
Aptamers, a New Therapeutic Opportunity for the Treatment of Multiple Myeloma



Abstract
:Simple Summary
Abstract
1. Multiple Myeloma and Monoclonal Antibodies

{kind=link}
{kind=link}
| Name | Target | Class | Mechanism | Phase | References |
|---|---|---|---|---|---|
| Daratumumab | CD38 | mAb | ADCC, CDC | Approved alone for RRMM non-responder to proteasome inhibitors and immunomodulatory agents; in combination with lenalidomide and dexamethasone for RRMM or non-autologous stem cell transplant eligible NDMM; in combination with bortezomib, prednisone and melphalan for non-autologous stem cell transplant eligible NDMM, in combination with bortezomib, thalidomide and dexamethasone for autologous stem cell transplant eligible NDMM, in combination with bortezomib/carfilzomib and dexamethasone for RRMM, in combination with pomalidomide and dexamethasone for RRMM with at least one prior treatment with lenalidomide and a proteasome inhibitor | [6,8] |
| Isatuximab | CD38 | mAb | ADCC, CDC | Approved in combination with pomalidomide and dexamethasone for RRMM | |
| Elotuzumab | SLAMF7 | mAb | ADCC | Approved in combination with lenalidomide, pomalidomide and dexamethasone for RRMM | |
| Belantamab mafodotin | BCMA | ADC (monomethyl auristatin-F) | ADCC, mitosis inhibition by microtubule disruption | Approved in monotherapy for RRMM | |
| MEDI2228 | BCMA | ADC (pyrrolobenzodiazepine) | DNA alkylation | Clinical Trial. Phase I | [9] |
| Tabalumab | BAFF | mAb | Soluble and membrane-bound BAFF neutralization | Clinical Trial. Phase II | [10,11] |
| Milatuzumab | CD74 | mAb | CD74 antagonist | Clinical Trial. Phase I/II | [12] |
| Siltuximab | IL-6 | mAb | IL-6 neutralization | Clinical Trial. Phase II | [13] |
