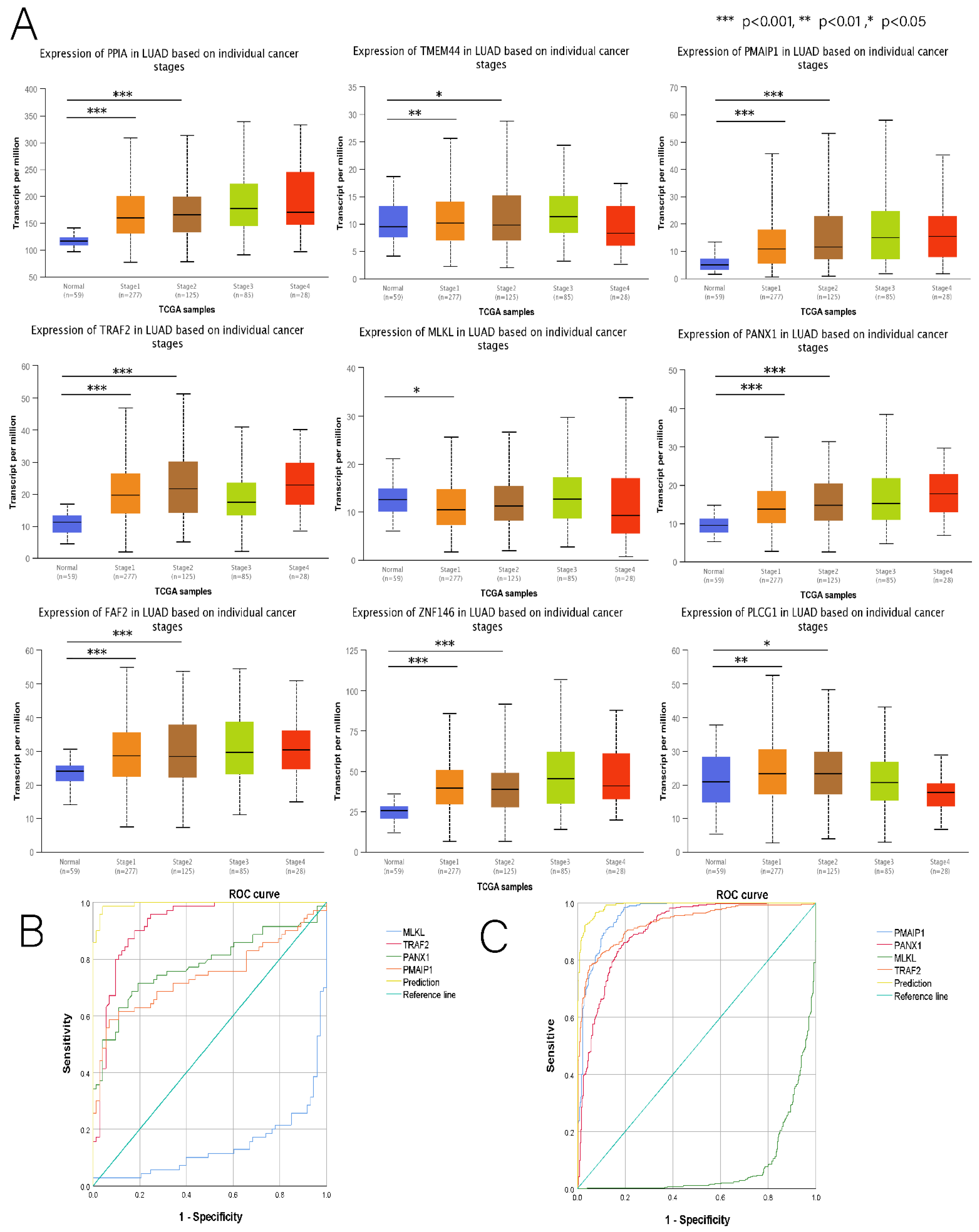
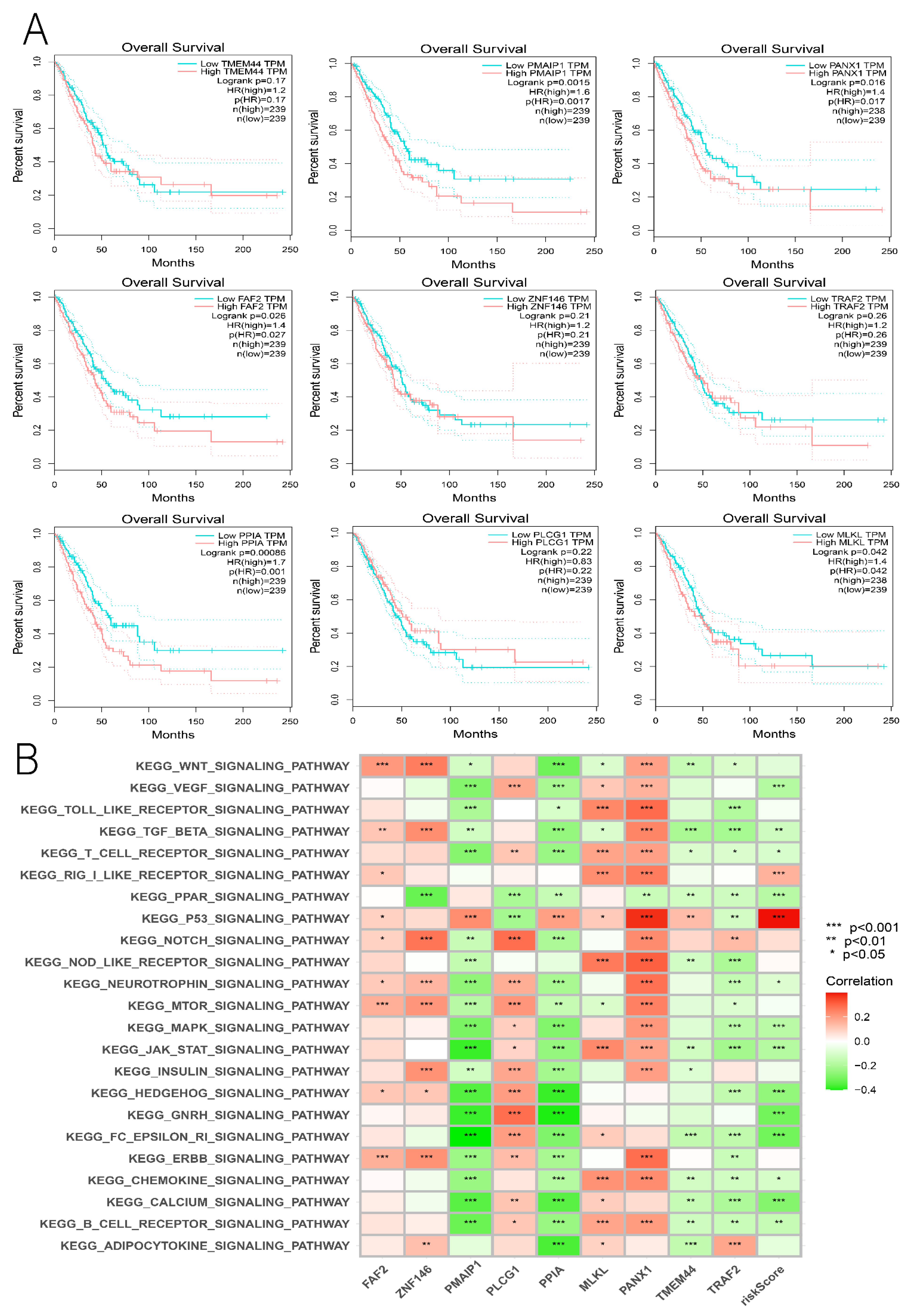
3.7. Comprehensive Analysis of Nine DENRGs in the Prognostic Model
3.8. Building LUAD Diagnostic Models
3.9. Hub Genes with Both Diagnosis and Prognosis
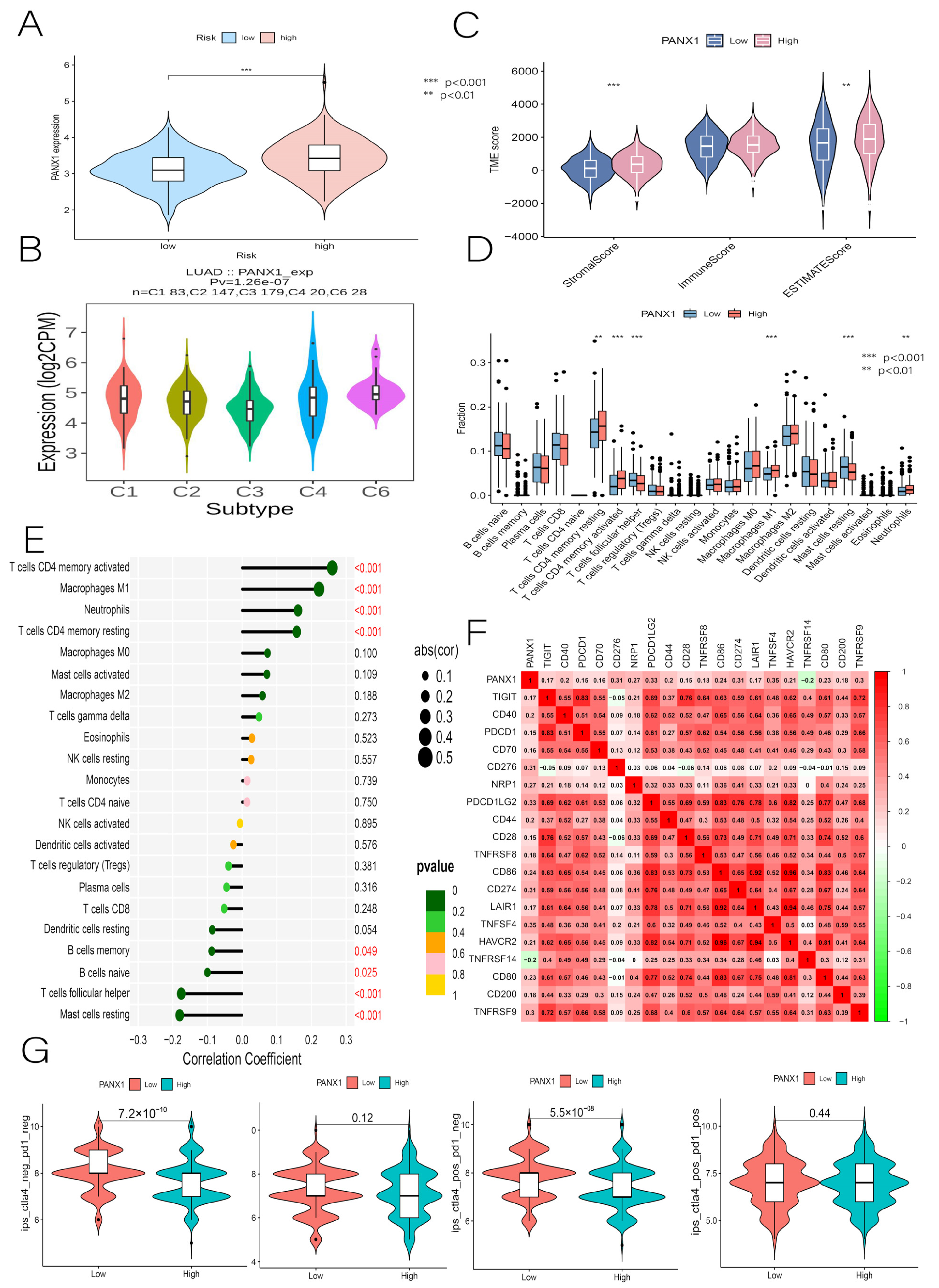
3.10. Immunocorrelation Analysis of PANX1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LUAD | Lung adenocarcinoma |
| NSCLC | Non-small cell lung cancer |
| NRGs | Necroptosis-related genes |
| DENRGs | Differentially expressed necroptosis genes |
| TCGA | The Cancer Genome Atlas |
| GEO | Gene Expression Omnibus |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GO | Gene Ontology |
| OS | Overall survival |
| K-M | Kaplan-Meier |
| ROC | Receiver operating characteristic |
| PCA | Principal component analysis |
| ICI | Immune checkpoint inhibitor |
| GTEx | Genotype-Tissue Expression |
| GSEA | Gene set enrichment analysis |
| C-index | Concordance index |
| DCA | Decision curve analysis |
| TME | Tumor microenvironment |
| ssGSEA | Single sample gene set enrichment analysis |
| ICs | Immune checkpoints |
| HPA | Human Protein Atlas |
| TIDE | The Tumor Immune Dysfunction and Exclusion |
| TCIA | The Cancer Immunome Atlas |
| OCLR | One-class logistic regression |
| GSVA | Gene set variation analysis |
References
- Cao, M.; Li, H.; Sun, D.; Chen, W. Cancer burden of major cancers in China: A need for sustainable actions. Cancer Commun. 2020, 40, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zou, K.; Feng, X.; Chen, M.; Li, C.; Tang, R.; Xuan, Y.; Luo, M.; Chen, W.; Qiu, H.; et al. Downregulation of NMI promotes tumor growth and predicts poor prognosis in human lung adenocarcinomas. Mol. Cancer 2017, 16, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Yamada, T.; Kita, K.; Taniguchi, H.; Arai, S.; Fukuda, K.; Terashima, M.; Ishimura, A.; Nishiyama, A.; Tanimoto, A.; et al. Transient IGF-1R inhibition combined with osimertinib eradicates AXL-low expressing EGFR mutated lung cancer. Nat. Commun. 2020, 11, 4607. [Google Scholar] [CrossRef]
