
Combination of STING and TLR 7/8 Agonists as Vaccine Adjuvants for Cancer Immunotherapy

 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Experimental
2.1. Materials
2.2. Animals and Cell Line
2.3. Culture of BMDCs
2.4. In Vitro BMDC Activation
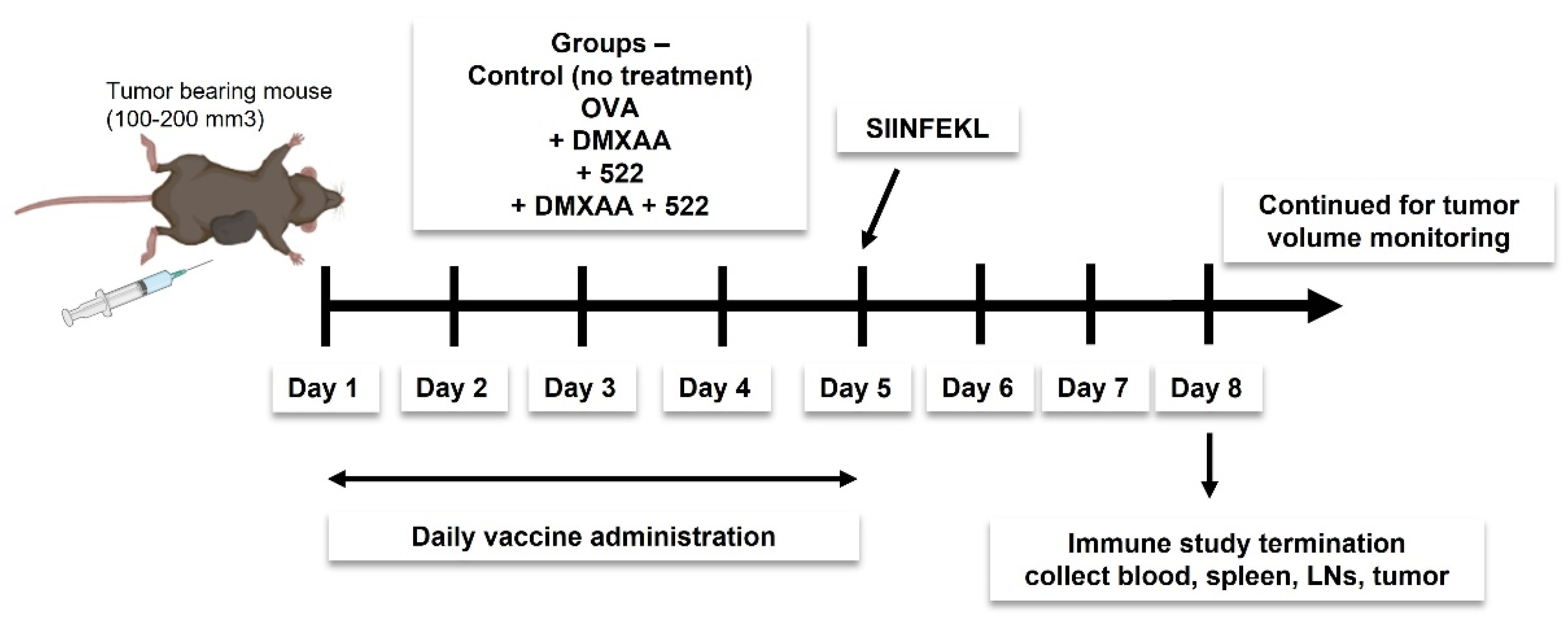
2.5. Immunization Protocol
2.6. In Vivo Antitumor Efficacy
2.7. In Vivo APC Activation and T Cell Proliferation Assay
2.8. Immunohistochemistry
2.9. Statistical Analyses
3. Results
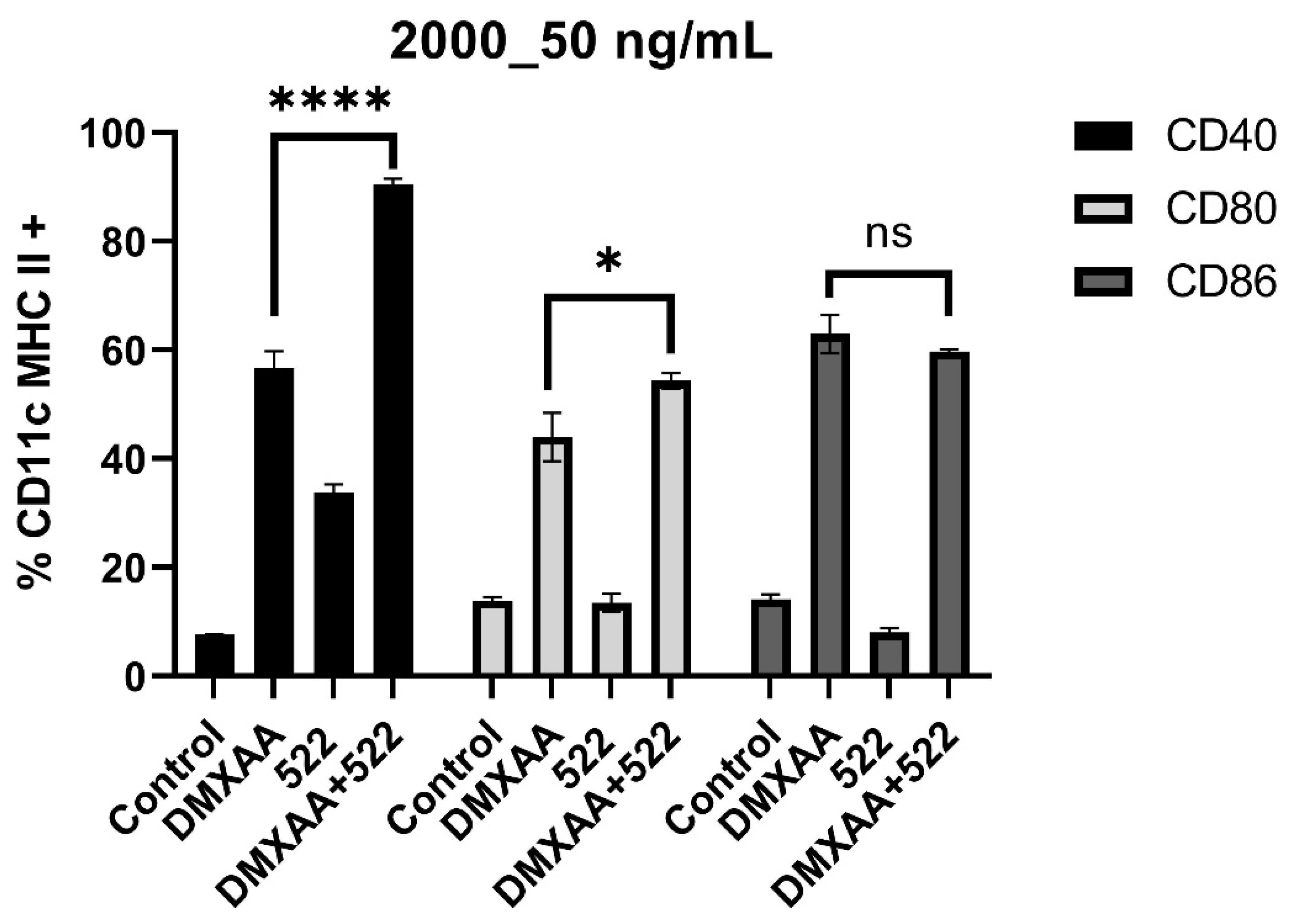
3.1. DMXAA and 522 Combination Enhances BMDC Activation
3.2. Therapeutic Vaccination with DMXAA + 522 Combination Enhances Tumor Inhibition and Improves Survival
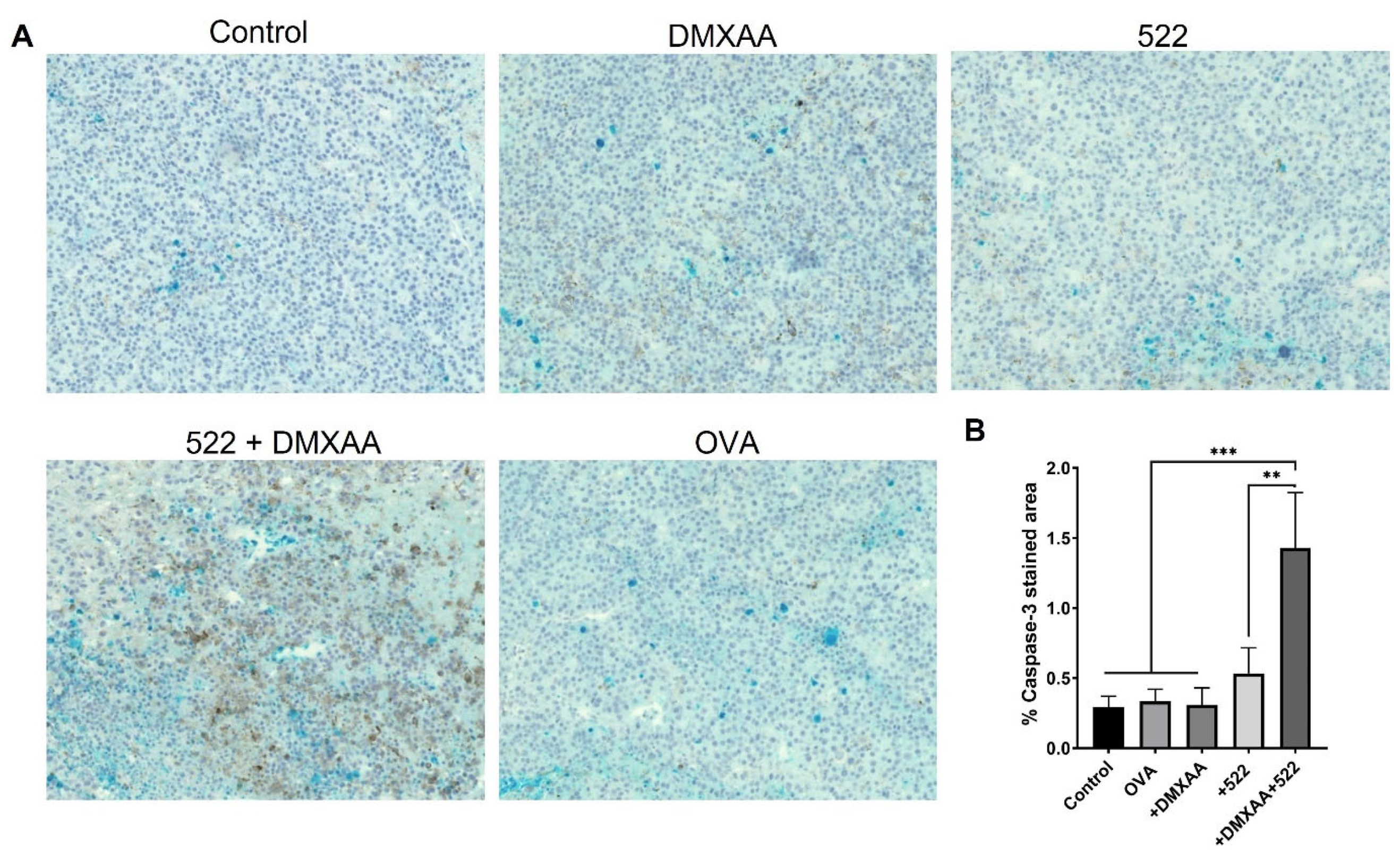
3.3. Immunohistology
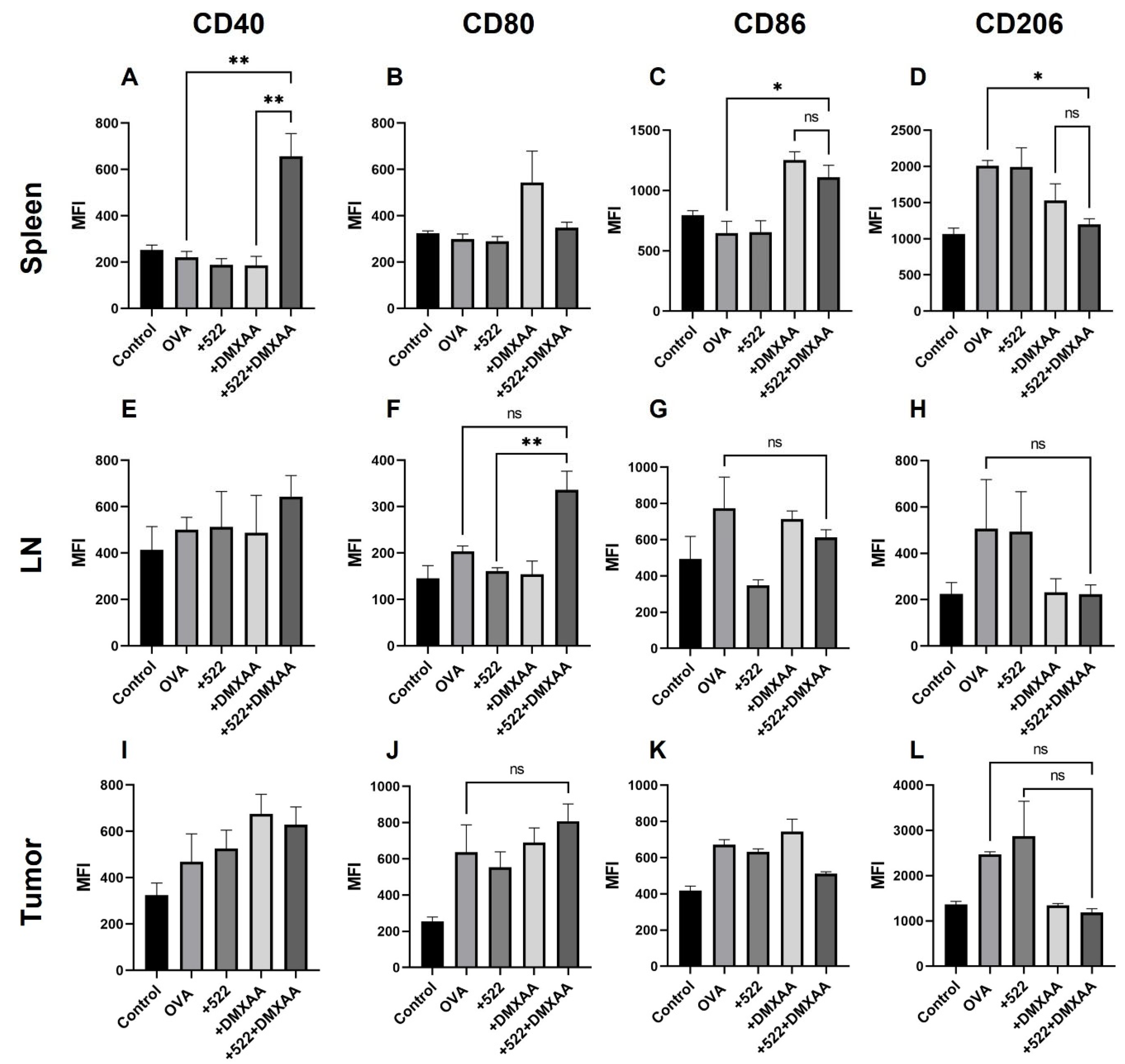
3.4. Activation of APCs In Vivo
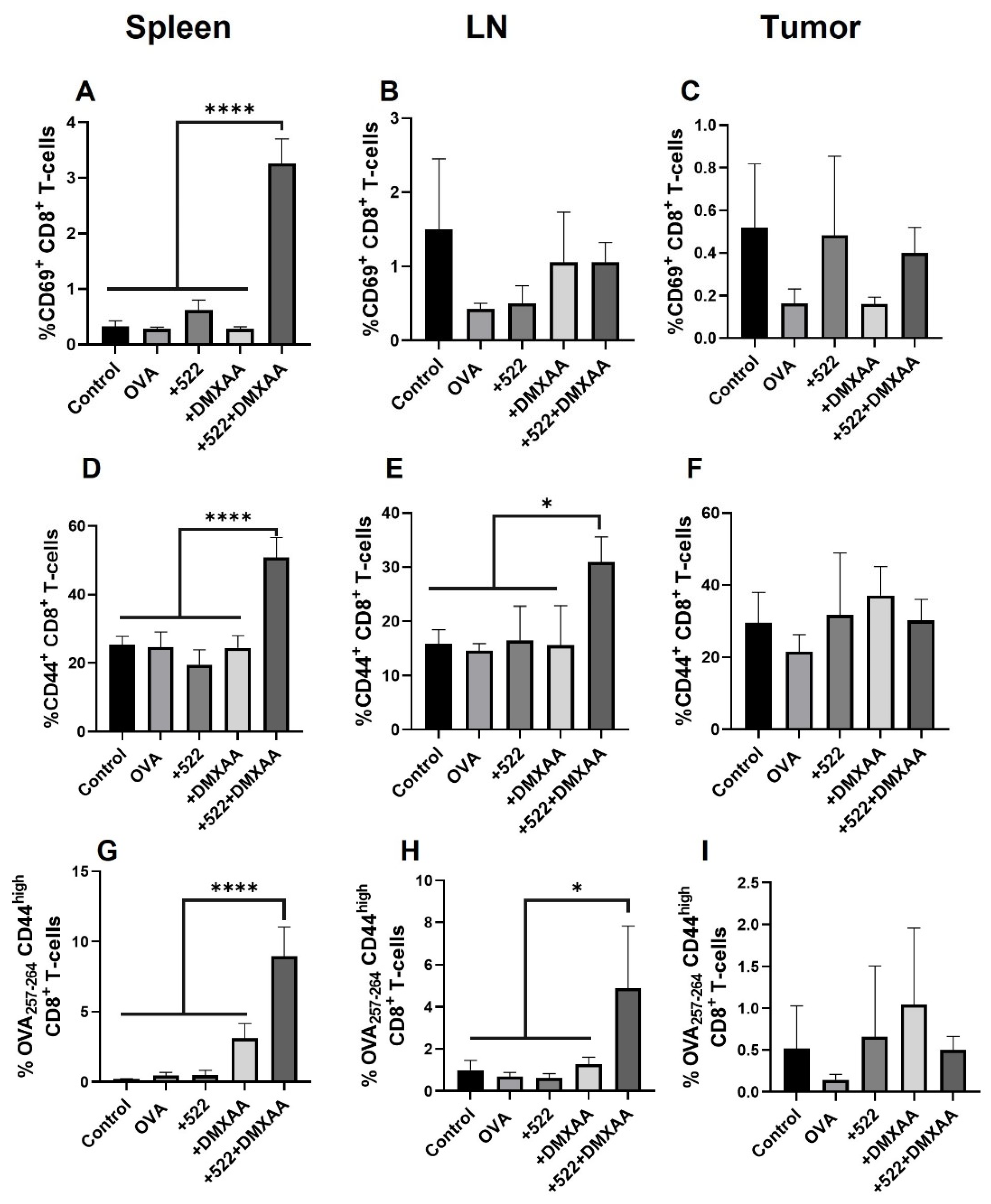
3.5. Antigen-Specific CD8+ T Cell Activation
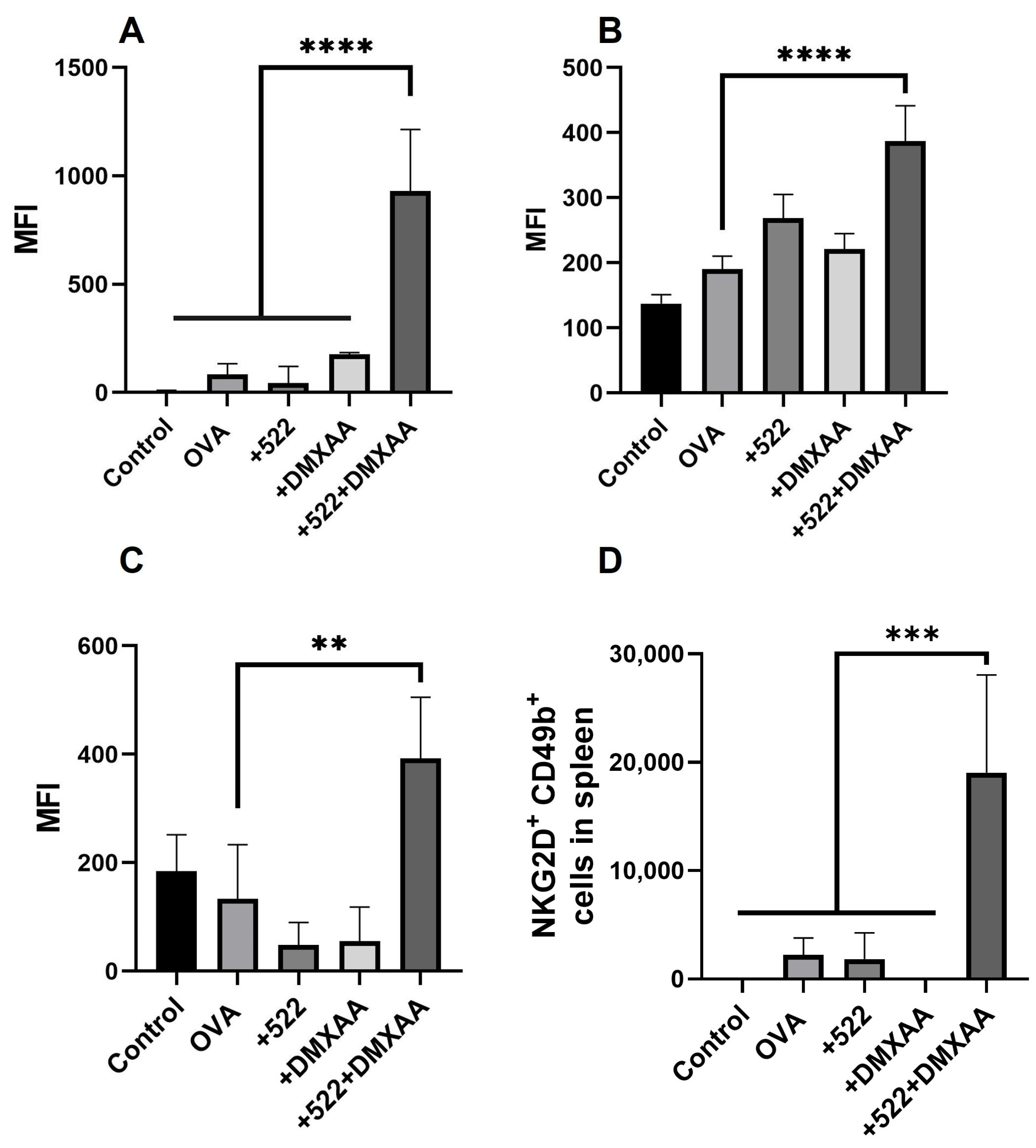
3.6. Natural Killer (NK) Cell Activation
4. Discussion