| BI-505 | ICAM1 | mAb | Macrophage dependent PCD | Clinical Trial. Phase II | [14] |
| Indatuximab ravtansine | CD138 | ADC (maytansinoid DM4) | Mitosis inhibition by impediment of tubulin polymerization and microtubule assembly | Clinical Trial. Phase I/IIa | [15] |
| Pembrolizumab | PD-L1 | Checkpoint inhibitor | Clinical Trial. Phase III | [16] | |
| Atezolizumab | PD-L1 | Checkpoint inhibitor | Clinical Trial. Phase Ib | [17] | |
| Nivolumab | PD-1 | Checkpoint inhibitor | Clinical Trial. Phase I | [18] | |
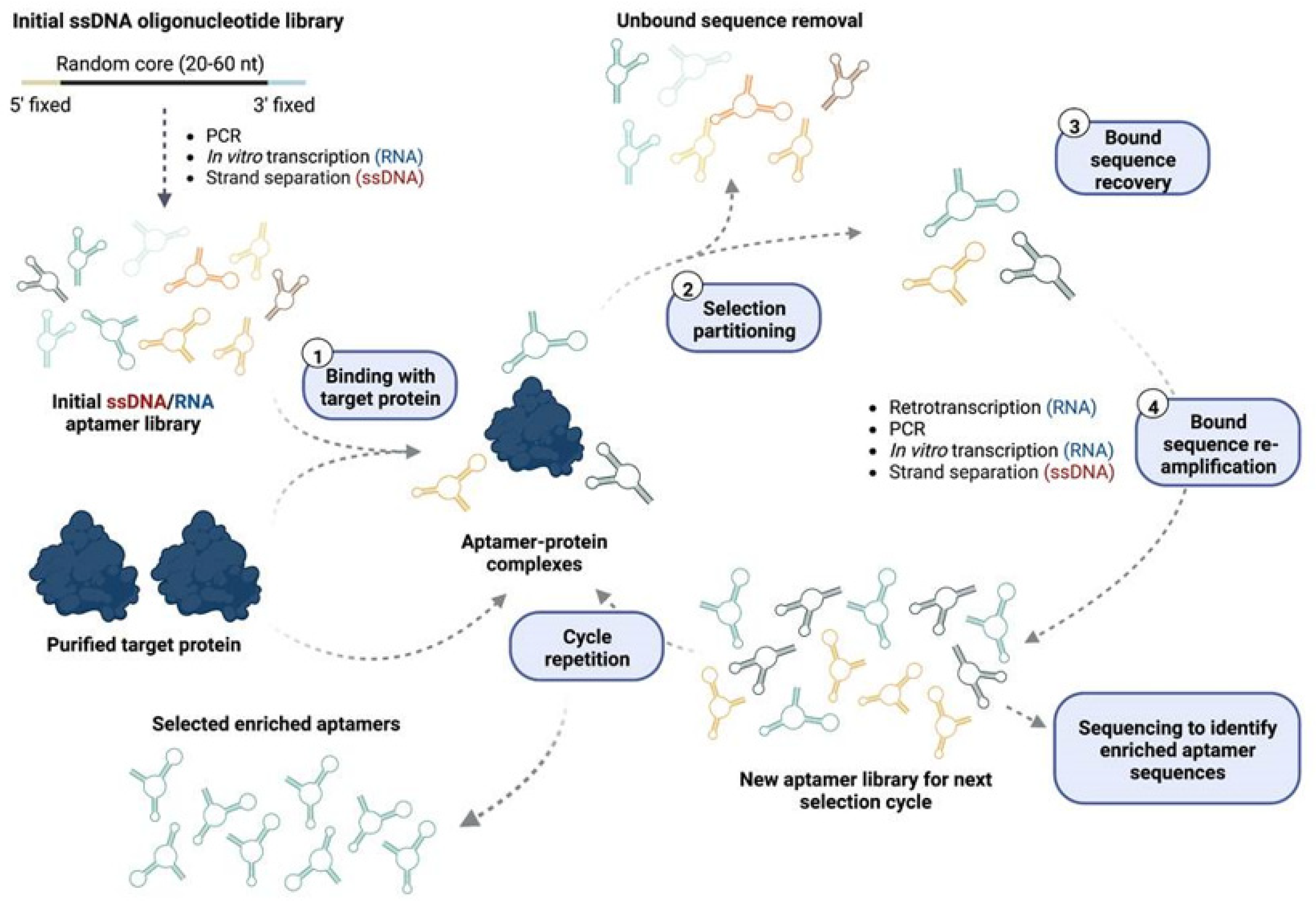
2. Aptamers
3. Current Aptamers for MM Precision Medicine
- Aptamer against AXII: Annexin A2 (AXII) is a calcium-dependent, phospholipid binding member of the annexin family. AXII is overexpressed in MM plasma cell membranes, with its expression being negatively correlated with patient survival [28]. The interaction of AXII with its receptor AXIIR enhances MM cell adhesion and growth in the BM microenvironment, potentially supporting the homing and growth of MM cells in the BM. AXII can be secreted by various cell types in the BM, promoting MM cell growth by creating a pro-tumorigenic niche, thus, targeting the AXII/AXIIR axis represents an attractive approach for the development of therapies targeted at the MM niche [29]. Zhou et al. [30] identified a ssDNA aptamer (wh6) that was able to bind AXII in the low nanomolar range through nine rounds of protein-based SELEX. They showed that the aptamer was able to specifically bind MM cells expressing AXII both in vitro and in vivo, and it could inhibit the AXII induced adhesion and progression in MM cell lines, indicating the suitability of the wh6 aptamer for targeted MM treatment.
