
The microRNA Lifecycle in Health and Cancer

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
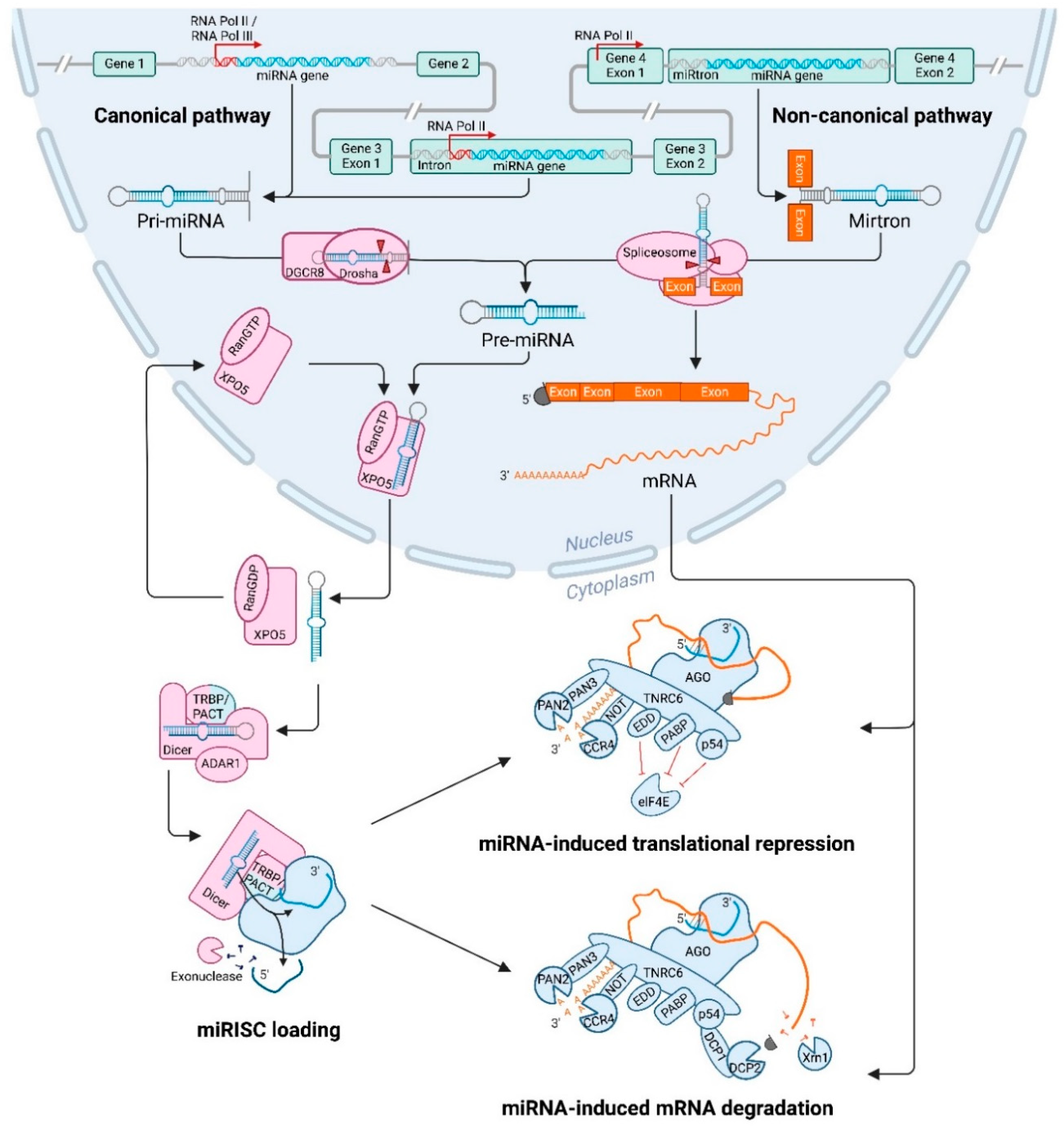
2. miRNA Biogenesis
2.1. miRNA Transcription
2.2. Processing of the Primary miRNA by Ribonuclease III Enzyme Drosha
2.3. Nuclear Export of Precursor microRNAs Mediated by Exportin-5
2.4. Processing of Precursor miRNA by Ribonuclease III Enzyme Dicer
2.5. Strand Selection and miRNA Induced Silencing Complex Formation
2.6. miRNA Isoforms
2.7. Non-Canonical Biogenesis Pathway
2.8. Biogenesis and Processing Rates
3. miRNA Target Regulation
3.1. Translational Repression and Messenger RNA Degradation
3.2. Translational Activation
3.3. Transcriptional Regulation
4. miRNA Subcellular Compartmentalization
4.1. Membrane Compartments
4.1.1. Nucleus
4.1.2. Mitochondria
4.1.3. Endoplasmic Reticulum
4.2. Membrane-Less Compartments
4.2.1. Stress Granules
4.2.2. Processing Bodies
5. miRNA Secretion and Uptake
5.1. Exosomes
5.2. Microvesicles
5.3. Apoptotic Bodies
6. miRNA Stability
6.1. Intrinsic Stability
6.2. Binding Proteins
6.3. Target-Directed miRNA Degradation or Protection
6.4. Adenosine to Inosine Transition
6.5. miRNA Methylation
6.6. Competing Endogenous RNAs
7. miRNA Dysregulation in Cancer
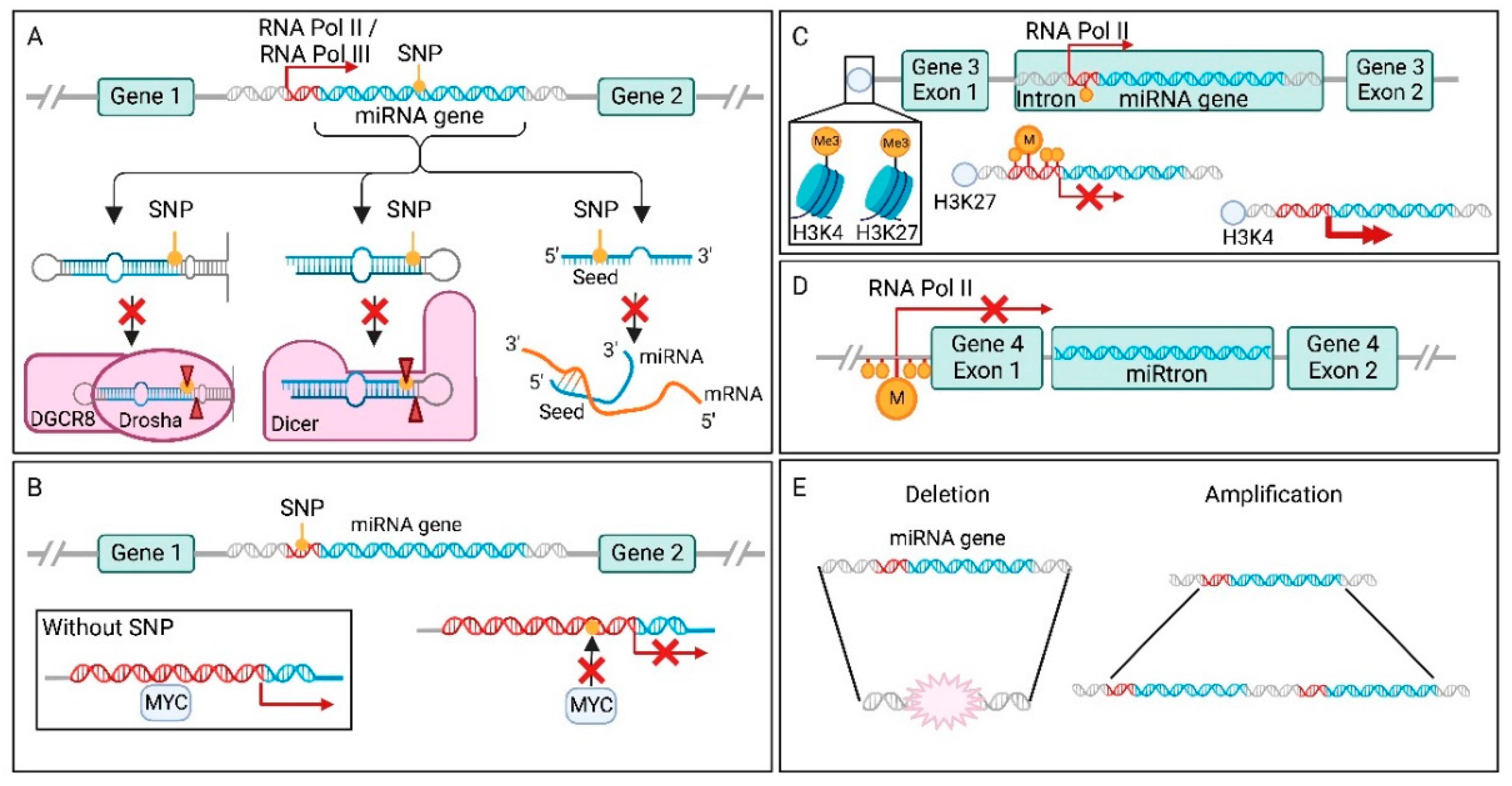
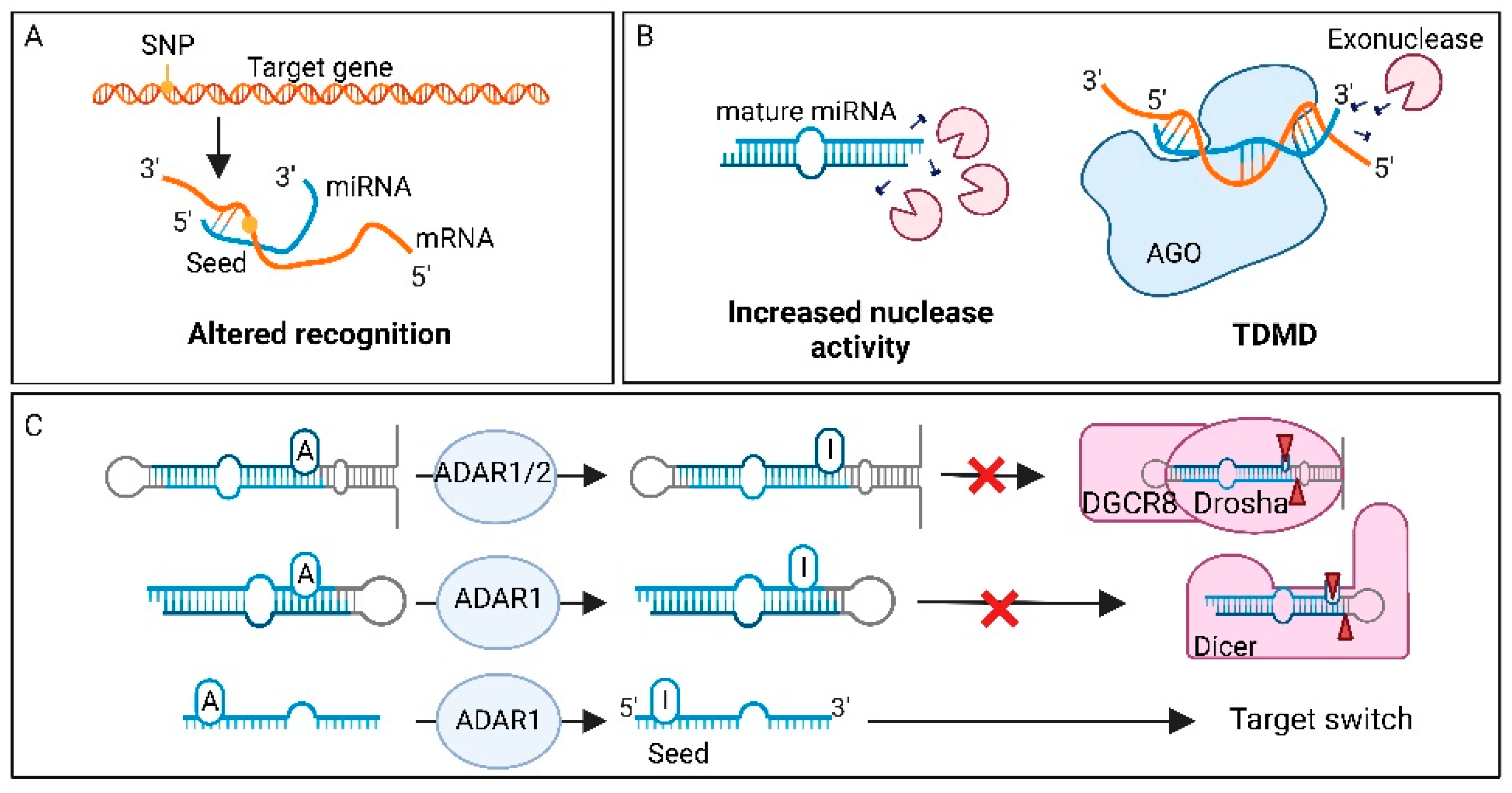
7.1. Single Nucleotide Polymorphisms in miRNA Genes
7.2. Single Nucleotide Polymorphisms in miRNA Promoter Elements and Transcription Factor Binding Sites
7.3. Epigenetic Changes to miRNA Genes in Cancer
7.4. miRNA Dysregulation through Host Genes
7.5. miRNA Dysregulation through Transcription Factors
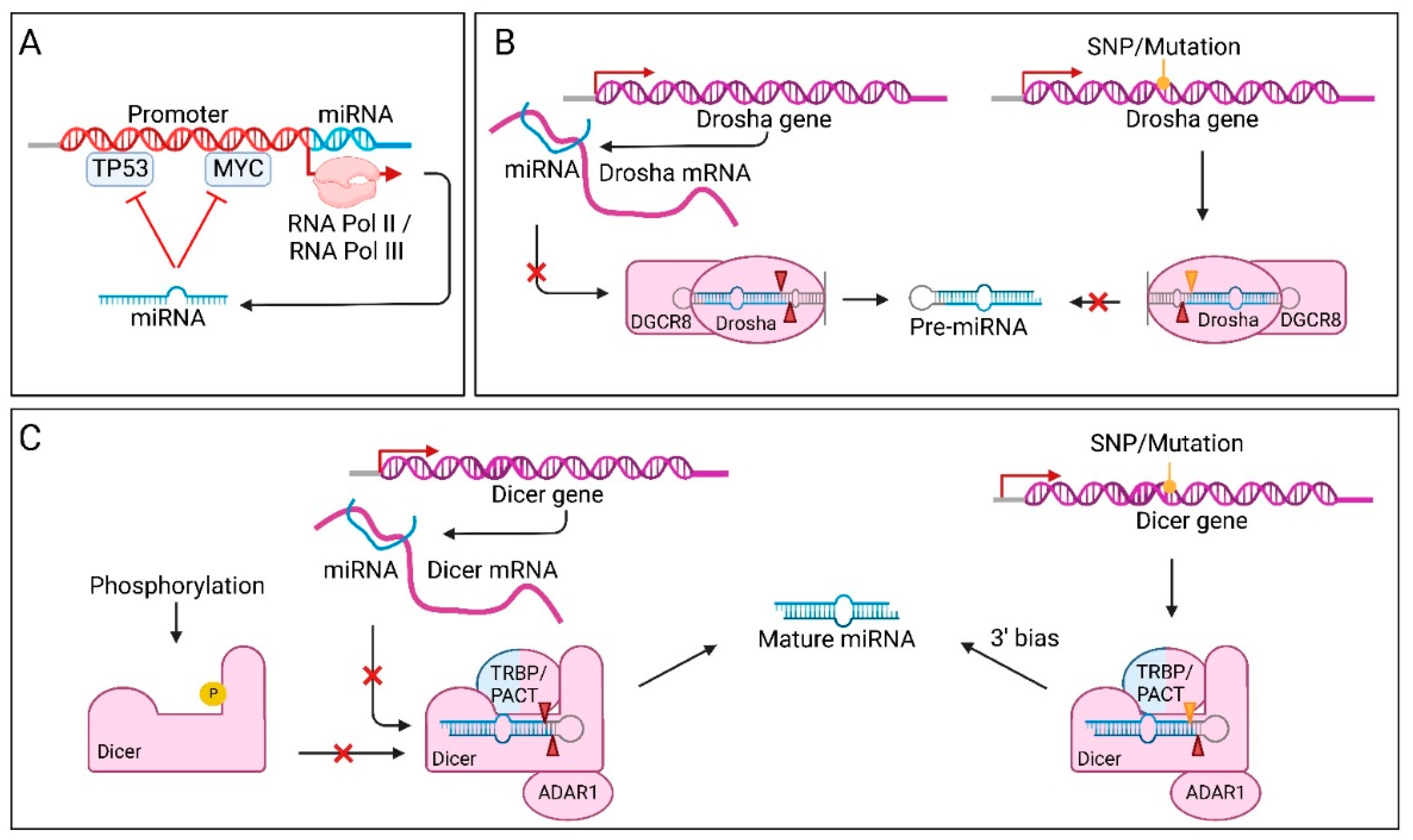
7.6. Factors Affecting miRNA Processing in Cancer
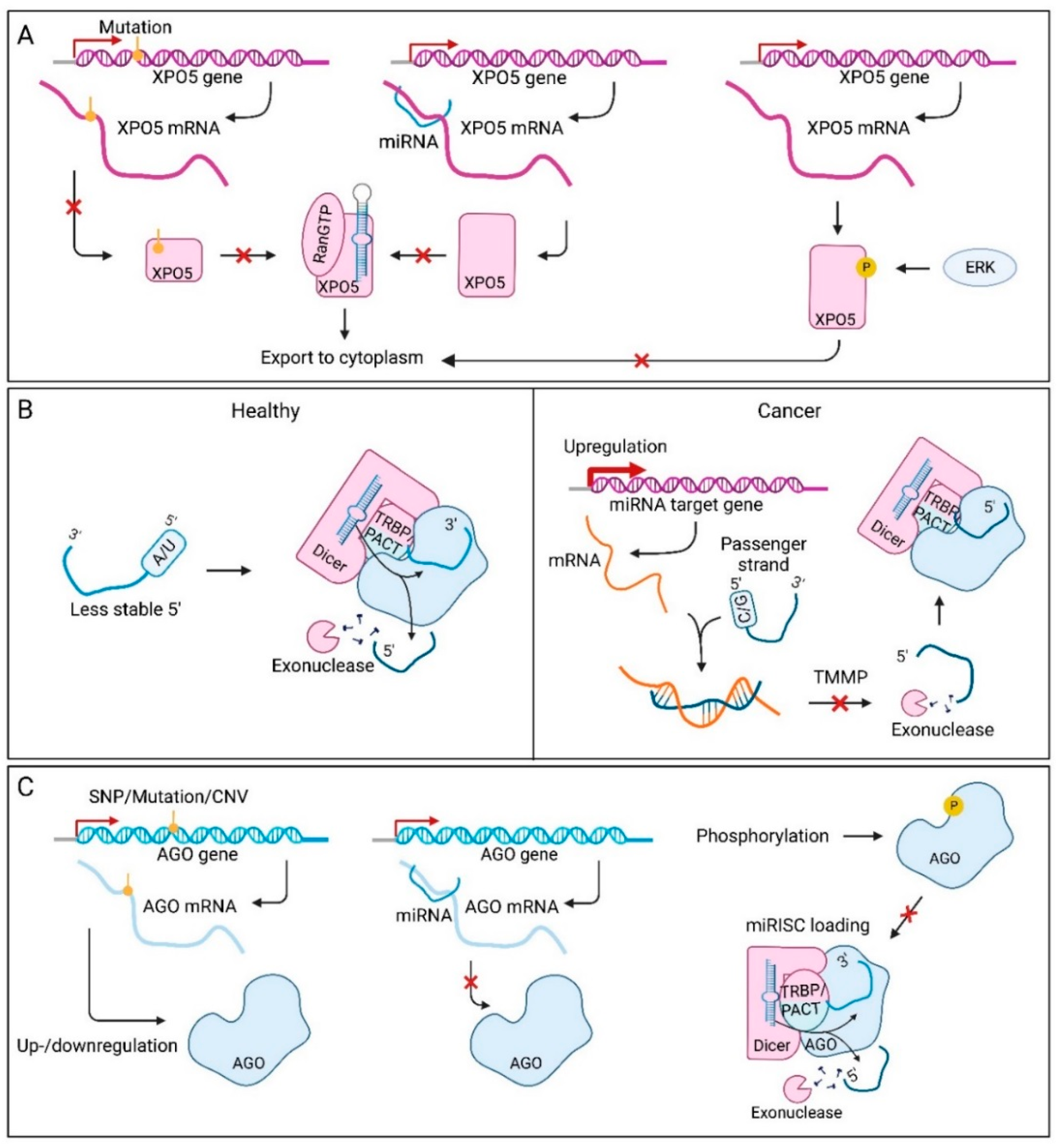
7.7. Factors Affecting Precursor microRNA Export in Cancer
7.8. Factors Affecting Strand Selection in Cancer
7.9. miRNA Dysregulation through miRNA-Induced Silencing Complex Assembly
7.10. miRNA Dysregulation through Target Genes
7.11. Factors Affecting miRNA Degradation in Cancer
7.12. Factors Affecting miRNA Editing in Cancer
7.13. Dysregulation of miRNAs by Long Non-Coding RNA
7.14. Dysregulation of miRNAs by Circular RNA
7.15. Dysregulation of miRNAs by Transfer RNA Derived Fragments
7.16. Self-Regulation of miRNA Expression
8. miRNAs as Cancer Biomarkers: Potential and Challenges
9. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional Regulation of the Heterochronic Gene Lin-14 by Lin-4 Mediates Temporal Pattern Formation in C. Elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Ambros, V. An Extensive Class of Small RNAs in Caenorhabditis Elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [Green Version]
- Caudy, A.A.; Myers, M.; Hannon, G.J.; Hammond, S.M. Fragile X-Related Protein and VIG Associate with the RNA Interference Machinery. Genes Dev. 2002, 16, 2491–2496. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.P.; Shih, I.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of Mammalian MicroRNA Targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most Mammalian MRNAs Are Conserved Targets of MicroRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial MicroRNA Target Predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef]
- Croce, C.M. Causes and Consequences of MicroRNA Dysregulation in Cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a Role in Cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA Expression Profiles Classify Human Cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Croce, C.M. MicroRNA Dysregulation in Cancer: Diagnostics, Monitoring and Therapeutics. A Comprehensive Review. EMBO Mol. Med. 2012, 4, 143–159. [Google Scholar] [CrossRef]
- Zhu, J.; Zheng, Z.; Wang, J.; Sun, J.; Wang, P.; Cheng, X.; Fu, L.; Zhang, L.; Wang, Z.; Li, Z. Different MiRNA Expression Profiles between Human Breast Cancer Tumors and Serum. Front. Genet. 2014, 5, 149. [Google Scholar] [CrossRef] [Green Version]
- Saini, H.K.; Sam, G.-J.; Enright, A.J. Genomic Analysis of Human MicroRNA Transcripts. Proc. Natl. Acad. Sci. USA 2007, 104, 17719–17724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinske, L.C.G.; Galante, P.A.F.; Kuo, W.P.; Ohno-Machado, L. A Potential Role for Intragenic MiRNAs on Their Hosts’ Interactome. BMC Genomics 2010, 11, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, K.; Hagen, J.W.; Duan, H.; Tyler, D.M.; Lai, E.C. The Mirtron Pathway Generates MicroRNA-Class Regulatory RNAs in Drosophila. Cell 2007, 130, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Shyr, Y.; Cai, J.; Liu, Q. Interplay between MiRNAs and Host Genes and Their Role in Cancer. Brief. Funct. Genomics 2019, 18, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Georgakilas, G.; Vlachos, I.S.; Paraskevopoulou, M.D.; Yang, P.; Zhang, Y.; Economides, A.N.; Hatzigeorgiou, A.G. MicroTSS: Accurate MicroRNA Transcription Start Site Identification Reveals a Significant Number of Divergent Pri-MiRNAs. Nat. Commun. 2014, 5, 5700. [Google Scholar] [CrossRef] [Green Version]
- Marsico, A.; Huska, M.R.; Lasserre, J.; Hu, H.; Vucicevic, D.; Musahl, A.; Orom, U.A.; Vingron, M. PROmiRNA: A New MiRNA Promoter Recognition Method Uncovers the Complex Regulation of Intronic MiRNAs. Genome Biol. 2013, 14, R84. [Google Scholar] [CrossRef] [Green Version]
- Monteys, A.M.; Spengler, R.M.; Wan, J.; Tecedor, L.; Lennox, K.A.; Xing, Y.; Davidson, B.L. Structure and Activity of Putative Intronic MiRNA Promoters. RNA 2010, 16, 495–505. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA Genes Are Transcribed by RNA Polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [Green Version]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA Polymerase III Transcribes Human MicroRNAs. Nat. Struct. Mol. Biol. 2006, 13, 1097–1101. [Google Scholar] [CrossRef]
- Lee, Y.; Jeon, K.; Lee, J.-T.; Kim, S.; Kim, V.N. MicroRNA Maturation: Stepwise Processing and Subcellular Localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The Nuclear RNase III Drosha Initiates MicroRNA Processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kolb, F.A.; Jaskiewicz, L.; Westhof, E.; Filipowicz, W. Single Processing Center Models for Human Dicer and Bacterial RNase III. Cell 2004, 118, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouleau, S.G.; Garant, J.-M.; Bolduc, F.; Bisaillon, M.; Perreault, J.-P. G-Quadruplexes Influence Pri-MicroRNA Processing. RNA Biol. 2018, 15, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Li, M.; Tucker, L.; Ramratnam, B. Glycogen Synthase Kinase 3 Beta (GSK3β) Phosphorylates the RNAase III Enzyme Drosha at S300 and S302. PLoS ONE 2011, 6, e20391. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wen, S.; Zheng, D.; Tucker, L.; Cao, L.; Pantazatos, D.; Moss, S.F.; Ramratnam, B. Acetylation of Drosha on the N-Terminus Inhibits Its Degradation by Ubiquitination. PLoS ONE 2013, 8, e72503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbert, K.M.; Pimienta, G.; DeGregorio, S.J.; Alexandrov, A.; Steitz, J.A. Phosphorylation of DGCR8 Increases Its Intracellular Stability and Induces a Progrowth MiRNA Profile. Cell Rep. 2013, 5, 1070–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, T.; Kikuchi, J.; Furukawa, Y. Histone Deacetylase 1 Enhances MicroRNA Processing via Deacetylation of DGCR8. EMBO Rep. 2012, 13, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Pedersen, J.S.; Kwon, S.C.; Belair, C.D.; Kim, Y.-K.; Yeom, K.-H.; Yang, W.-Y.; Haussler, D.; Blelloch, R.; Kim, V.N. Posttranscriptional Crossregulation between Drosha and DGCR8. Cell 2009, 136, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, M.; Kim, V.N. Regulation of MicroRNA Biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 Mediates the Nuclear Export of Pre-MicroRNAs and Short Hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, G.; Zhang, X.; Langley, R.R.; Liu, Y.; Hu, X.; Han, C.; Peng, G.; Ellis, L.M.; Jones, S.N.; Lu, X. DNA-Damage-Induced Nuclear Export of Precursor MicroRNAs Is Regulated by the ATM-AKT Pathway. Cell Rep. 2013, 3, 2100–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.L.; Cui, R.; Zhou, J.K.; Teng, K.Y.; Hsiao, Y.H.; Nakanishi, K.; Fassan, M.; Luo, Z.; Shi, G.; Tili, E.; et al. ERK Activation Globally Downregulates MiRNAs through Phosphorylating Exportin-5. Cancer Cell 2016, 30, 723–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-E.; Heo, I.; Tian, Y.; Simanshu, D.K.; Chang, H.; Jee, D.; Patel, D.J.; Kim, V.N. Dicer Recognizes the 5′ End of RNA for Efficient and Accurate Processing. Nature 2011, 475, 201–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacRae, I.J.; Zhou, K.; Li, F.; Repic, A.; Brooks, A.N.; Cande, W.Z.; Adams, P.D.; Doudna, J.A. Structural Basis for Double-Stranded RNA Processing by Dicer. Science 2006, 311, 195–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarthy, S.; Sternberg, S.H.; Kellenberger, C.A.; Doudna, J.A. Substrate-Specific Kinetics of Dicer-Catalyzed RNA Processing. J. Mol. Biol. 2010, 404, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ota, H.; Sakurai, M.; Gupta, R.; Valente, L.; Wulff, B.-E.; Ariyoshi, K.; Iizasa, H.; Davuluri, R.V.; Nishikura, K. ADAR1 Forms a Complex with Dicer to Promote MicroRNA Processing and RNA-Induced Gene Silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Hur, I.; Park, S.Y.; Kim, Y.K.; Mi, R.S.; Kim, V.N. The Role of PACT in the RNA Silencing Pathway. EMBO J. 2006, 25, 522. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP Recruits the Dicer Complex to Ago2 for MicroRNA Processing and Gene Silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Kim, J.; Yu, S.; Lee, Y.-Y.; Park, J.; Choi, R.J.; Yoon, S.-J.; Kang, S.-G.; Kim, V.N. A Mechanism for MicroRNA Arm Switching Regulated by Uridylation. Mol. Cell 2020, 78, 1224–1236.e5. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Agarwala, P.; Jayaraj, G.G.; Gargallo, R.; Maiti, S. The RNA Stem–Loop to G-Quadruplex Equilibrium Controls Mature MicroRNA Production inside the Cell. Biochemistry 2015, 54, 7067–7078. [Google Scholar] [CrossRef] [Green Version]
- Turchinovich, A.; Burwinkel, B. Distinct AGO1 and AGO2 Associated MiRNA Profiles in Human Cells and Blood Plasma. RNA Biol. 2012, 9, 1066–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, S.; Kobayashi, M.; Yoda, M.; Sakaguchi, Y.; Katsuma, S.; Suzuki, T.; Tomari, Y. Hsc70/Hsp90 Chaperone Machinery Mediates ATP-Dependent RISC Loading of Small RNA Duplexes. Mol. Cell 2010, 39, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Medley, J.C.; Panzade, G.; Zinovyeva, A.Y. MicroRNA Strand Selection: Unwinding the Rules. WIREs RNA 2021, 12, e1627. [Google Scholar] [CrossRef]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional SiRNAs and MiRNAs Exhibit Strand Bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Frank, F.; Sonenberg, N.; Nagar, B. Structural Basis for 5′-Nucleotide Base-Specific Recognition of Guide RNA by Human AGO2. Nature 2010, 465, 818–822. [Google Scholar] [CrossRef]
- Kwak, P.B.; Tomari, Y. The N Domain of Argonaute Drives Duplex Unwinding during RISC Assembly. Nat. Struct. Mol. Biol. 2012, 19, 145–151. [Google Scholar] [CrossRef]
- Paroo, Z.; Ye, X.; Chen, S.; Liu, Q. Phosphorylation of the Human Micro-RNA Generating Complex Mediates MAPK/Erk Signaling. Cell 2009, 139, 112. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Ye, C.; Ramirez, D.; Manjunath, N. Alternative Processing of Primary MicroRNA Transcripts by Drosha Generates 5′ End Variation of Mature MicroRNA. PLoS ONE 2009, 4, e7566. [Google Scholar] [CrossRef]
- Jang, Y.; Kim, M.A.; Kim, Y. Two Faces of Competition: Target-mediated Reverse Signalling in MicroRNA and Mitogen-activated Protein Kinase Regulatory Networks. IET Syst. Biol. 2017, 11, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Fasler, M.; Büssing, I.; Großhans, H. Target-Mediated Protection of Endogenous MicroRNAs in C. Elegans. Dev. Cell 2011, 20, 388–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berezikov, E.; Chung, W.J.; Willis, J.; Cuppen, E.; Lai, E.C. Mammalian Mirtron Genes. Mol. Cell 2007, 28, 328–336. [Google Scholar] [CrossRef] [Green Version]
- Westholm, J.O.; Lai, E.C. Mirtrons: MicroRNA Biogenesis via Splicing. Biochimie 2011, 93, 1897–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanta, A.; Chakrabarti, K. Dbr1 Functions in MRNA Processing, Intron Turnover and Human Diseases. Biochimie 2021, 180, 134–142. [Google Scholar] [CrossRef]
- Flynt, A.S.; Greimann, J.C.; Chung, W.-J.; Lima, C.D.; Lai, E.C. MicroRNA Biogenesis via Splicing and Exosome-Mediated Trimming in Drosophila. Mol. Cell 2010, 38, 900–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichholf, B.; Herzog, V.A.; Fasching, N.; Manzenreither, R.A.; Sowemimo, I.; Ameres, S.L. Time-Resolved Small RNA Sequencing Unravels the Molecular Principles of MicroRNA Homeostasis. Mol. Cell 2019, 75, 756–768.e7. [Google Scholar] [CrossRef] [PubMed]
- Kingston, E.R.; Bartel, D.P. Global Analyses of the Dynamics of Mammalian MicroRNA Metabolism. Genome Res. 2019, 29, 1777–1790. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global Quantification of Mammalian Gene Expression Control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Bortolamiol-Becet, D.; Hu, F.; Jee, D.; Wen, J.; Okamura, K.; Lin, C.-J.; Ameres, S.L.; Lai, E.C. Selective Suppression of the Splicing-Mediated MicroRNA Pathway by the Terminal Uridyltransferase Tailor. Mol. Cell 2015, 59, 217–228. [Google Scholar] [CrossRef]