- Shaurova, T.; Zhang, L.; Goodrich, D.W.; Hershberger, P.A. Understanding Lineage Plasticity as a Path to Targeted Therapy Failure in EGFR-Mutant Non-small Cell Lung Cancer. Front. Genet. 2020, 11, 281. [Google Scholar] [CrossRef]
- Xu, J.Y.; Zhang, C.; Wang, X.; Zhai, L.; Ma, Y.; Mao, Y.; Qian, K.; Sun, C.; Liu, Z.; Jiang, S.; et al. Integrative Proteomic Characterization of Human Lung Adenocarcinoma. Cell 2020, 182, 245–261.e217. [Google Scholar] [CrossRef]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef]
- Snyder, A.G.; Hubbard, N.W.; Messmer, M.N.; Kofman, S.B.; Hagan, C.E.; Orozco, S.L.; Chiang, K.; Daniels, B.P.; Baker, D.; Oberst, A. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci. Immunol. 2019, 4, eaaw2004. [Google Scholar] [CrossRef]
- Chan, F.K.; Luz, N.F.; Moriwaki, K. Programmed necrosis in the cross talk of cell death and inflammation. Annu. Rev. Immunol. 2015, 33, 79–106. [Google Scholar] [CrossRef]
- Han, W.; Li, L.; Qiu, S.; Lu, Q.; Pan, Q.; Gu, Y.; Luo, J.; Hu, X. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol. Cancer 2007, 6, 1641–1649. [Google Scholar] [CrossRef]
- Smyth, P.; Sessler, T.; Scott, C.J.; Longley, D.B. FLIP(L): The pseudo-caspase. FEBS J. 2020, 287, 4246–4260. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Nguyen, A.; Bakhshinyan, D.; Wei, J.; Hare, D.N.; MacNeill, K.L.; Wan, Y.; Oberst, A.; Bramson, J.L.; Nasir, J.A.; et al. De novo necroptosis creates an inflammatory environment mediating tumor susceptibility to immune checkpoint inhibitors. Commun. Biol. 2020, 3, 645. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Huang, S.; Shen, Z. Biomarkers for the detection of necroptosis. Cell Mol. Life Sci. 2016, 73, 2177–2181. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Wang, G.; Pei, K.; Mo, C.; Rong, Z.; Xu, G. A Novel Prognostic Model Based on Seven Necroptosis-Related miRNAs for Predicting the Overall Survival of Patients with Lung Adenocarcinoma. BioMed Res. Int. 2022, 2022, 3198590. [Google Scholar] [CrossRef]
- Lu, Y.; Luo, X.; Wang, Q.; Chen, J.; Zhang, X.; Li, Y.; Chen, Y.; Li, X.; Han, S. A Novel Necroptosis-Related lncRNA Signature Predicts the Prognosis of Lung Adenocarcinoma. Front. Genet. 2022, 13, 862741. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.; Wang, J.; Liu, N.; Zheng, X.; Li, Y.; Ma, Y.; Zheng, H.; Chen, X.; Fan, C.; Zhang, R.; et al. Integration of Single-Cell RNA Sequencing and Bulk RNA Sequencing Data to Establish and Validate a Prognostic Model for Patients with Lung Adenocarcinoma. Front. Genet. 2022, 13, 833797. [Google Scholar] [CrossRef]
- Yu, Z.; Jiang, N.; Su, W.; Zhuo, Y. Necroptosis: A Novel Pathway in Neuroinflammation. Front. Pharmacol. 2021, 12, 701564. [Google Scholar] [CrossRef]
- Belli-Marin, J.F.C.; Bocchi, E.A.; Ayub-Ferreira, S.; Junior, N.C.; Guimaraes, G.V. Effects of beta-blocker therapy on exercise oscillatory ventilation in reduced ejection fraction heart failure patients: A case series study. Biomed. Pharmacol. 2022, 152, 113106. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauss, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells—Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Park, H.H.; Kim, H.R.; Park, S.Y.; Hwang, S.M.; Hong, S.M.; Park, S.; Kang, H.C.; Morgan, M.J.; Cha, J.H.; Lee, D.; et al. RIPK3 activation induces TRIM28 derepression in cancer cells and enhances the anti-tumor microenvironment. Mol. Cancer 2021, 20, 107. [Google Scholar] [CrossRef]