- BCMA targeted aptamer: B-cell maturation antigen (BCMA) is a member of the tumor necrosis factor (TNF) receptor superfamily, which is preferentially expressed by late-stage B lymphocytes while showing minimal expression in hematopoietic stem cells [31]. Under physiological conditions, the binding of its specific ligands BAFF and APRIL induces the activation of both canonical and non-canonical NF-κB pathways, promoting long-lived plasma cells survival. However, BCMA overexpression and increased activation are associated with MM progression in terms of the upregulation of NF-κB pathways and subsequent overexpression of critical genes for MM growth and survival [31,32]. In this direction, Catuogno et al. [32] selected a BCMA-targeted internalizing RNA aptamer (apt69.T) through a variation of the cell-SELEX approach. In vitro approaches using MM cell lines showed that the selected apt69.T aptamer was able to readily bind BCMA and inhibit the APRIL dependent downstream signaling pathway. Furthermore, it was able to internalize rapidly and successfully deliver therapeutic oligonucleotides to MM cells. For that, the BCMA aptamer was conjugated to miRNA and miRNA antagonists using a sticky-end based approach, leading to consequent upregulation of miR-137 and downregulation of miR-222 in MM cell lines. The upregulation of the tumor suppressor miR-137 was able to reduce MM cell viability, highlighting the feasibility of using MM-specific aptamers for the effective delivery of therapeutic oligonucleotides to MM cells.
- Aptamer against C-MET: C-MET is a transmembrane tyrosine kinase known to be the receptor of the hepatocyte growth factor (HGF) cytokine. In MM, C-MET expression gradually increases during disease development, its high expression being correlated with poor MM patient outcomes [33]. Upon HGF binding, C-MET dimerizes, resulting in kinase auto-phosphorylation and the creation of a multi-substrate docking site necessary for the induction of downstream signaling cascades which ultimately contribute to MM development by promoting cell growth, migration and angiogenesis while inhibiting apoptosis [33]. SL1 [34] is the truncated version of the original CLN0003 ssDNA aptamer, which was selected against purified C-MET through a filter SELEX approach for the recognition of C-MET overexpressing tumors [35]. Accordingly, Zhang et al. [33] demonstrated that targeting C-MET via the SL1 aptamer [34,35] could be a potential therapeutic approach in MM, showing that SL1 was able to inhibit HGF-dependent C-MET signaling and suppress MM cell growth in vitro. Furthermore, SL1 showed synergism with bortezomib, highlighting the potential for novel combination therapies in MM.
- Conjugated CD38-doxorubicin aptamer: CD38 is a cell surface glycoprotein which is highly and homogeneously expressed in MM cells, with minimal expression on normal myeloid and lymphoid cells. This highly versatile molecule contributes to MM development by acting as a receptor for proliferative signaling, as an adhesion molecule or as an ectoenzyme in the catabolism of NAD+ and NADP [36,37]; thus, in recent years, it has become one of the main targets for anti-MM targeted therapy development. Wen et al. [38] were able to identify a CD38 specific ssDNA aptamer via a hybrid protein- and cell-based SELEX approach. This aptamer was subsequently non-covalently conjugated to doxorubicin for the generation of CD38-specific aptamer–drug conjugates (ApDC). The ApDCs were readily internalized by MM cells, and after a pH-dependent release of the cargo in lysosomes, doxorubicin was able to exert its specific antitumor activity by inhibiting tumor growth without toxicity in both MM in vitro and in vivo models.