- Yi, R.; Doehle, B.P.; Qin, Y.; Macara, I.G.; Cullen, B.R. Overexpression of Exportin 5 Enhances RNA Interference Mediated by Short Hairpin RNAs and MicroRNAs. RNA 2005, 11, 220–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, S.; Izaurralde, E. Towards a Molecular Understanding of MicroRNA-Mediated Gene Silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Handrick, R.; Aschrafi, A.; Otte, K. Unveiling the Principle of MicroRNA-Mediated Redundancy in Cellular Pathway Regulation. RNA Biol. 2015, 12, 238–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffries, C.D.; Fried, H.M.; Perkins, D.O. Nuclear and Cytoplasmic Localization of Neural Stem Cell MicroRNAs. RNA 2011, 17, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from Repression to Activation: MicroRNAs Can Up-Regulate Translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [Green Version]
- Elkayam, E.; Faehnle, C.R.; Morales, M.; Sun, J.; Li, H.; Joshua-Tor, L. Multivalent Recruitment of Human Argonaute by GW182. Mol. Cell 2017, 67, 646–658.e3. [Google Scholar] [CrossRef]
- Bazzini, A.; Lee, M.T.; Giraldez, A.J. Ribosome Profiling Shows That MiR-430 Reduces Translation before Causing MRNA Decay in Zebrafish. Science 2012, 336, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Pillai, R.S.; Bhattacharyya, S.N.; Artus, C.G.; Zoller, T.; Cougot, N.; Basyuk, E.; Bertrand, E.; Filipowicz, W. Inhibition of Translational Initiation by Let-7 MicroRNA in Human Cells. Science 2005, 309, 1573–1576. [Google Scholar] [CrossRef] [Green Version]
- Petersen, C.P.; Bordeleau, M.-E.; Pelletier, J.; Sharp, P.A. Short RNAs Repress Translation after Initiation in Mammalian Cells. Mol. Cell 2006, 21, 533–542. [Google Scholar] [CrossRef]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 Mediates RNA Cleavage Targeted by MiRNAs and SiRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef]
- Kiriakidou, M.; Tan, G.S.; Lamprinaki, S.; De Planell-Saguer, M.; Nelson, P.T.; Mourelatos, Z. An MRNA M7G Cap Binding-like Motif within Human Ago2 Represses Translation. Cell 2007, 129, 1141–1151. [Google Scholar] [CrossRef] [Green Version]
- Eulalio, A.; Huntzinger, E.; Izaurralde, E. GW182 Interaction with Argonaute Is Essential for MiRNA-Mediated Translational Repression and MRNA Decay. Nat. Struct. Mol. Biol. 2008, 15, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N. The Mechanics of MiRNA-Mediated Gene Silencing: A Look under the Hood of MiRISC. Nat. Struct. Mol. Biol. 2012, 19, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Mathonnet, G.; Sundermeier, T.; Mathys, H.; Zipprich, J.T.; Svitkin, Y.V.; Rivas, F.; Jinek, M.; Wohlschlegel, J.; Doudna, J.A.; et al. Mammalian MiRNA RISC Recruits CAF1 and PABP to Affect PABP-Dependent Deadenylation. Mol. Cell 2009, 35, 868–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behm-Ansmant, I.; Rehwinkel, J.; Doerks, T.; Stark, A.; Bork, P.; Izaurralde, E. MRNA Degradation by MiRNAs and GW182 Requires Both CCR4:NOT Deadenylase and DCP1:DCP2 Decapping Complexes. Genes Dev. 2006, 20, 1885–1898. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, T.; Zekri, L.; Braun, J.E.; Izaurralde, E. MiRISC Recruits Decapping Factors to MiRNA Targets to Enhance Their Degradation. Nucleic Acids Res. 2013, 41, 8692–8705. [Google Scholar] [CrossRef] [Green Version]
- da Sacco, L.; Masotti, A. Recent Insights and Novel Bioinformatics Tools to Understand the Role of MicroRNAs Binding to 5′ Untranslated Region. Int. J. Mol. Sci. 2012, 14, 480–495. [Google Scholar] [CrossRef] [Green Version]
- Truesdell, S.S.; Mortensen, R.D.; Seo, M.; Schroeder, J.C.; Lee, J.H.; Letonqueze, O.; Vasudevan, S.V. MicroRNA-Mediated MRNA Translation Activation in Quiescent Cells and Oocytes Involves Recruitment of a Nuclear MicroRNP. Sci. Reports 2012, 2, srep00842. [Google Scholar] [CrossRef] [Green Version]
- Ørom, U.A.; Nielsen, F.C.; Lund, A.H. MicroRNA-10a Binds the 5′UTR of Ribosomal Protein MRNAs and Enhances Their Translation. Mol. Cell 2008, 30, 460–471. [Google Scholar] [CrossRef]
- Ghanbarian, H.; Aghamiri, S.; Eftekhary, M.; Wagner, N.; Wagner, K.-D. Small Activating RNAs: Towards the Development of New Therapeutic Agents and Clinical Treatments. Cells 2021, 10, 591. [Google Scholar] [CrossRef]
- Nishi, K.; Nishi, A.; Nagasawa, T.; Ui-Tei, K. Human TNRC6A Is an Argonaute-Navigator Protein for MicroRNA-Mediated Gene Silencing in the Nucleus. RNA 2013, 19, 17–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Li, L.; Wang, D.; Zhang, C.-Y.; Zen, K. Importin 8 Regulates the Transport of Mature MicroRNAs into the Cell Nucleus. J. Biol. Chem. 2014, 289, 10270–10275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, H.-W.; Wentzel, E.A.; Mendell, J.T. A Hexanucleotide Element Directs MicroRNA Nuclear Import. Science 2007, 315, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Jie, M.; Feng, T.; Huang, W.; Zhang, M.; Feng, Y.; Jiang, H.; Wen, Z. Subcellular Localization of MiRNAs and Implications in Cellular Homeostasis. Genes 2021, 12, 856. [Google Scholar] [CrossRef] [PubMed]
- Makarova, J.A.; Shkurnikov, M.U.; Wicklein, D.; Lange, T.; Samatov, T.R.; Turchinovich, A.A.; Tonevitsky, A.G. Intracellular and Extracellular MicroRNA: An Update on Localization and Biological Role. Prog. Histochem. Cytochem. 2016, 51, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Sripada, L.; Tomar, D.; Singh, R. Mitochondria: One of the Destinations of MiRNAs. Mitochondrion 2012, 12, 593–599. [Google Scholar] [CrossRef]
- Gao, S.; Chen, C.; Wu, J.; Tan, Y.; Yu, K.; Xing, C.-Y.; Ye, A.; Yin, L.; Jiang, L. Synergistic Apoptosis Induction in Leukemic Cells by MiR-15a/16-1 and Arsenic Trioxide. Biochem. Biophys. Res. Commun. 2010, 403, 203–208. [Google Scholar] [CrossRef]
- Das, S.; Ferlito, M.; Kent, O.A.; Fox-Talbot, K.; Wang, R.; Liu, D.; Raghavachari, N.; Yang, Y.; Wheelan, S.J.; Murphy, E.; et al. Nuclear MiRNA Regulates the Mitochondrial Genome in the Heart. Circ. Res. 2012, 110, 1596–1603. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Tian, Y.; Tang, D.; Zou, S.; Liu, G.; Song, J.; Zhang, G.; Du, X.; Huang, W.; He, B.; et al. An Endoplasmic Reticulum Stress–MicroRNA-26a Feedback Circuit in NAFLD. Hepatology 2021, 73, 1327–1345. [Google Scholar] [CrossRef]
- Emde, A.; Eitan, C.; Liou, L.; Libby, R.T.; Rivkin, N.; Magen, I.; Reichenstein, I.; Oppenheim, H.; Eilam, R.; Silvestroni, A.; et al. Dysregulated Mi RNA Biogenesis Downstream of Cellular Stress and ALS-causing Mutations: A New Mechanism for ALS. EMBO J. 2015, 34, 2633–2651. [Google Scholar] [CrossRef]
- Luo, Y.; Na, Z.; Slavoff, S.A. P-Bodies: Composition, Properties, and Functions. Biochemistry 2018, 57, 2424–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozgur, S.; Chekulaeva, M.; Stoecklin, G. Human Pat1b Connects Deadenylation with MRNA Decapping and Controls the Assembly of Processing Bodies. Mol. Cell. Biol. 2010, 30, 4308–4323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, A.J.; MacRae, I.J. The RNA-Induced Silencing Complex: A Versatile Gene-Silencing Machine *. J. Biol. Chem. 2009, 284, 17897–17901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, R.; Sheth, U. P Bodies and the Control of MRNA Translation and Degradation. Mol. Cell 2007, 25, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Cui, Q. The Relationship of Human Tissue MicroRNAs with Those from Body Fluids. Sci. Rep. 2020, 10, 5644. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zhang, S.; Weber, J.; Baxter, D.; Galas, D.J. Export of MicroRNAs and MicroRNA-Protective Protein by Mammalian Cells. Nucleic Acids Res. 2010, 38, 7248–7259. [Google Scholar] [CrossRef] [Green Version]