5. Conclusion and Future Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janeway, C.A. Approaching the asymptote? Evolution and revolution in immunology. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989; pp. 1–13. [Google Scholar]
- Janeway, C.A. Pillars article: Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 1989. 54: 1–13. J. Immunol. 2013, 191, 4475–4487. [Google Scholar]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Bortoluci, K.R.; Medzhitov, R. Control of infection by pyroptosis and autophagy: Role of TLR and NLR. Cell. Mol. Life Sci. 2010, 67, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.; Yang, F.C. A Review of Topical Imiquimod in the Management of Basal Cell Carcinoma, Actinic Keratoses, and Other Skin Lesions. Clin. Medicine. Ther. 2009, 1, CMT.S1969. [Google Scholar] [CrossRef]
- Lebwohl, M.; Dinehart, S.; Whiting, D.; Lee, P.K.; Tawfik, N.; Jorizzo, J.; Lee, J.H.; Fox, T.L. Imiquimod 5% cream for the treatment of actinic keratosis: Results from two phase III, randomized, double-blind, parallel group, vehicle-controlled trials. J. Am. Acad. Dermatol. 2004, 50, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Frega, G.; Wu, Q.; Le Naour, J.; Vacchelli, E.; Galluzzi, L.; Kroemer, G.; Kepp, O. Trial Watch: Experimental TLR7/TLR8 agonists for oncological indications. OncoImmunology 2020, 9, 1796002. [Google Scholar] [CrossRef]
- Guerini, D. STING Agonists/Antagonists: Their Potential as Therapeutics and Future Developments. Cells 2022, 11, 1159. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Chen, Z.J. Antiviral innate immunity pathways. Cell Res. 2006, 16, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Chen, Z.J. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci. Signal. 2012, 5, ra20. [Google Scholar] [CrossRef] [Green Version]
- Brault, M.; Olsen, T.M.; Martinez, J.; Stetson, D.B.; Oberst, A. Intracellular nucleic acid sensing triggers necroptosis through synergistic type I IFN and TNF signaling. J. Immunol. 