- RNA aptamer for CXCL12: CXCL12, also known as stromal cell-derived factor-1 (SDF-1), is a chemoattractant chemokine that, upon binding to its receptors CXCR4 and CXCR7, induces the adhesion and homing of MM cells to the protective BM niche [39,40], and therefore, it is considered one of the major players in cell adhesion-mediated drug resistance (CAM-DR) [40]. MM cells present high levels of CXCL12, CXCR4 and CXCR7; therefore, CXCL12 neutralization represents an attractive option to modulate the BM niche for MM therapy and overcome CAM-DR [39,40].
| Target | Class | Identification | Internalization in mm Cells | Therapeutic Application | Effect | Stage | Reference |
|---|---|---|---|---|---|---|---|
| AXII | ssDNA | Recombinant protein SELEX | Not determined | Aptamer alone. | Inhibition of MM cell-line adhesion and proliferation | Preclinical | [40] |
| BCMA | RNA | Cell-SELEX | Yes | Aptamer alone. | Inhibition of BCMA pathway in vitro. | Preclinical | [42] |
| Aptamer miRNA (miR-137) chimera. | Upregulation of tumor suppressor miR-137 leading to reduced viability in vitro. | ||||||
| Aptamer anti-miRNA (anti-miR-222) chimera. | Inhibition of oncogenic miR-222 in vitro. | ||||||
| C-MET | ssDNA | Recombinant protein SELEX | Not determined | Aptamer alone. | Suppression of HGF-induced C-MET activation, inhibition of MM cell line proliferation and increased apoptosis, inhibition of cell migration and adhesion. | Preclinical | [20,43,44] |
| Combination therapy with bortezomib. | Synergy with bortezomib. | ||||||
| CD38 | ssDNA | Hybrid protein and cell SELEX | Yes | Aptamer-Doxorubicin conjugate. | Inhibition of MM cell-line proliferation in vitro, tumor inhibition in xenograft models | Preclinical | [45] |
| CXCL12 (NOX-A12) | spiegelmer | Protein SELEX against target enantiomer | No | Aptamer alone. | Inhibition of CXCR4 and CXCR7 activity in vitro, reduction of tumor growth in vivo, release of plasma cells into circulation in vivo. | Preclinical | [46,47,48] |
| Combination therapy with bortezomib. | Synergy with bortezomib in vivo. | Preclinical | |||||
| Combination therapy with dexamethasone and bortezomib. | Plasma cell mobilization. | Clinical Trial. Phase II completed (NTC01521533) |
4. Future Directions
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Braggio, E.; Kortüm, K.M.; Stewart, A.K. SnapShot: Multiple Myeloma. Cancer Cell 2015, 28, 678–678.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.; et al. Continued Improvement in Survival in Multiple Myeloma: Changes in Early Mortality and Outcomes in Older Patients. Leukemia 2014, 28, 1122–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajkumar, S.V. Multiple Myeloma: 2020 Update on Diagnosis, Risk-Stratification and Management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; Van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple Myeloma. Nat. Rev. Dis. Prim. 2017, 3, 1–20. [Google Scholar] [CrossRef]
- Rasche, L.; Hudecek, M.; Einsele, H. What Is the Future of Immunotherapy in Multiple Myeloma? Blood 2020, 136, 2491–2497. [Google Scholar] [CrossRef]
- Zanwar, S.; Nandakumar, B.; Kumar, S. Immune-Based Therapies in the Management of Multiple Myeloma. Blood Cancer J. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Sperling, A.S.; Anderson, K.C. Facts and Hopes in Multiple Myeloma Immunotherapy. Clin. Cancer Res. 2021, 27, 4468–4477. [Google Scholar] [CrossRef]