- Yue, B.; Yang, H.; Wang, J.; Ru, W.; Wu, J.; Huang, Y.; Lan, X.; Lei, C.; Chen, H. Exosome Biogenesis, Secretion and Function of Exosomal MiRNAs in Skeletal Muscle Myogenesis. Cell Prolif. 2020, 53, e12857. [Google Scholar] [CrossRef]
- Bayraktar, R.; Van Roosbroeck, K.; Calin, G.A. Cell-to-cell Communication: MicroRNAs as Hormones. Mol. Oncol. 2017, 11, 1673–1686. [Google Scholar] [CrossRef] [Green Version]
- Squadrito, M.L.; Baer, C.; Burdet, F.; Maderna, C.; Gilfillan, G.D.; Lyle, R.; Ibberson, M.; De Palma, M. Endogenous RNAs Modulate MicroRNA Sorting to Exosomes and Transfer to Acceptor Cells. Cell Rep. 2014, 8, 1432–1446. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, S.; Li, L.; Li, M.; Guo, C.; Yao, J.; Mi, S. Exosome and Exosomal MicroRNA: Trafficking, Sorting, and Function. Genom. Proteom. Bioinform. 2015, 13, 17–24. [Google Scholar] [CrossRef]
- Zheng, D.; Huo, M.; Li, B.; Wang, W.; Piao, H.; Wang, Y.; Zhu, Z.; Li, D.; Wang, T.; Liu, K. The Role of Exosomes and Exosomal MicroRNA in Cardiovascular Disease. Front. Cell Dev. Biol. 2021, 8, 616161. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Wei, Y.; Wang, D.; Zhang, C.-Y.; Zen, K.; Li, L. Argonaute 2 in Cell-Secreted Microvesicles Guides the Function of Secreted MiRNAs in Recipient Cells. PLoS ONE 2014, 9, e103599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzenbach, H.; Gahan, P. MicroRNA Shuttle from Cell-To-Cell by Exosomes and Its Impact in Cancer. Non-Coding RNA 2019, 5, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarroya-Beltri, C.; Gutiérrez-Vázquez, C.; Sánchez-Cabo, F.; Pérez-Hernández, D.; Vázquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sánchez-Madrid, F. Sumoylated HnRNPA2B1 Controls the Sorting of MiRNAs into Exosomes through Binding to Specific Motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Chen, Q.; Lin, L.; Sha, C.; Li, T.; Liu, Y.; Yin, X.; Xu, Y.; Chen, L.; Gao, W.; et al. Regulation of Exosome Production and Cargo Sorting. Int. J. Biol. Sci. 2021, 17, 163–177. [Google Scholar] [CrossRef]
- Lee, Y.S.; Pressman, S.; Andress, A.P.; Kim, K.; White, J.L.; Cassidy, J.J.; Li, X.; Lubell, K.; Lim, D.H.; Cho, I.S.; et al. Silencing by Small RNAs Is Linked to Endosomal Trafficking. Nat. Cell Biol. 2009, 11, 1150–1156. [Google Scholar] [CrossRef] [Green Version]
- Corradi, E.; Dalla Costa, I.; Gavoci, A.; Iyer, A.; Roccuzzo, M.; Otto, T.A.; Oliani, E.; Bridi, S.; Strohbuecker, S.; Santos-Rodriguez, G.; et al. Axonal Precursor MiRNAs Hitchhike on Endosomes and Locally Regulate the Development of Neural Circuits. EMBO J. 2020, 39, e102513. [Google Scholar] [CrossRef]
- Rana, S.; Yue, S.; Stadel, D.; Zöller, M. Toward Tailored Exosomes: The Exosomal Tetraspanin Web Contributes to Target Cell Selection. Int. J. Biochem. Cell Biol. 2012, 44, 1574–1584. [Google Scholar] [CrossRef]
- Tian, T.; Zhu, Y.-L.; Zhou, Y.-Y.; Liang, G.-F.; Wang, Y.-Y.; Hu, F.-H.; Xiao, Z.-D. Exosome Uptake through Clathrin-Mediated Endocytosis and Macropinocytosis and Mediating MiR-21 Delivery *. J. Biol. Chem. 2014, 289, 22258–22267. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, Biologic Function and Clinical Potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Shu, Z.; Tan, J.; Miao, Y.; Zhang, Q. The Role of Microvesicles Containing MicroRNAs in Vascular Endothelial Dysfunction. J. Cell. Mol. Med. 2019, 23, 7933–7945. [Google Scholar] [CrossRef] [PubMed]
- Sohel, M.H. Extracellular/Circulating MicroRNAs: Release Mechanisms, Functions and Challenges. Achiev. Life Sci. 2016, 10, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Zernecke, A.; Bidzhekov, K.; Noels, H.; Shagdarsuren, E.; Gan, L.; Denecke, B.; Hristov, M.; Köppel, T.; Jahantigh, M.N.; Lutgens, E.; et al. Delivery of MicroRNA-126 by Apoptotic Bodies Induces CXCL12-Dependent Vascular Protection. Sci. Signal. 2009, 2, ra81. [Google Scholar] [CrossRef] [PubMed]
- Rissland, O.S.; Hong, S.-J.; Bartel, D.P. MicroRNA Destabilization Enables Dynamic Regulation of the MiR-16 Family in Response to Cell-Cycle Changes. Mol. Cell 2011, 43, 993–1004. [Google Scholar] [CrossRef] [Green Version]
- van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of Stress-Dependent Cardiac Growth and Gene Expression by a MicroRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef] [Green Version]
- Chan, L.Y.; Mugler, C.F.; Heinrich, S.; Vallotton, P.; Weis, K. Non-Invasive Measurement of MRNA Decay Reveals Translation Initiation as the Major Determinant of MRNA Stability. eLife 2018, 7, e32536. [Google Scholar] [CrossRef]
- Coenen-Stass, A.M.L.; Pauwels, M.J.; Hanson, B.; Martin Perez, C.; Conceição, M.; Wood, M.J.A.; Mäger, I.; Roberts, T.C. Extracellular MicroRNAs Exhibit Sequence-Dependent Stability and Cellular Release Kinetics. RNA Biol. 2019, 16, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 Complexes Carry a Population of Circulating MicroRNAs Independent of Vesicles in Human Plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [Green Version]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs Are Transported in Plasma and Delivered to Recipient Cells by High-Density Lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Ameres, S.L.; Horwich, M.D.; Hung, J.H.; Xu, J.; Ghildiyal, M.; Weng, Z.; Zamore, P.D. Target RNA-Directed Trimming and Tailing of Small Silencing RNAs. Science 2010, 328, 1534–1539. [Google Scholar] [CrossRef]
- Konno, M.; Koseki, J.; Asai, A.; Yamagata, A.; Shimamura, T.; Motooka, D.; Okuzaki, D.; Kawamoto, K.; Mizushima, T.; Eguchi, H.; et al. Distinct Methylation Levels of Mature MicroRNAs in Gastrointestinal Cancers. Nat. Commun. 2019, 10, 3888. [Google Scholar] [CrossRef] [Green Version]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A CeRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, E.C.; Wiel, C.; Rubin, G.M. Complementary MiRNA Pairs Suggest a Regulatory Role for MiRNA:MiRNA Duplexes. RNA 2004, 10, 171–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Qin, Y.-W.; Brewer, G.; Jing, Q. MicroRNA Degradation and Turnover: Regulating the Regulators. Wiley Interdiscip. Rev. RNA 2012, 3, 593–600. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.-J.; Paik, J.-H.; Zhang, H.; Shukla, S.A.; Mortensen, R.; Hu, J.; Ying, H.; Hu, B.; Hurt, J.; Farny, N.; et al. STAR RNA-Binding Protein Quaking Suppresses Cancer via Stabilization of Specific MiRNA. Genes Dev. 2012, 26, 1459–1472. [Google Scholar] [CrossRef] [Green Version]
- Sheu-Gruttadauria, J.; Pawlica, P.; Klum, S.M.; Wang, S.; Yario, T.A.; Schirle Oakdale, N.T.; Steitz, J.A.; MacRae, I.J. Structural Basis for Target-Directed MicroRNA Degradation. Mol. Cell 2019, 75, 1243–1255.e7. [Google Scholar] [CrossRef]