2018, 200, 2748–2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Dai, T.; He, X.; Zhang, Z.; Xie, F.; Wang, S.; Zhang, L.; Zhou, F. The interactions between cGAS-STING pathway and pathogens. Signal Transduct. Target. Ther. 2020, 5, 91. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Niu, L.; Larson, P.; Kucaba, T.A.; Murphy, K.A.; James, B.R.; Ferguson, D.M.; Griffith, T.S.; Panyam, J. Polymeric nanoparticles encapsulating novel TLR7/8 agonists as immunostimulatory adjuvants for enhanced cancer immunotherapy. Biomaterials 2018, 164, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Schiaffo, C.E.; Shi, C.; Xiong, Z.; Olin, M.; Ohlfest, J.R.; Aldrich, C.C.; Ferguson, D.M. Structure–Activity Relationship Analysis of Imidazoquinolines with Toll-like Receptors 7 and 8 Selectivity and Enhanced Cytokine Induction. J. Med. Chem. 2014, 57, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Baguley, B.C.; Ching, L.-M. DMXAA: An antivascular agent with multiple host responses. Int. J. Radiat. Oncol. Biol. Phys. 2002, 54, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.; Burdette, D.L.; Sharma, S.; Bhat, N.; Thompson, M.; Jiang, Z.; Rathinam, V.A.K.; Monks, B.; Jin, T.; Xiao, T.S.; et al. Mouse, but not Human STING, Binds and Signals in Response to the Vascular Disrupting Agent 5,6-Dimethylxanthenone-4-Acetic Acid. J. Immunol. 2013, 190, 5216–5225. [Google Scholar] [CrossRef] [Green Version]
- Daei Farshchi Adli, A.; Jahanban-Esfahlan, R.; Seidi, K.; Samandari-Rad, S.; Zarghami, N. An overview on Vadimezan (DMXAA): The vascular disrupting agent. Chem. Biol. Drug Des. 2018, 91, 996–1006. [Google Scholar] [CrossRef]
- Larson, P.; Kucaba, T.A.; Xiong, Z.; Olin, M.; Griffith, T.S.; Ferguson, D.M. Design and Synthesis of N1-Modified Imidazoquinoline Agonists for Selective Activation of Toll-like Receptors 7 and 8. ACS Med. Chem. Lett. 2017, 8, 1148–1152. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Bottomly, K. Signals and signs for lymphocyte responses. Cell 1994, 76, 275–285. [Google Scholar] [CrossRef]
