
Emerging Role of Targeted Therapy in Metastatic Pancreatic Adenocarcinoma

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. First-Line Chemotherapy
3. Second-Line Chemotherapy
4. Biomarker-Driven Therapy
4.1. Homologous Recombination Deficiency
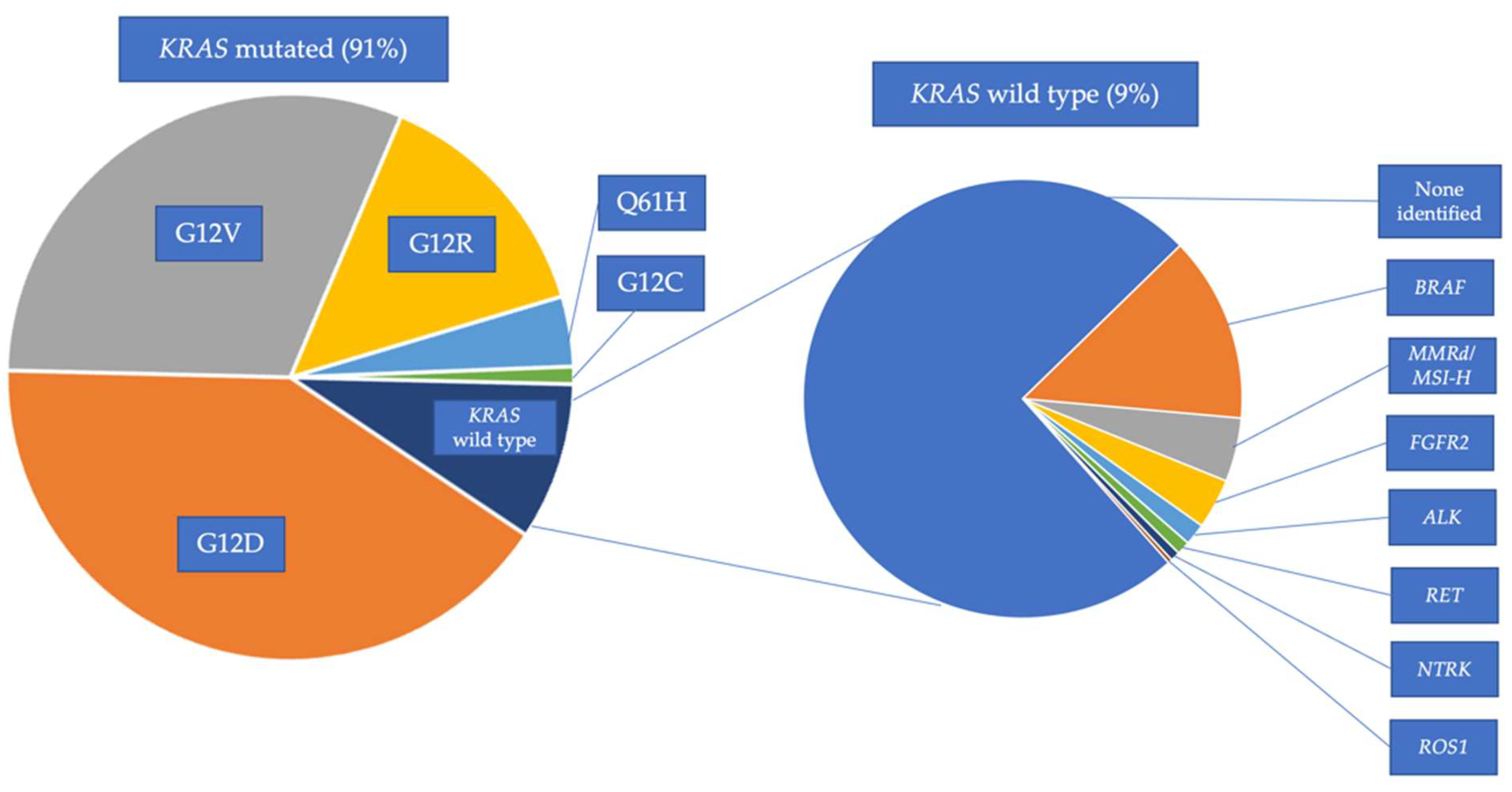
4.2. KRAS Mutated PDAC
4.3. KRAS Wild-Type (WT) PDAC
4.4. BRAF Alterations
4.5. FGFR2 Fusions
4.6. ALK Fusions
4.7. NTRK Fusions
4.8. NRG1 Fusions
4.9. RET Fusions
4.10. ROS1 Alterations
4.11. MMR Deficient PDAC
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, S.; Kamgar, M.; Mahipal, A. Systemic Therapy of Metastatic Pancreatic Adenocarcinoma: Current Status, Challenges, and Opportunities. Cancers 2022, 14, 2588. [Google Scholar] [CrossRef]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic Adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef]
- Rubinson, D.A.; Wolpin, B.M. Therapeutic Approaches for Metastatic Pancreatic Adenocarcinoma. Hematol. Oncol. Clin. N. Am. 2015, 29, 761–776. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients with Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef]
- Sohal, D.P.; Mangu, P.B.; Khorana, A.; Shah, M.A.; Philip, P.A.; O’Reilly, E.M.; Uronis, H.E.; Ramanathan, R.K.; Crane, C.H.; Engebretson, A.; et al. Metastatic Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2016, 34, 2784–2796. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.P.S.; Kennedy, E.B.; Cinar, P.; Conroy, T.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Lau, M.W.; Johnson, T.; Krishnamurthi, S.; et al. Metastatic Pancreatic Cancer: ASCO Guideline Update. J. Clin. Oncol. 2020, 38, 3217–3230. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.P.S.; Kennedy, E.B.; Khorana, A.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Krishnamurthi, S.; Moravek, C.; O’Reilly, E.M.; Philip, P.A.; et al. Metastatic Pancreatic Cancer: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2018, 36, 2545–2556. [Google Scholar] [CrossRef] [PubMed]
- Pijnappel, E.N.; Dijksterhuis, W.P.; van der Geest, L.G.; de Vos-Geelen, J.; de Groot, J.W.B.; Homs, M.Y.; Creemers, G.-J.; Mohammad, N.H.; Besselink, M.G.; van Laarhoven, H.W.; et al. First- and Second-Line Palliative Systemic Treatment Outcomes in a Real-World Metastatic Pancreatic Cancer Cohort. J. Natl. Compr. Cancer Netw. 2021, 20, 443–450.e3. [Google Scholar] [CrossRef]