- Franssen, L.E.; Stege, C.A.M.; Zweegman, S.; van de Donk, N.W.C.J.; Nijhof, I.S. Resistance Mechanisms towards CD38−directed Antibody Therapy in Multiple Myeloma. J. Clin. Med. 2020, 9, 1195. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Rossi, J. Aptamers as Targeted Therapeutics: Current Potential and Challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [Green Version]
- Tuerk, C.; Gold, L. Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In Vitro Selection of RNA Molecules That Bind Specific Ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Komarova, N.; Kuznetsov, A. Inside the Black Box: What Makes Selex Better? Molecules 2019, 24, 3598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, S.; Herrera, A.; Rossi, J.J.; Zhou, J. Current Advances in Aptamers for Cancer Diagnosis and Therapy. Cancers 2018, 10, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Hu, L.; Zhang, B.T.; Lu, A.; Wang, Y.; Yu, Y.; Zhang, G. Artificial Intelligence in Aptamer–Target Binding Prediction. Int. J. Mol. Sci. 2021, 22, 3605. [Google Scholar] [CrossRef]
- Hamula, C.L.A.; Peng, H.; Wang, Z.; Newbigging, A.M.; Tyrrell, G.J.; Li, X.-F.; Le, X.C. The Effects of SELEX Conditions on the Resultant Aptamer Pools in the Selection of Aptamers Binding to Bacterial Cells. J. Mol. Evol. 2015, 81, 194–209. [Google Scholar] [CrossRef]
- Takahashi, M.; Wu, X.; Ho, M.; Chomchan, P.; Rossi, J.J.; Burnett, J.C.; Zhou, J. High Throughput Sequencing Analysis of RNA Libraries Reveals the Influences of Initial Library and PCR Methods on SELEX Efficiency. Sci. Rep. 2016, 6, 33697. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Liu, G.; Kai, M. DNA Aptamers in the Diagnosis and Treatment of Human Diseases. Molecules 2015, 20, 20979–20997. [Google Scholar] [CrossRef]
- Sola, M.; Menon, A.P.; Moreno, B.; Meraviglia-Crivelli, D.; Soldevilla, M.M.; Cartón-García, F.; Pastor, F. Aptamers Against Live Targets: Is In Vivo SELEX Finally Coming to the Edge? Mol. Ther.-Nucleic Acids 2020, 21, 192–204. [Google Scholar] [CrossRef]
- Nuzzo, S.; Roscigno, G.; Affinito, A.; Ingenito, F.; Quintavalle, C.; Condorelli, G. Potential and Challenges of Aptamers as Specific Carriers of Therapeutic Oligonucleotides for Precision Medicine in Cancer. Cancers 2019, 11, 1521. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Li, H.; Xu, L.; Deng, Z.; Han, W.; Liu, Y.; Jiang, W.; Zu, Y. Oligonucleotide Aptamer-Mediated Precision Therapy of Hematological Malignancies. Mol. Ther.-Nucleic Acids 2018, 13, 164–175. [Google Scholar] [CrossRef]
- Esposito, C.L.; Catuogno, S.; Condorelli, G.; Ungaro, P.; De Franciscis, V. Aptamer Chimeras for Therapeutic Delivery: The Challenging Perspectives. Genes 2018, 9, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor, F.; Kolonias, D.; Giangrande, P.H.; Gilboa, E. Induction of Tumour Immunity by Targeted Inhibition of Nonsense-Mediated MRNA Decay. Nature 2010, 465, 227–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, C.L.; Nuzzo, S.; Kumar, S.A.; Rienzo, A.; Lawrence, C.L.; Pallini, R.; Shaw, L.; Alder, J.E.; Ricci-Vitiani, L.; Catuogno, S.; et al. A Combined MicroRNA-Based Targeted Therapeutic Approach to Eradicate Glioblastoma Stem-like Cells. J. Control. Release 2016, 238, 43–57. [Google Scholar] [CrossRef]