- Kim, H.; Sehgal, D.; Kucaba, T.A.; Ferguson, D.M.; Griffith, T.S.; Panyam, J. Acidic pH-responsive polymer nanoparticles as a TLR7/8 agonist delivery platform for cancer immunotherapy. Nanoscale 2018, 10, 20851–20862. [Google Scholar] [CrossRef]
- Azarov, I.; Peskov, K.; Helmlinger, G.; Kosinsky, Y. Role of T Cell-To-Dendritic Cell Chemoattraction in T Cell Priming Initiation in the Lymph Node: An Agent-Based Modeling Study. Front. Immunol. 2019, 10, 1289. [Google Scholar] [CrossRef] [PubMed]
- Casey, K.A.; Fraser, K.A.; Schenkel, J.M.; Moran, A.; Abt, M.C.; Beura, L.K.; Lucas, P.J.; Artis, D.; Wherry, E.J.; Hogquist, K.; et al. Antigen-Independent Differentiation and Maintenance of Effector-like Resident Memory T Cells in Tissues. J. Immunol. 2012, 188, 4866–4875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Information from www.bio-rad-antibodies.com. Available online: https://www.bio-rad-antibodies.com/nkcells-minireview.html?JSESSIONID_STERLING=2D89989EE5205549D7376A8FDE068D11.ecommerce1&evCntryLang=US-en&cntry=US&thirdPartyCookieEnabled=true (accessed on 7 July 2022).