- Pusceddu, S.; Ghidini, M.; Torchio, M.; Corti, F.; Tomasello, G.; Niger, M.; Prinzi, N.; Nichetti, F.; Coinu, A.; Di Bartolomeo, M.; et al. Comparative Effectiveness of Gemcitabine plus Nab-Paclitaxel and FOLFIRINOX in the First-Line Setting of Metastatic Pancreatic Cancer: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 484. [Google Scholar] [CrossRef] [Green Version]
- Carrato, A.; Pazo-Cid, R.; Macarulla, T.; Gallego, J.; Jiménez-Fonseca, P.; Rivera, F.; Cano, M.T.; Garrote, M.R.; Pericay, C.; Diaz, I.; et al. Sequential nab-paclitaxel/gemcitabine followed by modified FOLFOX for first-line metastatic pancreatic cancer: The SEQUENCE trial. J. Clin. Oncol. 2022, 40, 4022. [Google Scholar] [CrossRef]
- Oettle, H.; Riess, H.; Stieler, J.M.; Heil, G.; Schwaner, I.; Seraphin, J.; Görner, M.; Mölle, M.; Greten, T.F.; Lakner, V.; et al. Second-Line Oxaliplatin, Folinic Acid, and Fluorouracil Versus Folinic Acid and Fluorouracil Alone for Gemcitabine-Refractory Pancreatic Cancer: Outcomes from the CONKO-003 Trial. J. Clin. Oncol. 2014, 32, 2423–2429. [Google Scholar] [CrossRef]
- Gill, S.; Ko, Y.-J.; Cripps, C.; Beaudoin, A.; Dhesy-Thind, S.; Zulfiqar, M.; Zalewski, P.; Do, T.; Cano, P.; Lam, W.Y.H.; et al. PANCREOX: A Randomized Phase III Study of Fluorouracil/Leucovorin with or without Oxaliplatin for Second-Line Advanced Pancreatic Cancer in Patients Who Have Received Gemcitabine-Based Chemotherapy. J. Clin. Oncol. 2016, 34, 3914–3920. [Google Scholar] [CrossRef]
- Hecht, J.R.; Lonardi, S.; Bendell, J.; Sim, H.-W.; Macarulla, T.; Lopez, C.D.; Van Cutsem, E.; Martin, A.J.M.; Park, J.O.; Greil, R.; et al. Randomized Phase III Study of FOLFOX Alone or with Pegilodecakin as Second-Line Therapy in Patients with Metastatic Pancreatic Cancer That Progressed After Gemcitabine (SEQUOIA). J. Clin. Oncol. 2021, 39, 1108–1118. [Google Scholar] [CrossRef]
- Chung, V.; McDonough, S.; Philip, P.A.; Cardin, D.; Wang-Gillam, A.; Hui, L.; Tejani, M.A.; Seery, T.E.; Dy, I.A.; Al Baghdadi, T.; et al. Effect of Selumetinib and MK-2206 vs Oxaliplatin and Fluorouracil in Patients with Metastatic Pancreatic Cancer After Prior Therapy: SWOG S1115 Study Randomized Clinical Trial. JAMA Oncol. 2017, 3, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Wang-Gillam, A.; Li, C.-P.; Bodoky, G.; Dean, A.; Shan, Y.-S.; Jameson, G.; Macarulla, T.; Lee, K.