- Wang, X.; Avsec, D.; Obreza, A.; Yousefi, S.; Mlinaric-Rascan, I.; Simon, H.U. A Putative Serine Protease is Required to Initiate the RIPK3-MLKL-Mediated Necroptotic Death Pathway in Neutrophils. Front. Pharmacol. 2020, 11, 614928. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef] [PubMed]
- Galvano, A.; Gristina, V.; Malapelle, U.; Pisapia, P.; Pepe, F.; Barraco, N.; Castiglia, M.; Perez, A.; Rolfo, C.; Troncone, G.; et al. The prognostic impact of tumor mutational burden (TMB) in the first-line management of advanced non-oncogene addicted non-small-cell lung cancer (NSCLC): A systematic review and meta-analysis of randomized controlled trials. ESMO Open 2021, 6, 100124. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Ricci, A.D.; Brandi, G. PD-L1, TMB, MSI, and Other Predictors of Response to Immune Checkpoint Inhibitors in Biliary Tract Cancer. Cancers 2021, 13, 558. [Google Scholar] [CrossRef] [PubMed]
- Brueckl, W.M.; Ficker, J.H.; Zeitler, G. Clinically relevant prognostic and predictive markers for immune-checkpoint-inhibitor (ICI) therapy in non-small cell lung cancer (NSCLC). BMC Cancer 2020, 20, 1185. [Google Scholar] [CrossRef]
- Tsai, H.F.; Hsu, P.N. Cancer immunotherapy by targeting immune checkpoints: Mechanism of T cell dysfunction in cancer immunity and new therapeutic targets. J. Biomed. Sci. 2017, 24, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahluwalia, P.; Ahluwalia, M.; Mondal, A.K.; Sahajpal, N.; Kota, V.; Rojiani, M.V.; Rojiani, A.M.; Kolhe, R. Immunogenomic Gene Signature of Cell-Death Associated Genes with Prognostic Implications in Lung Cancer. Cancers 2021, 13, 155. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Ciuleanu, T.-E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Jia, R.; Sui, Z.; Zhang, H.; Yu, Z. Identification and Validation of Immune-Related Gene Signature for Predicting Lymph Node Metastasis and Prognosis in Lung Adenocarcinoma. Front. Mol. Biosci. 2021, 8, 679031. [Google Scholar] [CrossRef] [PubMed]