- Khong, H.; Overwijk, W.W. Adjuvants for peptide-based cancer vaccines. J. Immunother. Cancer 2016, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- DeMaria, P.J.; Bilusic, M. Cancer Vaccines. Hematol. Oncol. Clin. N. Am. 2019, 33, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; García-Martínez, E.; Pitter, M.R.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Toll-like receptor agonists in cancer immunotherapy. OncoImmunology 2018, 7, e1526250. [Google Scholar] [CrossRef]
- Wang, S.; Campos, J.; Gallotta, M.; Gong, M.; Crain, C.; Naik, E.; Coffman, R.L.; Guiducci, C. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8<sup>+</sup> T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7240–E7249. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Zhang, C.-d.; Wu, X.-h. Therapeutic cancer vaccines: From initial findings to prospects. Immunol. Lett. 2018, 196, 11–21. [Google Scholar] [CrossRef]
- de Titta, A.; Ballester, M.; Julier, Z.; Nembrini, C.; Jeanbart, L.; van der Vlies, A.J.; Swartz, M.A.; Hubbell, J.A. Nanoparticle conjugation of CpG enhances adjuvancy for cellular immunity and memory recall at low dose. Proc. Natl. Acad. Sci. USA 2013, 110, 19902–19907. [Google Scholar] [CrossRef] [Green Version]
- Kundi, M. New hepatitis B vaccine formulated with an improved adjuvant system. Expert Rev. Vaccines 2007, 6, 133–140. [Google Scholar] [CrossRef]
- Khanna, V.; Kim, H.; Zhang, W.; Larson, P.; Shah, M.; Griffith, T.S.; Ferguson, D.; Panyam, J. Novel TLR 7/8 agonists for improving NK cell mediated antibody-dependent cellular cytotoxicity (ADCC). Sci. Rep. 2021, 11, 3346. [Google Scholar] [CrossRef]
- Su, T.; Zhang, Y.; Valerie, K.; Wang, X.-Y.; Lin, S.; Zhu, G. STING activation in cancer immunotherapy. Theranostics 2019, 9, 7759. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flood, B.A.; Higgs, E.F.; Li, S.; Luke, J.J.; Gajewski, T.F. STING pathway agonism as a cancer therapeutic. Immunol. Rev. 2019, 290, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Temizoz, B.; Kuroda, E.; Ohata, K.; Jounai, N.; Ozasa, K.; Kobiyama, K.; Aoshi, T.; Ishii, K.J. TLR9 and STING agonists synergistically induce innate and adaptive type-II IFN. Eur. J. Immunol. 2015, 45, 1159–1169. [Google Scholar] [CrossRef]
- Deb, P.; Dai, J.; Singh, S.; Kalyoussef, E.; Fitzgerald-Bocarsly, P. Triggering of the cGAS–STING Pathway in Human Plasmacytoid Dendritic Cells Inhibits TLR9-Mediated IFN Production. J. Immunol. 2020, 205, 223–236. [Google Scholar] [CrossRef]
- Baguley, B.C. Antivascular therapy of cancer: DMXAA. Lancet Oncol. 2003, 4, 141–148. [Google Scholar] [CrossRef]
- Jr, P.N.L.; Douillard, J.-Y.; Nakagawa, K.; Pawel, J.v.; McKeage, M.J.; Albert, I.; Losonczy, G.; Reck, M.; Heo, D.-S.; Fan, X.; et al. Randomized Phase III Placebo-Controlled Trial of Carboplatin and Paclitaxel With or Without the Vascular Disrupting Agent Vadimezan (ASA404) in Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2011, 29, 2965–2971. [Google Scholar] [CrossRef]
- Head, M.; Jameson, M.B. The development of the tumor vascular-disrupting agent ASA404 (vadimezan, DMXAA): Current status and future opportunities. Expert Opin. Investig. Drugs 2010, 19, 295–304. [Google Scholar] [CrossRef]
- Lasek, W.; Zagożdżon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, F.; Montfort, A.; Marcheteau, E.; Imbert, C.; Gilhodes, J.; Filleron, T.; Rochaix, P.; Andrieu-Abadie, N.; Levade, T.; Meyer, N.; et al. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 2017, 8, 2256. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.K.; Gracias, D.T.; Croft, M. TNF activity and T cells. Cytokine 2018, 101, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Rong, L.; Zhao, X.; Li, X.; Liu, X.; Deng, J.; Wu, H.; Xu, X.; Erben, U.; Wu, P. TNF signaling drives myeloid-derived suppressor cell accumulation. J. Clin. Investig. 2012, 122, 4094–4104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Karin, M. The wolf in sheep’s clothing: The role of interleukin-6 in immunity, inflammation and cancer. Trends Mol. Med. 2008, 14, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Mikucki, M.E.; Fisher, D.T.; Ku, A.W.; Appenheimer, M.M.; Muhitch, J.B.; Evans, S.S. Preconditioning thermal therapy: Flipping the switch on IL-6 for anti-tumour immunity. Int. J. Hyperth. 2013, 29, 464–473. [Google Scholar] [CrossRef] [Green Version]