- Song, J.B.; Song, J.; Mo, B.X.; Chen, X.M. Uridylation and Adenylation of RNAs. Sci. China Life Sci. 2015, 58, 1057. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Shao, T.J.; Bofill-De Ros, X.; Lian, C.; Villanueva, P.; Dai, L.; Gu, S. AGO-Bound Mature MiRNAs Are Oligouridylated by TUTs and Subsequently Degraded by DIS3L2. Nat. Commun. 2020, 11, 2765. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Bjerke, G.A.; Muhlrad, D.; Yi, R.; Parker, R. The RNase PARN Controls the Levels of Specific MiRNAs That Contribute to P53 Regulation. Mol. Cell 2019, 73, 1204–1216.e4. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; LaVigne, C.A.; Jones, B.T.; Zhang, H.; Gillett, F.; Mendell, J.T. A Ubiquitin Ligase Mediates Target-Directed MicroRNA Decay Independently of Tailing and Trimming. Science 2020, 370, eabc9546. [Google Scholar] [CrossRef]
- Burns, D.M.; D’Ambrogio, A.; Nottrott, S.; Richter, J.D. CPEB and Two Poly(A) Polymerases Control MiR-122 Stability and P53 MRNA Translation. Nature 2011, 473, 105–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of MicroRNA Processing and Expression through RNA Editing by ADAR Deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mingardi, J.; Musazzi, L.; De Petro, G.; Barbon, A. MiRNA Editing: New Insights into the Fast Control of Gene Expression in Health and Disease. Mol. Neurobiol. 2018, 55, 7717–7727. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of Silencing Targets by Adenosine-to-Inosine Editing of MiRNAs. Science 2007, 315, 1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, Y.; Zinshteyn, B.; Chendrimada, T.P.; Shiekhattar, R.; Nishikura, K. RNA Editing of the MicroRNA-151 Precursor Blocks Cleavage by the Dicer–TRBP Complex. EMBO Rep. 2007, 8, 763–769. [Google Scholar] [CrossRef]
- Heale, B.S.E.; Keegan, L.P.; McGurk, L.; Michlewski, G.; Brindle, J.; Stanton, C.M.; Caceres, J.F.; O’Connell, M.A. Editing Independent Effects of ADARs on the MiRNA/SiRNA Pathways. EMBO J. 2009, 28, 3145–3156. [Google Scholar] [CrossRef]
- Elbarbary, R.A.; Miyoshi, K.; Myers, J.R.; Du, P.; Ashton, J.M.; Tian, B.; Maquat, L.E. Tudor-SN–Mediated Endonucleolytic Decay of Human Cell MicroRNAs Promotes G1/S Phase Transition. Science 2017, 356, 859–862. [Google Scholar] [CrossRef] [Green Version]
- Paraskevopoulou, M.D.; Georgakilas, G.; Kostoulas, N.; Reczko, M.; Maragkakis, M.; Dalamagas, T.M.; Hatzigeorgiou, A.G. DIANA-LncBase: Experimentally Verified and Computationally Predicted MicroRNA Targets on Long Non-Coding RNAs. Nucleic Acids Res. 2013, 41, D239–D245. [Google Scholar] [CrossRef]
- Blythe, A.J.; Fox, A.H.; Bond, C.S. The Ins and Outs of LncRNA Structure: How, Why and What Comes Next? Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 46–58. [Google Scholar] [CrossRef] [Green Version]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs Are Abundant, Conserved, and Associated with ALU Repeats. RNA 2013, 19, 141. [Google Scholar] [CrossRef]
- Haussecker, D.; Huang, Y.; Lau, A.; Parameswaran, P.; Fire, A.Z.; Kay, M.A. Human TRNA-Derived Small RNAs in the Global Regulation of RNA Silencing. RNA 2010, 16, 673–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasler, D.; Meister, G. From TRNA to MiRNA: RNA-Folding Contributes to Correct Entry into Noncoding RNA Pathways. FEBS Lett. 2016, 590, 2354–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zisoulis, D.G.; Kai, Z.S.; Chang, R.K.; Pasquinelli, A.E. Autoregulation of MicroRNA Biogenesis by Let-7 and Argonaute. Nature 2012, 486, 541–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, R.; Li, L.; Zhu, D.; Hou, D.; Cao, T.; Gu, H.; Zhang, J.; Chen, J.; Zhang, C.-Y.; Zen, K. Mouse MiRNA-709 Directly Regulates MiRNA-15a/16-1 Biogenesis at the Posttranscriptional Level in the Nucleus: Evidence for a MicroRNA Hierarchy System. Cell Res. 2012, 22, 504–515. [Google Scholar] [CrossRef]
- Sylvestre, Y.; De Guire, V.; Querido, E.; Mukhopadhyay, U.K.; Bourdeau, V.; Major, F.; Ferbeyre, G.; Chartrand, P. An E2F/MiR-20a Autoregulatory Feedback Loop. J. Biol. Chem. 2007, 282, 2135–2143. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Chu, H.; Wang, M.; Gu, X.; Shi, D.; Lan, M.; Zhong, D.; Du, M.; Li, P.; Tong, N.; et al. Genetic Variation in DROSHA 3′UTR Regulated by Hsa-MiR-27b Is Associated with Bladder Cancer Risk. PLoS ONE 2013, 8, e81524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toraih, E.A.; Hussein, M.H.; Al Ageeli, E.; Riad, E.; AbdAllah, N.B.; Helal, G.M.; Fawzy, M.S. Structure and Functional Impact of Seed Region Variant in MIR-499 Gene Family in Bronchial Asthma. Respir. Res. 2017, 18, 169. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Jiang, Y.; Zhou, J.; Liu, S.; Qin, N.; Du, J.; Jin, G.; Hu, Z.; Ma, H.; Shen, H.; et al. Evaluation of CpG-SNPs in MiRNA Promoters and Risk of Breast Cancer. Gene 2018, 651, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human MicroRNA Genes Are Frequently Located at Fragile Sites and Genomic Regions Involved in Cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The Widespread Regulation of MicroRNA Biogenesis, Function and Decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Nicoloso, M.S.; Sun, H.; Spizzo, R.; Kim, H.; Wickramasinghe, P.; Shimizu, M.; Wojcik, S.E.; Ferdin, J.; Kunej, T.; Xiao, L.; et al. Single-Nucleotide Polymorphisms inside MicroRNA Target Sites Influence Tumor Susceptibility. Cancer Res. 2010, 70, 2789–2798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeone, I.; Rubolino, C.; Noviello, T.M.R.; Farinello, D.; Cerulo, L.; Marzi, M.J.; Nicassio, F. Prediction and Pan-Cancer Analysis of Mammalian Transcripts Involved in Target Directed MiRNA Degradation. Nucleic Acids Res. 2022, 50, 2019–2035. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, X.; Yu, S.; Jeong, K.J.; Zhou, Z.; Han, L.; Tsang, Y.H.; Li, J.; Chen, H.; Mangala, L.S.; et al. Systematic Characterization of A-to-I RNA Editing Hotspots in MicroRNAs across Human Cancers. Genome Res. 2017, 27, 1112–1125. [Google Scholar] [CrossRef] [Green Version]
- Rakheja, D.; Chen, K.S.; Liu, Y.; Shukla, A.A.; Schmid, V.; Chang, T.C.; Khokhar, S.; Wickiser, J.E.; Karandikar, N.J.; Malter, J.S.; et al. Somatic Mutations in DROSHA and DICER1 Impair MicroRNA Biogenesis through Distinct Mechanisms in Wilms Tumours. Nat. Commun. 2014, 5, 4802. [Google Scholar] [CrossRef] [Green Version]
- Melo, S.A.; Moutinho, C.; Ropero, S.; Calin, G.A.; Rossi, S.; Spizzo, R.; Fernandez, A.F.; Davalos, V.; Villanueva, A.; Montoya, G.; et al. A Genetic Defect in Exportin-5 Traps Precursor MicroRNAs in the Nucleus of Cancer Cells. Cancer Cell 2010, 18, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Oh, J.E.; Kim, Y.R.; Park, S.W.; Kang, M.R.; Kim, S.S.; Ahn, C.H.; Yoo, N.J.; Lee, S.H. Somatic Mutations and Losses of Expression of MicroRNA Regulation-Related Genes AGO2 and TNRC6A in Gastric and Colorectal Cancers. J. Pathol. 2010, 221, 139–146. [Google Scholar] [CrossRef]