- Zanfardino, M.; Pane, K.; Mirabelli, P.; Salvatore, M.; Franzese, M. TCGA-TCIA Impact on Radiogenomics Cancer Research: A Systematic Review. Int. J. Mol. Sci. 2019, 20, 6033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.; Di Maio, M. Platinum-based chemotherapy in advanced non-small-cell lung cancer: Optimal number of treatment cycles. Expert Rev. Anticancer 2016, 16, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. p53 signaling in cancer progression and therapy. Cancer Cell Int. 2021, 21, 703. [Google Scholar] [CrossRef]
- Shi, D.; Jiang, P. A Different Facet of p53 Function: Regulation of Immunity and Inflammation During Tumor Development. Front. Cell Dev. Biol. 2021, 9, 762651. [Google Scholar] [CrossRef]
- Nouri-Nejad, D.; O’Donnell, B.L.; Patil, C.S.; Sanchez-Pupo, R.E.; Johnston, D.; Sayedyahossein, S.; Jurcic, K.; Lau, R.; Gyenis, L.; Litchfield, D.W.; et al. Pannexin 1 mutation found in melanoma tumor reduces phosphorylation, glycosylation, and trafficking of the channel-forming protein. Mol. Biol. Cell 2021, 32, 376–390. [Google Scholar] [CrossRef]
- Sayedyahossein, S.; Huang, K.; Li, Z.; Zhang, C.; Kozlov, A.M.; Johnston, D.; Nouri-Nejad, D.; Dagnino, L.; Betts, D.H.; Sacks, D.B.; et al. Pannexin 1 binds beta-catenin to modulate melanoma cell growth and metabolism. J. Biol. Chem. 2021, 296, 100478. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, X.; Liang, Q.; An, Y.; Wei, M.; Shi, W. Inhibiting of circ-TLK1 inhibits the progression of glioma through down-regulating PANX1 via targeting miR-17-5p. J. Mol. Histol. 2021, 52, 1007–1020. [Google Scholar] [CrossRef]