- Drzyzga, A.; Cichoń, T.; Czapla, J.; Jarosz-Biej, M.; Pilny, E.; Matuszczak, S.; Wojcieszek, P.; Urbaś, Z.; Smolarczyk, R. The Proper Administration Sequence of Radiotherapy and Anti-Vascular Agent—DMXAA Is Essential to Inhibit the Growth of Melanoma Tumors. Cancers 2021, 13, 3924. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.-R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [Green Version]
- Smolarczyk, R.; Cichoń, T.; Pilny, E.; Jarosz-Biej, M.; Poczkaj, A.; Kułach, N.; Szala, S. Combination of anti-vascular agent-DMXAA and HIF-1α inhibitor-digoxin inhibits the growth of melanoma tumors. Sci. Rep. 2018, 8, 7355. [Google Scholar] [CrossRef] [Green Version]
- Rauca, V.-F.; Patras, L.; Luput, L.; Licarete, E.; Toma, V.-A.; Porfire, A.; Mot, A.C.; Rakosy-Tican, E.; Sesarman, A.; Banciu, M. Remodeling tumor microenvironment by liposomal codelivery of DMXAA and simvastatin inhibits malignant melanoma progression. Sci. Rep. 2021, 11, 22102. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Steinhagen, F.; Kinjo, T.; Bode, C.; Klinman, D.M. TLR-based immune adjuvants. Vaccine 2011, 29, 3341–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baguley, B.C.; Ching, L.-M. Immunomodulatory Actions of Xanthenone Anticancer Agents. BioDrugs 1997, 8, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Wang, Y.; Liu, X.; Li, L.; Opejin, A.; Hsueh, E.C.; Luo, H.; Wang, T.; Hawiger, D.; Peng, G. TLR7 Signaling Regulates Th17 Cells and Autoimmunity: Novel Potential for Autoimmune Therapy. J. Immunol. 2017, 199, 941–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelvanambi, M.; Fecek, R.J.; Taylor, J.L.; Storkus, W.J. STING agonist-based treatment promotes vascular normalization and tertiary lymphoid structure formation in the therapeutic melanoma microenvironment. J. Immunother. Cancer 2021, 9, e001906. [Google Scholar] [CrossRef]
- Kim, H.; Khanna, V.; Kucaba, T.A.; Zhang, W.; Sehgal, D.; Ferguson, D.M.; Griffith, T.S.; Panyam, J. TLR7/8 Agonist-Loaded Nanoparticles Augment NK Cell-Mediated Antibody-Based Cancer Immunotherapy. Mol. Pharm. 2020, 17, 2109–2124. [Google Scholar] [CrossRef]
- Ni, L.; Lu, J. Interferon gamma in cancer immunotherapy. Cancer Med. 2018, 7, 4509–4516. [Google Scholar] [CrossRef]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e399. [Google Scholar] [CrossRef] [Green Version]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Interferon-γ: Teammate or opponent in the tumour microenvironment? Nat. Rev. Immunol. 2022, 22, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; McAllister, D.; Vonderhaar, E.P.; Palen, K.; Riese, M.J.; Gershan, J.; Johnson, B.D.; Dwinell, M.B. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J. Immunother. Cancer 2019, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Jassar, A.; Mishalian, I.; Wang, L.C.; Kapoor, V.; Cheng, G.; Sun, J.; Singhal, S.; Levy, L.; Albelda, S.M. Using macrophage activation to augment immunotherapy of established tumours. Br. J. Cancer 2013, 108, 1288–1297. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhatnagar, S.; Revuri, V.; Shah, M.; Larson, P.; Shao, Z.; Yu, D.; Prabha, S.; Griffith, T.S.; Ferguson, D.; Panyam, J. Combination of STING and TLR 7/8 Agonists as Vaccine Adjuvants for Cancer Immunotherapy. Cancers 2022, 14, 6091. https://doi.org/10.3390/cancers14246091
Bhatnagar S, Revuri V, Shah M, Larson P, Shao Z, Yu D, Prabha S, Griffith TS, Ferguson D, Panyam J. Combination of STING and TLR 7/8 Agonists as Vaccine Adjuvants for Cancer Immunotherapy. Cancers. 2022; 14(24):6091. https://doi.org/10.3390/cancers14246091
Chicago/Turabian StyleBhatnagar, Shubhmita, Vishnu Revuri, Manan Shah, Peter Larson, Zekun Shao, Daohai Yu, Swayam Prabha, Thomas S. Griffith, David Ferguson, and Jayanth Panyam. 2022. "Combination of STING and TLR 7/8 Agonists as Vaccine Adjuvants for Cancer Immunotherapy" Cancers 14, no. 24: 6091. https://doi.org/10.3390/cancers14246091
APA StyleBhatnagar, S., Revuri, V., Shah, M., Larson, P., Shao, Z., Yu, D., Prabha, S., Griffith, T. S., Ferguson, D., & Panyam, J. (2022). Combination of STING and TLR 7/8 Agonists as Vaccine Adjuvants for Cancer Immunotherapy. Cancers, 14(24), 6091. https://doi.org/10.3390/cancers14246091