- Catuogno, S.; Rienzo, A.; Di Vito, A.; Esposito, C.L.; de Franciscis, V. Selective Delivery of Therapeutic Single Strand AntimiRs by Aptamer-Based Conjugates. J. Control. Release 2015, 210, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Soldevilla, M.M.; de Caso, D.; Menon, A.P.; Pastor, F. Aptamer-IRNAs as Therapeutics for Cancer Treatment. Pharmaceuticals 2018, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Gonzalez, M.; Willner, I. Aptamer-Functionalized Micro- And Nanocarriers for Controlled Release. ACS Appl. Mater. Interfaces 2021, 13, 9520–9541. [Google Scholar] [CrossRef]
- Yan, A.C.; Levy, M. Aptamer-Mediated Delivery and Cell-Targeting Aptamers: Room for Improvement. Nucleic Acid Ther. 2018, 28, 194–199. [Google Scholar] [CrossRef]
- Seckinger, A.; Mei, T.; Ro, A.; Jauch, A.; Schnettler, R.; Ewerbeck, V.; Goldschmidt, H.; Klein, B.; Hose, D. Clinical and Prognostic Role of Annexin A2 in Multiple Myeloma. Blood 2012, 120, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, S.; Kurihara, N.; Shiozawa, Y.; Joseph, J.; Taichman, R.; Galson, D.L.; Roodman, G.D. Annexin II Interactions with the Annexin II Receptor Enhance Multiple Myeloma Cell Adhesion and Growth in the Bone Marrow Microenvironment. Blood 2012, 119, 1888–1896. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Zhang, Y.; Zeng, Y.; Peng, M.; Li, H.; Sun, S.; Ma, B.; Wang, Y.; Ye, M.; Liu, J. Screening and Characterization of an Annexin A2 Binding Aptamer That Inhibits the Proliferation of Myeloma Cells. Biochimie 2018, 151, 150–158. [Google Scholar] [CrossRef]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-Cell Maturation Antigen (BCMA) in Multiple Myeloma: Rationale for Targeting and Current Therapeutic Approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catuogno, S.; Di Martino, M.T.; Nuzzo, S.; Esposito, C.L.; Tassone, P.; de Franciscis, V. An Anti-BCMA RNA Aptamer for MiRNA Intracellular Delivery. Mol. Ther.-Nucleic Acids 2019, 18, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gao, H.; Zhou, W.; Sun, S.; Zeng, Y.; Zhang, H.; Liang, L.; Xiao, X.; Song, J.; Ye, M.; et al. Targeting C-Met Receptor Tyrosine Kinase by the DNA Aptamer SL1 as a Potential Novel Therapeutic Option for Myeloma. J. Cell. Mol. Med. 2018, 22, 5978–5990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueki, R.; Sando, S. A DNA Aptamer to C-Met Inhibits Cancer Cell Migration. Chem. Commun. 2014, 50, 13131–13134. [Google Scholar] [CrossRef]
- Boltz, A.; Piater, B.; Toleikis, L.; Guenther, R.; Kolmar, H.; Hock, B. Bi-Specific Aptamers Mediating Tumor Cell Lysis. J. Biol. Chem. 2011, 286, 21896–21905. [Google Scholar] [CrossRef] [Green Version]
- Giudice, V.; Mensitieri, F.; Izzo, V.; Filippelli, A.; Selleri, C. Aptamers and Antisense Oligonucleotides for Diagnosis and Treatment of Hematological Diseases. Int. J. Mol. Sci. 2020, 21, 3252. [Google Scholar] [CrossRef]