- Newie, I.; Søkilde, R.; Persson, H.; Jacomasso, T.; Gorbatenko, A.; Borg, Å.; de Hoon, M.; Pedersen, S.F.; Rovira, C. HER2-Encoded Mir-4728 Forms a Receptor-Independent Circuit with MiR-21-5p through the Non-Canonical Poly(A) Polymerase PAPD5. Sci. Rep. 2016, 6, 35664. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zheng, Y.; Zhou, L.; Jin, J.; Deng, Y.; Yao, J.; Yang, P.; Yao, L.; Wu, Y.; Zhai, Z.; et al. MiR-499 Rs3746444 and MiR-196a-2 Rs11614913 Are Associated with the Risk of Glioma, but Not the Prognosis. Mol. Ther. Nucleic Acids 2020, 22, 340–351. [Google Scholar] [CrossRef]
- Yang, H.; Dinney, C.P.; Ye, Y.; Zhu, Y.; Grossman, H.B.; Wu, X. Evaluation of Genetic Variants in MicroRNA-Related Genes and Risk of Bladder Cancer. Cancer Res. 2008, 68, 2530–2537. [Google Scholar] [CrossRef]
- Hu, Y.-Y.; Jiang, G.-B.; Song, Y.-F.; Zhan, A.-L.; Deng, C.; Niu, Y.-M.; Zhou, L.; Duan, Q.-W. Association between the Pri-MiR-26a-1 Rs7372209 C>T Polymorphism and Cancer Susceptibility: Multivariate Analysis and Trial Sequential Analysis. Aging 2020, 12, 19060–19072. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chen, J.; Tian, T.; Zhou, X.; Gu, H.; Xu, L.; Zeng, Y.; Miao, R.; Jin, G.; Ma, H.; et al. Genetic Variants of MiRNA Sequences and Non–Small Cell Lung Cancer Survival. J. Clin. Invest. 2008, 118, 2600–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalho-Carvalho, J.; Martins, J.B.; Cekaite, L.; Sveen, A.; Torres-Ferreira, J.; Graça, I.; Costa-Pinheiro, P.; Eilertsen, I.A.; Antunes, L.; Oliveira, J.; et al. Epigenetic Disruption of MiR-130a Promotes Prostate Cancer by Targeting SEC23B and DEPDC1. Cancer Lett. 2017, 385, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent Deletions and Down-Regulation of Micro- RNA Genes MiR15 and MiR16 at 13q14 in Chronic Lymphocytic Leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.T.; Boyd, J.C.; Frierson, H.F. Loss of Heterozygosity at 13q14 and 13q21 in High Grade, High Stage Prostate Cancer. Prostate 2001, 49, 166–171. [Google Scholar] [CrossRef]
- Pekarsky, Y.; Balatti, V.; Croce, C.M. BCL2 and MiR-15/16: From Gene Discovery to Treatment. Cell Death Differ. 2018, 25, 21–26. [Google Scholar] [CrossRef]
- Palmbos, P.L.; Wang, L.; Yang, H.; Wang, Y.; Leflein, J.; Ahmet, M.L.; Wilkinson, J.E.; Kumar-Sinha, C.; Ney, G.M.; Tomlins, S.A.; et al. ATDC/TRIM29 Drives Invasive Bladder Cancer Formation through MiRNA-Mediated and Epigenetic Mechanisms. Cancer Res. 2015, 75, 5155–5166. [Google Scholar] [CrossRef] [Green Version]
- Eskandari, F.; Teimoori, B.; Rezaei, M.; Mohammadpour-Gharehbagh, A.; Narooei-Nejad, M.; Mehrabani, M.; Salimi, S. Relationships between Dicer 1 Polymorphism and Expression Levels in the Etiopathogenesis of Preeclampsia. J. Cell. Biochem. 2018, 119, 5563–5570. [Google Scholar] [CrossRef]
- Li, X.I.; Tian, X.; Zhang, B.O.; Chen, J. Polymorphisms in MicroRNA-Related Genes Are Associated with Survival of Patients with T-Cell Lymphoma. Oncologist 2014, 19, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.N.; Kim, J.O.; Lee, S.M.; Park, H.; Lee, J.H.; Rim, K.S.; Hwang, S.G.; Kim, N.K. Variation in the Dicer and RAN Genes Are Associated with Survival in Patients with Hepatocellular Carcinoma. PLoS ONE 2016, 11, e0162279. [Google Scholar] [CrossRef]
- Srinivasan, G.; Williamson, E.A.; Kong, K.; Jaiswal, A.S.; Huang, G.; Kim, H.S.; Schärer, O.; Zhao, W.; Burma, S.; Sung, P.; et al. MiR223-3p Promotes Synthetic Lethality in BRCA1-Deficient Cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 17438–17443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, K.-W.; Leung, C.-M.; Lo, Y.-H.; Chen, T.-W.; Chan, W.-C.; Yu, S.-Y.; Tu, Y.-T.; Lam, H.-C.; Li, S.-C.; Ger, L.-P.; et al. Arm Selection Preference of MicroRNA-193a Varies in Breast Cancer. Sci. Rep. 2016, 6, 28176. [Google Scholar] [CrossRef] [Green Version]
- Krishna, B.; Jana, S.; Panda, A.; Horne, D.; Awasthi, S.; Salgia, R.; Singhal, S. Association of TGF-Β1 Polymorphisms with Breast Cancer Risk: A Meta-Analysis of Case–Control Studies. Cancers 2020, 12, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, D.; Park, E.S.; Fisher, P.B. Defining the Mechanism by Which IFN-β Dowregulates c-Myc Expression in Human Melanoma Cells: Pivotal Role for Human Polynucleotide Phosphorylase (HPNPaseold-35). Cell Death Differ. 2006, 13, 1541–1553. [Google Scholar] [CrossRef] [Green Version]
- Das, S.K.; Sokhi, U.K.; Bhutia, S.K.; Azab, B.; Su, Z.Z.; Sarkar, D.; Fisher, P.B. Human Polynucleotide Phosphorylase Selectively and Preferentially Degrades MicroRNA-221 in Human Melanoma Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11948–11953. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Wang, J.; Yang, X.; Yu, H.; Zhou, R.; Lu, H.-C.; Yuan, W.-B.; Lu, J.; Zhou, Z.; Lu, Q.; et al. METTL3 Promote Tumor Proliferation of Bladder Cancer by Accelerating Pri-MiR221/222 Maturation in M6A-Dependent Manner. Mol. Cancer 2019, 18, 110. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Zhang, J. Long Non-Coding RNA HOTAIR Functions as MiRNA Sponge to Promote the Epithelial to Mesenchymal Transition in Esophageal Cancer. Biomed. Pharmacother. 2017, 90, 888–896. [Google Scholar] [CrossRef]
- Li, X.; Yan, X.; Wang, F.; Yang, Q.; Luo, X.; Kong, J.; Ju, S. Down-regulated LncRNA SLC25A5-AS1 Facilitates Cell Growth and Inhibits Apoptosis via MiR-19a-3p/PTEN/PI3K/AKT Signalling Pathway in Gastric Cancer. J. Cell. Mol. Med. 2019, 23, 2920. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Li, Y.; Huang, K.; Wagar, N.; Shim, H. Regulation of MiR-19 to Breast Cancer Chemoresistance Through Targeting PTEN. Pharm. Res. 2011, 28, 3091. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Tian, P.; Yang, C.; Liang, Z.; Li, M.; Sims, M.; Lu, L.; Zhang, Z.; Li, H.; Pfeffer, L.M.; et al. Silencing the Double-Stranded RNA Binding Protein DGCR8 Inhibits Ovarian Cancer Cell Proliferation, Migration, and Invasion. Pharm. Res. 2015, 32, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA Circles Function as Efficient MicroRNA Sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Kefas, B.; Godlewski, J.; Comeau, L.; Li, Y.; Abounader, R.; Hawkinson, M.; Lee, J.; Fine, H.; Chiocca, E.A.; Lawler, S.; et al. MicroRNA-7 Inhibits the Epidermal Growth Factor Receptor and the Akt Pathway and Is Down-Regulated in Glioblastoma. Cancer Res. 2008, 68, 3566–3572. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yang, J.; Fei, X.; Wang, X.; Wang, K. CircRNA CiRS-7: A Novel Oncogene in Multiple Cancers. Int. J. Biol. Sci. 2021, 17, 379–389. [Google Scholar] [CrossRef]
- Huang, B.; Yang, H.; Cheng, X.; Wang, D.; Fu, S.; Shen, W.; Zhang, Q.; Zhang, L.; Xue, Z.; Li, Y.; et al. TRF/MiR-1280 Suppresses Stem Cell–like Cells and Metastasis in Colorectal Cancer. Cancer Res. 2017, 77, 3194–3206. [Google Scholar] [CrossRef] [Green Version]