- Shi, G.; Liu, C.; Yang, Y.; Song, L.; Liu, X.; Wang, C.; Peng, Z.; Li, H.; Zhong, L. Panx1 promotes invasion-metastasis cascade in hepatocellular carcinoma. J. Cancer 2019, 10, 5681–5688. [Google Scholar] [CrossRef] [Green Version]
- Vultaggio-Poma, V.; Sarti, A.C.; Di Virgilio, F. Extracellular ATP: A Feasible Target for Cancer Therapy. Cells 2020, 9, 2496. [Google Scholar] [CrossRef]
- Taruno, A. ATP Release Channels. Int. J. Mol. Sci. 2018, 19, 808. [Google Scholar] [CrossRef] [Green Version]
- Alomari, O.A.; Makhadmeh, S.N.; Al-Betar, M.A.; Alyasseri, Z.A.A.; Doush, I.A.; Abasi, A.K.; Awadallah, M.A.; Zitar, R.A. Gene selection for microarray data classification based on Gray Wolf Optimizer enhanced with TRIZ-inspired operators. Knowl.-Based Syst. 2021, 223, 107034. [Google Scholar] [CrossRef]
- Tang, C.; Zheng, X.; Liu, X.; Zhang, W.; Zhang, J.; Xiong, J.; Wang, L. Cross-View Locality Preserved Diversity and Consensus Learning for Multi-View Unsupervised Feature Selection. IEEE Trans. Knowl. Data Eng. 2022, 34, 4705–4716. [Google Scholar] [CrossRef]
- Tang, C.; Liu, X.; Zhu, X.; Xiong, J.; Li, M.; Xia, J.; Wang, X.; Wang, L. Feature Selective Projection with Low-Rank Embedding and Dual Laplacian Regularization. IEEE Trans. Knowl. Data Eng. 2020, 32, 1747–1760. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCGA-LUAD | GEO-GSE75037 | GEO-GSE19188 | ||
|---|---|---|---|---|
| Variable | Category | Numbers | ||
| Gender | Male | 352 | 48 | 66 |
| Female | 242 | 118 | 22 | |
| Diagnostic Age | ≤65 | 265 | 58 | Unknown |
| >65 | 310 | 108 | Unknown | |
| Stage | I | 174 | 50 | 40 |
| II | 324 | 20 | Unknown | |
| III | 71 | 11 | Unknown | |
| IV | 23 | 2 | Unknown | |
| T | T1 | 23 | Unknown | Unknown |
| T2 | 93 | Unknown | Unknown | |
| T3 | 200 | Unknown | Unknown | |
| T4 | 119 | Unknown | Unknown | |
| M | M0 | 445 | Unknown | Unknown |