- Van De Donk, N.W.C.J.; Richardson, P.G.; Malavasi, F. CD38 Antibodies in Multiple Myeloma: Back to the Future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Tao, W.; Hao, S.; Iyer, S.P.; Zu, Y. A Unique Aptamer-Drug Conjugate for Targeted Therapy of Multiple Myeloma. Leukemia 2016, 30, 987–991. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Sacco, A.; Purschke, W.G.; Moschetta, M.; Buchner, K.; Maasch, C.; Zboralski, D.; Zöllner, S.; Vonhoff, S.; Mishima, Y.; et al. SDF-1 Inhibition Targets the Bone Marrow Niche for Cancer Therapy. Cell Rep. 2014, 9, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Waldschmidt, J.M.; Simon, A.; Wider, D.; Müller, S.J.; Follo, M.; Ihorst, G.; Decker, S.; Lorenz, J.; Chatterjee, M.; Azab, A.K.; et al. CXCL12 and CXCR7 Are Relevant Targets to Reverse Cell Adhesion-Mediated Drug Resistance in Multiple Myeloma. Br. J. Haematol. 2017, 179, 36–49. [Google Scholar] [CrossRef]
- Hoellenriegel, J.; Zboralski, D.; Maasch, C.; Rosin, N.Y.; Wierda, W.G.; Keating, M.J.; Kruschinski, A.; Burger, J.A. The Spiegelmer NOX-A12, a Novel CXCL12 Inhibitor, Interferes with Chronic Lymphocytic Leukemia Cell Motility and Causes Chemosensitization. Blood 2014, 123, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Vater, A.; Klussmann, S. Turning Mirror-Image Oligonucleotides into Drugs: The Evolution of Spiegelmer® Therapeutics. Drug Discov. Today 2015, 20, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, H.; Weisel, K.; Petrucci, M.T.; Leleu, X.; Cafro, A.M.; Laurent, G.; Zojer, N.; Foa, R.; Greil, R.; Yakoub-Agha, I.; et al. Final Results from the Phase IIa Study of the Anti-CXCL12 Spiegelmer® Olaptesed Pegol (NOX-A12) in Combination with Bortezomib and Dexamethasone in Patients with Multiple Myeloma. Blood 2014, 124, 2111. [Google Scholar] [CrossRef]
- Bausch-Fluck, D.; Goldmann, U.; Müller, S.; van Oostrum, M.; Müller, M.; Schubert, O.T.; Wollscheid, B. The in Silico Human Surfaceome. Proc. Natl. Acad. Sci. USA 2018, 115, E10988–E10997. [Google Scholar] [CrossRef] [Green Version]
- Rose, M.; Cardon, T.; Aboulouard, S.; Hajjaji, N.; Kobeissy, F.; Duhamel, M.; Fournier, I.; Salzet, M. Surfaceome Proteomic of Glioblastoma Revealed Potential Targets for Immunotherapy. Front. Immunol. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Ferguson, I.D.; Patiño-Escobar, B.; Tuomivaara, S.T.; Lin, Y.-H.T.; Nix, M.A.; Leung, K.K.; Kasap, C.; Ramos, E.; Nieves Vasquez, W.; Talbot, A.; et al. The Surfaceome of Multiple Myeloma Cells Suggests Potential Immunotherapeutic Strategies and Protein Markers of Drug Resistance. Nat. Commun. 2022, 13, 4121. [Google Scholar] [CrossRef]
- Yoon, S.; Rossi, J.J. Aptamers: Uptake Mechanisms and Intracellular Applications. Adv. Drug Deliv. Rev. 2018, 134, 22–35. [Google Scholar] [CrossRef]
- Mondragón, E.; Maher, L.J. Anti-Transcription Factor RNA Aptamers as Potential Therapeutics. Nucleic Acid Ther. 2016, 26, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Bausch-Fluck, D.; Milani, E.S.; Wollscheid, B. Surfaceome Nanoscale Organization and Extracellular Interaction Networks. Curr. Opin. Chem. Biol. 2019, 48, 26–33. [Google Scholar] [CrossRef]
- Leung, K.K.; Wilson, G.M.; Kirkemo, L.L.; Riley, N.M.; Coon, J.J.; Wells, J.A. Broad and Thematic Remodeling of the Surfaceome and Glycoproteome on Isogenic Cells Transformed with Driving Proliferative Oncogenes. Proc. Natl. Acad. Sci. USA 2020, 117, 7764–7775. [Google Scholar] [CrossRef]