- Forrest, A.R.R.; Kanamori-Katayama, M.; Tomaru, Y.; Lassmann, T.; Ninomiya, N.; Takahashi, Y.; De Hoon, M.J.L.; Kubosaki, A.; Kaiho, A.; Suzuki, M.; et al. Induction of MicroRNAs, Mir-155, Mir-222, Mir-424 and Mir-503, Promotes Monocytic Differentiation through Combinatorial Regulation. Leukemia 2009, 24, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.S.; Su, J.L.; Cha, S.T.; Tarn, W.Y.; Wang, M.Y.; Hsu, H.C.; Lin, M.T.; Chu, C.Y.; Hua, K.T.; Chen, C.N.; et al. MiR-107 Promotes Tumor Progression by Targeting the Let-7 MicroRNA in Mice and Humans. J. Clin. Invest. 2011, 121, 3442–3455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bao, W.; Liu, Y.; Wang, S.; Xu, S.; Li, X.; Li, Y.; Wu, S. MiR-98-5p Contributes to Cisplatin Resistance in Epithelial Ovarian Cancer by Suppressing MiR-152 Biogenesis via Targeting Dicer1. Cell Death Dis. 2018, 9, 447. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting Oncogenic Myc as a Strategy for Cancer Treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, M.; Li, Z.; Aau, M.; Wong, C.H.; Yang, X.; Yu, Q. Myc/MiR-378/TOB2/Cyclin D1 Functional Module Regulates Oncogenic Transformation. Oncogene 2011, 30, 2242–2251. [Google Scholar] [CrossRef] [Green Version]
- Furuta, M.; Kozaki, K.I.; Tanaka, S.; Arii, S.; Imoto, I.; Inazawa, J. MiR-124 and MiR-203 Are Epigenetically Silenced Tumor-Suppressive MicroRNAs in Hepatocellular Carcinoma. Carcinogenesis 2010, 31, 766–776. [Google Scholar] [CrossRef]
- Dakhlallah, D.; Batte, K.; Wang, Y.; Cantemir-Stone, C.Z.; Yan, P.; Nuovo, G.; Mikhail, A.; Hitchcock, C.L.; Wright, V.P.; Nana-Sinkam, S.P.; et al. Epigenetic Regulation of MiR-17∼92 Contributes to the Pathogenesis of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2013, 187, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalgi, R.; Lieber, D.; Oren, M.; Pilpel, Y. Global and Local Architecture of the Mammalian MicroRNA–Transcription Factor Regulatory Network. PLOS Comput. Biol. 2007, 3, e131. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. C-Myc-Regulated MicroRNAs Modulate E2F1 Expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef]
- Ke, H.L.; Chen, M.; Ye, Y.; Hildebrandt, M.A.T.; Wu, W.J.; Wei, H.; Huang, M.; Chang, D.W.; Dinney, C.P.; Wu, X. Genetic Variations in Micro-RNA Biogenesis Genes and Clinical Outcomes in Non-Muscle-Invasive Bladder Cancer. Carcinogenesis 2013, 34, 1006–1011. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Lee, J.H.; Park, J.W.; Kwon, T.K.; Baek, S.K.; Hwang, I.; Kim, S. An Essential MicroRNA Maturing Microprocessor Complex Component DGCR8 Is Up-Regulated in Colorectal Carcinomas. Clin. Exp. Med. 2014, 14, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Le, C.T.; Nguyen, T.L.; Nguyen, T.D.; Nguyen, T.A. Human Disease-Associated Single Nucleotide Polymorphism Changes the Orientation of DROSHA on Pri-Mir-146a. RNA 2020, 26, 1777–1786. [Google Scholar] [CrossRef]
- Heravi-Moussavi, A.; Anglesio, M.S.; Cheng, S.-W.G.; Senz, J.; Yang, W.; Prentice, L.; Fejes, A.P.; Chow, C.; Tone, A.; Kalloger, S.E.; et al. Recurrent Somatic DICER1 Mutations in Nonepithelial Ovarian Cancers. N. Engl. J. Med. 2012, 366, 234–242. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Wang, Y.; Yang, W.; Senz, J.; Wan, A.; Heravi-Moussavi, A.; Salamanca, C.; Maines-Bandiera, S.; Huntsman, D.G.; Morin, G.B. Cancer-Associated Somatic DICER1 Hotspot Mutations Cause Defective MiRNA Processing and Reverse-Strand Expression Bias to Predominantly Mature 3p Strands through Loss of 5p Strand Cleavage. J. Pathol. 2013, 229, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Drake, M.; Furuta, T.; Suen, K.M.; Gonzalez, G.; Liu, B.; Kalia, A.; Ladbury, J.E.; Fire, A.Z.; Skeath, J.B.; Arur, S. A Requirement for ERK-Dependent Dicer Phosphorylation in Coordinating Oocyte-to-Embryo Transition in C. Elegans. Dev. Cell 2014, 31, 614–628. [Google Scholar] [CrossRef] [Green Version]
- Shigeyasu, K.; Okugawa, Y.; Toden, S.; Boland, C.R.; Goel, A. Exportin-5 Functions as an Oncogene and a Potential Therapeutic Target in Colorectal Cancer. Clin. Cancer Res. 2017, 23, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Leaderer, D.; Hoffman, A.E.; Zheng, T.; Fu, A.; Weidhaas, J.; Paranjape, T.; Zhu, Y. Genetic and Epigenetic Association Studies Suggest a Role of MicroRNA Biogenesis Gene Exportin-5 (XPO5) in Breast Tumorigenesis. Int. J. Mol. Epidemiol. Genet. 2011, 2, 9. [Google Scholar]
- Li, J.; Chen, Y.; Qin, X.; Wen, J.; Ding, H.; Xia, W.; Li, S.; Su, X.; Wang, W.; Li, H.; et al. MiR-138 Downregulates MiRNA Processing in HeLa Cells by Targeting RMND5A and Decreasing Exportin-5 Stability. Nucleic Acids Res. 2014, 42, 458–474. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Pu, W.; Sun, H.L.; Zhou, J.K.; Fan, X.; Zheng, Y.; He, J.; Liu, X.; Xia, Z.; Liu, L.; et al. Pin1 Impairs MicroRNA Biogenesis by Mediating Conformation Change of XPO5 in Hepatocellular Carcinoma. Cell Death Differ. 2018 259 2018, 25, 1612–1624. [Google Scholar] [CrossRef]
- Li, J.; Zhou, J.; Mu, X.; Shen, S.; Xu, X.; Luo, Y.; Luo, Y.; Ming, Y.; Wu, Y.; Peng, Y. Regulation of XPO5 Phosphorylation by PP2A in Hepatocellular Carcinoma. MedComm 2022, 3, e125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, J.; Yang, N.; Greshock, J.; Megraw, M.S.; Giannakakis, A.; Liang, S.; Naylor, T.L.; Barchetti, A.; Ward, M.R.; et al. MicroRNAs Exhibit High Frequency Genomic Alterations in Human Cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 9136–9141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Meng, J.; Zhai, Y.; Zhang, H.; Yu, L.; Wang, Z.; Zhang, X.; Cao, P.; Chen, X.; Han, Y.; et al. Argonaute 2 and Nasopharyngeal Carcinoma: A Genetic Association Study and Functional Analysis. BMC Cancer 2015, 15, 862. [Google Scholar] [CrossRef] [Green Version]
- Leonov, G.; Shah, K.; Yee, D.; Timmis, J.; Sharp, T.V.; Lagos, D. Suppression of AGO2 by MiR-132 as a Determinant of MiRNA-Mediated Silencing in Human Primary Endothelial Cells. Int. J. Biochem. Cell Biol. 2015, 69, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Xia, W.; Khotskaya, Y.B.; Huo, L.; Nakanishi, K.; Lim, S.O.; Du, Y.; Wang, Y.; Chang, W.C.; Chen, C.H.; et al. EGFR Modulates MicroRNA Maturation in Response to Hypoxia through Phosphorylation of AGO2. Nature 2013, 497, 383–387. [Google Scholar] [CrossRef] [Green Version]
- Abdelhaleem, M. Over-Expression of RNA Helicases in Cancer. Anticancer Res. 2004, 24, 3951–3954. [Google Scholar]