| M1 | 16 | Unknown | Unknown | |
| N | N0 | 383 | Unknown | Unknown |
| N1 | 127 | Unknown | Unknown | |
| N2 | 67 | Unknown | Unknown | |
| N3 | 5 | Unknown | Unknown | |
| Fustat | Alive | 354 | 83 | 86 |
| Dead | 240 | 83 | 24 | |
| Train-GSE75037 | Normal | LUAD | All | Accuracy |
| 4-mRNA negative | 70 | 3 | 73 | 95.9 |
| 4-mRNA positive | 2 | 68 | 70 | 97.1 |
| All accuracy | 70 | 68 | 143 | 96.5 |
| Test-TCGALUAD | Normal | LUAD | All | Accuracy |
| 4-mRNA negative | 321 | 26 | 347 | 92.5 |
| 4-mRNA positive | 14 | 368 | 382 | 96.3 |
| All accuracy | 321 | 368 | 729 | 94.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Wang, Y.; Zhou, X.; Zhang, Z.; Ju, H.; Diao, X.; Wu, J.; Zhang, J. Construction of a Prognostic and Early Diagnosis Model for LUAD Based on Necroptosis Gene Signature and Exploration of Immunotherapy Potential. Cancers 2022, 14, 5153. https://doi.org/10.3390/cancers14205153
Zhang B, Wang Y, Zhou X, Zhang Z, Ju H, Diao X, Wu J, Zhang J. Construction of a Prognostic and Early Diagnosis Model for LUAD Based on Necroptosis Gene Signature and Exploration of Immunotherapy Potential. Cancers. 2022; 14(20):5153. https://doi.org/10.3390/cancers14205153
Chicago/Turabian StyleZhang, Baizhuo, Yudong Wang, Xiaozhu Zhou, Zhen Zhang, Haoyu Ju, Xiaoqi Diao, Jiaoqi Wu, and Jing Zhang. 2022. "Construction of a Prognostic and Early Diagnosis Model for LUAD Based on Necroptosis Gene Signature and Exploration of Immunotherapy Potential" Cancers 14, no. 20: 5153. https://doi.org/10.3390/cancers14205153
APA StyleZhang, B., Wang, Y., Zhou, X., Zhang, Z., Ju, H., Diao, X., Wu, J., & Zhang, J. (2022). Construction of a Prognostic and Early Diagnosis Model for LUAD Based on Necroptosis Gene Signature and Exploration of Immunotherapy Potential. Cancers, 14(20), 5153. https://doi.org/10.3390/cancers14205153