- Bushweller, J.H. Targeting Transcription Factors in Cancer—From Undruggable to Reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Vallet, S.; Sacco, A.; Roccaro, A.; Lentzsch, S.; Podar, K. Targeting Transcription Factors in Multiple Myeloma: Evolving Therapeutic Strategies. Expert Opin. Investig. Drugs 2019, 28, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Tan, G.; Jiang, X.; Wu, K.; Tan, W.; Tan, Y. Suppression of FOXM1 Transcriptional Activities via a Single-Stranded DNA Aptamer Generated by SELEX. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Salamanca, H.H.; Antonyak, M.A.; Cerione, R.A.; Shi, H.; Lis, J.T. Inhibiting Heat Shock Factor 1 in Human Cancer Cells with a Potent RNA Aptamer. PLoS ONE 2014, 9, e96330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebruska, L.L.; Maher, L.J. Selection and Characterization of an RNA Decoy for Transcription Factor NF-ΚB. Biochemistry 1999, 38, 3168–3174. [Google Scholar] [CrossRef]
- Cassiday, L.A.; Maher, L.J. Yeast Genetic Selections to Optimize RNA Decoys for Transcription Factor NF-ΚB. Proc. Natl. Acad. Sci. USA 2003, 100, 3930–3935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurster, S.E.; Maher, L.J. Selection and Characterization of Anti-NF-ΚB P65 RNA Aptamers. RNA 2008, 14, 1037–1047. [Google Scholar] [CrossRef] [Green Version]
- Barton, J.L.; Bunka, D.H.J.; Knowling, S.E.; Lefevre, P.; Warren, A.J.; Bonifer, C.; Stockley, P.G. Characterization of RNA Aptamers That Disrupt the RUNX1–CBFβ/DNA Complex. Nucleic Acids Res. 2009, 37, 6818–6830. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.-H.; Wu, C.-F.; Rajasekaran, N.; Shin, Y.K. Loss of Tumor Suppressor Gene Function in Human Cancer: An Overview. Cell. Physiol. Biochem. 2018, 51, 2647–2693. [Google Scholar] [CrossRef]
- Chen, L.; Rashid, F.; Shah, A.; Awan, H.M.; Wu, M.; Liu, A.; Wang, J.; Zhu, T.; Luo, Z.; Shan, G. The Isolation of an RNA Aptamer Targeting to P53 Protein with Single Amino Acid Mutation. Proc. Natl. Acad. Sci. USA 2015, 112, 10002–10007. [Google Scholar] [CrossRef]
- Robert, F.; Roman, W.; Bramoullé, A.; Fellmann, C.; Roulston, A.; Shustik, C.; Porco, J.A.; Shore, G.C.; Sebag, M.; Pelletier, J. Translation Initiation Factor EIF4F Modifies the Dexamethasone Response in Multiple Myeloma. Proc. Natl. Acad. Sci. USA 2014, 111, 13421–13426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oguro, A.; Ohtsu, T.; Svitkin, Y.V.; Sonenberg, N.; Nakamura, Y. RNA Aptamers to Initiation Factor 4A Helicase Hinder Cap-Dependent Translation by Blocking ATP Hydrolysis. Rna 2003, 9, 394–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.H. Targeting Transcription Factors for Cancer Treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amundarain, A.; Pastor, F.; Prósper, F.; Agirre, X. Aptamers, a New Therapeutic Opportunity for the Treatment of Multiple Myeloma. Cancers 2022, 14, 5471. https://doi.org/10.3390/cancers14215471
Amundarain A, Pastor F, Prósper F, Agirre X. Aptamers, a New Therapeutic Opportunity for the Treatment of Multiple Myeloma. Cancers. 2022; 14(21):5471. https://doi.org/10.3390/cancers14215471
Chicago/Turabian StyleAmundarain, Ane, Fernando Pastor, Felipe Prósper, and Xabier Agirre. 2022. "Aptamers, a New Therapeutic Opportunity for the Treatment of Multiple Myeloma" Cancers 14, no. 21: 5471. https://doi.org/10.3390/cancers14215471
APA StyleAmundarain, A., Pastor, F., Prósper, F., & Agirre, X. (2022). Aptamers, a New Therapeutic Opportunity for the Treatment of Multiple Myeloma. Cancers, 14(21), 5471. https://doi.org/10.3390/cancers14215471