- Pinto, Y.; Buchumenski, I.; Levanon, E.Y.; Eisenberg, E. Human Cancer Tissues Exhibit Reduced A-to-I Editing of MiRNAs Coupled with Elevated Editing of Their Targets. Nucleic Acids Res. 2018, 46, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Guo, J.; Fan, Z. Interactions between M6A Modification and MiRNAs in Malignant Tumors. Cell Death Dis. 2021, 12, 598. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sharpless, N.E. Detecting and Characterizing Circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- Hasler, D.; Lehmann, G.; Murakawa, Y.; Klironomos, F.; Jakob, L.; Grässer, F.A.; Rajewsky, N.; Landthaler, M.; Meister, G. The Lupus Autoantigen La Prevents Mis-Channeling of TRNA Fragments into the Human MicroRNA Pathway. Mol. Cell 2016, 63, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Zhang, J.; Zen, K.; Zhang, C.Y.; Chen, X. Nuclear MicroRNAs and Their Unconventional Role in Regulating Non-Coding RNAs. Protein Cell 2013, 4, 325–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sempere, L.F.; Azmi, A.S.; Moore, A. microRNA-based Diagnostic and Therapeutic Applications in Cancer Medicine. WIREs RNA 2021, 12, e1662. [Google Scholar] [CrossRef]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. MiRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilad, S.; Meiri, E.; Yogev, Y.; Benjamin, S.; Lebanony, D.; Yerushalmi, N.; Benjamin, H.; Kushnir, M.; Cholakh, H.; Melamed, N.; et al. Serum MicroRNAs Are Promising Novel Biomarkers. PLoS ONE 2008, 3, e3148. [Google Scholar] [CrossRef] [Green Version]
- Tiberio, P.; Callari, M.; Angeloni, V.; Daidone, M.G.; Appierto, V. Challenges in Using Circulating MiRNAs as Cancer Biomarkers. Biomed Res. Int. 2015, 2015, 731479. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, M.B.; Kao, S.C.; Edelman, J.J.; Armstrong, N.J.; Vallely, M.P.; van Zandwijk, N.; Reid, G. Haemolysis during Sample Preparation Alters MicroRNA Content of Plasma. PLoS ONE 2011, 6, e24145. [Google Scholar] [CrossRef]
- Shrestha, S.; Hsu, S.; Huang, W.; Huang, H.; Chen, W.; Weng, S.; Huang, H. A Systematic Review of MicroRNA Expression Profiling Studies in Human Gastric Cancer. Cancer Med. 2014, 3, 878–888. [Google Scholar] [CrossRef]
- Herrera-Espejo, S.; Santos-Zorrozua, B.; Álvarez-González, P.; Lopez-Lopez, E.; Garcia-Orad, Á. A Systematic Review of MicroRNA Expression as Biomarker of Late-Onset Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 8376–8391. [Google Scholar] [CrossRef] [PubMed]
- Francavilla, A.; Gagliardi, A.; Piaggeschi, G.; Tarallo, S.; Cordero, F.; Pensa, R.G.; Impeduglia, A.; Caviglia, G.P.; Ribaldone, D.G.; Gallo, G.; et al. Faecal MiRNA Profiles Associated with Age, Sex, BMI, and Lifestyle Habits in Healthy Individuals. Sci. Rep. 2021, 11, 20645. [Google Scholar] [CrossRef] [PubMed]
- Diener, C.; Keller, A.; Meese, E. Emerging Concepts of MiRNA Therapeutics: From Cells to Clinic. Trends Genet. 2022, 38, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The Multilayered Complexity of CeRNA Crosstalk and Competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Zhang, D.-H.; Wu, N.; Xiao, J.-H.; Wang, X.; Ma, W. CeRNA in Cancer: Possible Functions and Clinical Implications. J. Med. Genet. 2015, 52, 710–718. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The Role of MicroRNAs in Human Cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Fan, M.; Zhang, X.; Huang, F.; Wu, K.; Zhang, J.; Liu, J.; Huang, Z.; Luo, H.; Tao, L.; et al. Cellular MicroRNAs Up-Regulate Transcription via Interaction with Promoter TATA-Box Motifs. RNA 2014, 20, 1878–1889. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lifecycle Step | microRNA | Cause | Direct Effect | Cancer Type | References |
|---|---|---|---|---|---|
| Translational repression | mir-499a-3p | SNP | Altered targetome | Glioma | [147,159] |
| Processing of the pri-miRNA | mir-26a | SNP | Decreased pri-miRNA processing by Drosha | BLCA, OSCC, ESCC, LC, CC, BRCA, CRC | [160,161] |
| Processing of the pre-miRNA | mir-196a-3p | SNP | Increased pre-miRNA processing by Dicer | NSCLC | [162] |
| Transcription | mir-378 | SNP | Decreased miRNA expression | BRCA | [148,162] |
| Transcription | mir-130a | Hypermethylation miRNA promoter | Decreased miRNA expression | PCa | [163] |
| Transcription | mir-15/16 | CNV | Decreased miRNA expression | B-CLL, PCa | [164,165,166] |
| Transcription | mir-29 | Upregulation transcription factor | Decreased miRNA expression | BLCA | [167] |
| Transcription | miR-4728 | Host gene overexpression | Increased miRNA expression | BRCA, GC | [158] |
| Processing of the pri-miRNA | let-7 | Drosha mutant | Decreased miRNA processing by Drosha | Wilms tumour | [155] |
| Processing of the pri-miRNA | miR-27b | Drosha SNP | Decreased post-translational repression of Drosha mRNA | BLCA | [146] |
| Processing of the pre-miRNA | miR-3622a-5p, miR-5582-5p | Dicer SNP | Increased post-translational repression of Dicer mRNA | TCL, HCC | [168,169,170] |
| Strand selection | miR-223 | Altered NHEJ | Arm switching (3p to 5p) | BRCA | [171] |
| Strand selection | miR-193a | Increased expression target genes (TMMP) | Arm switching (5p to 3p) | BRCA | [172] |
| Translational repression | miR-187 | SNP in target gene | Increased target mRNA repression | BRCA | [152,173] |
| Degradation | miR-106, miR-221/222 | Increased expression exoribonuclease | Increased degradation | MEL | [174,175] |
| Modification | miR-200b | A to I editing | Altered targetome | HNSCC, KIRP, THCA, UCEC | [154] |
| Methylation | miR-221/222 | m6A modification | Increased processing | BLCA | [176] |
| ceRNAs | miR-148a | Upregulation lncRNA | Decreased expression | ESCC | [177] |
| ceRNAs | miR-19a-3p | Downregulation lncRNA | Increased expression | GC, BRCA | [178,179,180] |
| ceRNAs | miR-7 | Upregulation circRNA | Decreased expression | LC, HCC, MEL, CRC, BRCA | [181,182,183] |
| ceRNAs | miR-1280 | Downregulation tRNALeu /miR-1280 | Increased target mRNA repression | CRC | [141,184] |
| Self-regulation | miR-424, miR-503, mir-9 | Disruption interaction between mature and pri-miRNA | Increased miR-9 | AML | [185] |
| Self-regulation | miR-107 let-7 | Upregulation miR-107 | Degradation let-7 | BRCA | [186] |
| Self-regulation | miR-20a | Upregulation miRNA | Negative autoregulatory loop through downregulation of transcription factor | PCa | [145] |
| Self-regulation | miR-98-5p | Upregulation miRNA | Global decrease in Dicer processing | OV | [187] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Rooij, L.A.; Mastebroek, D.J.; ten Voorde, N.; van der Wall, E.; van Diest, P.J.; Moelans, C.B. The microRNA Lifecycle in Health and Cancer. Cancers 2022, 14, 5748. https://doi.org/10.3390/cancers14235748
de Rooij LA, Mastebroek DJ, ten Voorde N, van der Wall E, van Diest PJ, Moelans CB. The microRNA Lifecycle in Health and Cancer. Cancers. 2022; 14(23):5748. https://doi.org/10.3390/cancers14235748
Chicago/Turabian Stylede Rooij, Laura Adriana, Dirk Jan Mastebroek, Nicky ten Voorde, Elsken van der Wall, Paul Joannes van Diest, and Cathy Beatrice Moelans. 2022. "The microRNA Lifecycle in Health and Cancer" Cancers 14, no. 23: 5748. https://doi.org/10.3390/cancers14235748
APA Stylede Rooij, L. A., Mastebroek, D. J., ten Voorde, N., van der Wall, E., van Diest, P. J., & Moelans, C. B. (2022). The microRNA Lifecycle in Health and Cancer. Cancers, 14(23), 5748. https://doi.org/10.3390/cancers14235748