-H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, E.G.; Guthrie, K.A.; Philip, P.A.; Swisher, E.M.; Jalikis, F.; Pishvaian, M.J.; Berlin, J.; Noel, M.S.; Suga, J.M.; Garrido-Laguna, I.; et al. Randomized Phase II Study of PARP Inhibitor ABT-888 (Veliparib) with Modified FOLFIRI versus FOLFIRI as Second-line Treatment of Metastatic Pancreatic Cancer: SWOG S1513. Clin. Cancer Res. 2021, 27, 6314–6322. [Google Scholar] [CrossRef] [PubMed]
- Hammel, P.; El-Hariry, I.; Macarulla, T.; Garcia-Carbonero, R.; Metges, J.-P.; Bouché, O.; Portales, F.; Cid, R.A.P.; Mineur, L.; Gracian, A.M.C.; et al. Trybeca-1: A randomized, phase 3 study of eryaspase in combination with chemotherapy versus chemotherapy alone as second-line treatment in patients with advanced pancreatic adenocarcinoma (NCT03665441). J. Clin. Oncol. 2022, 40, 518. [Google Scholar] [CrossRef]
- Huffman, B.M.; Mallick, A.B.; Horick, N.K.; Wang-Gillam, A.; Hosein, P.J.; Morse, M.; Beg, M.S.; Murphy, J.E.; Schlechter, B.L.; Sanoff, H.; et al. Abstract A019: A multicenter randomized phase II study of gemcitabine and nab-paclitaxel versus gemcitabine and nab-paclitaxel with a MUC5AC antibody (NPC-1C) in advanced pancreatic cancer previously treated with FOLFIRINOX (NCT01834235). Cancer Res. 2022, 82, A019. [Google Scholar] [CrossRef]
- Xu, Z.; Hu, K.; Bailey, P.; Springfeld, C.; Roth, S.; Kurilov, R.; Brors, B.; Gress, T.; Buchholz, M.; An, J.; et al. Clinical Impact of Molecular Subtyping of Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 743908. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Nowak, J.A.; Camarda, N.D.; Moffitt, R.A.; Ghazani, A.A.; Hazar-Rethinam, M.; Raghavan, S.; Kim, J.; Brais, L.K.; Ragon, D.; et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. 2018, 8, 1096–1111. [Google Scholar] [CrossRef] [Green Version]
- Hosein, A.N.; Dougan, S.K.; Aguirre, A.J.; Maitra, A. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat. Cancer 2022, 3, 272–286. [Google Scholar] [CrossRef]
- Philip, P.A.; Azar, I.; Xiu, J.; Hall, M.J.; Hendifar, A.E.; Lou, E.; Hwang, J.J.; Gong, J.; Feldman, R.; Ellis, M.; et al. Molecular Characterization of KRAS Wild-type Tumors in Patients with Pancreatic Adenocarcinoma. Clin. Cancer Res. 2022, 28, 2704–2714. [Google Scholar] [CrossRef]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted with Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253.e4. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Lyons, E.; DeArbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sahai, V.; Sohal, D.P.S.; et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: A retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020, 21, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Marciano, N.D.; Kroening, G.; Dayyani, F.; Zell, J.A.; Lee, F.-C.; Cho, M.; Valerin, J.G. BRCA-Mutated Pancreatic Cancer: From Discovery to Novel Treatment Paradigms. Cancers 2022, 14, 2453. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.M.; Aguirre, A.J.; Shapiro, G.I.; D’Andrea, A.D. Biomarker-Guided Development of DNA Repair Inhibitors. Mol. Cell 2020, 78, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Park, W.; Chen, J.; Chou, J.F.; Varghese, A.M.; Yu, K.H.; Wong, W.; Capanu, M.; Balachandran, V.; McIntyre, C.A.; El Dika, I.; et al. Genomic Methods Identify Homologous Recombination Deficiency in Pancreas Adenocarcinoma and Optimize Treatment Selection. Clin. Cancer Res. 2020, 26, 3239–3247. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Chittenden, A.B.; Morales-Oyarvide, V.; Rubinson, D.A.; Dunne, R.F.; Kozak, M.M.; Qian, Z.R.; Welch, M.W.; Brais, L.K.; Da Silva, A.; et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet. Med. 2019, 21, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Barenboim, A.; Lahat, G.; Nachmany, I.; Goykhman, Y.; Shacham-Shmueli, E.; Halpern, N.; Brazowski, E.; Geva, R.; Wolf, I.; et al. Increased Rate of Complete Pathologic Response After Neoadjuvant FOLFIRINOX for BRCA Mutation Carriers with Borderline Resectable Pancreatic Cancer. Ann. Surg. Oncol. 2020, 27, 3963–3970. [Google Scholar] [CrossRef]
- Yadav, S.; Kasi, P.M.; Bamlet, W.R.; Ho, T.P.; Polley, E.C.; Hu, C.; Hart, S.N.; Rabe, K.G.; Boddicker, N.J.; Gnanaolivu, R.D.; et al. Effect of Germline Mutations in Homologous Recombination Repair Genes on Overall Survival of Patients with Pancreatic Adenocarcinoma. Clin. Cancer Res. 2020, 26, 6505–6512. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin with or without Veliparib in Patients with Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef]
- Heinemann, V.; Quietzsch, D.; Gieseler, F.; Gonnermann, M.; Schönekäs, H.; Rost, A.; Neuhaus, H.; Haag, C.; Clemens, M.; Heinrich, B.; et al. Randomized Phase III Trial of Gemcitabine Plus Cisplatin Compared with Gemcitabine Alone in Advanced Pancreatic Cancer. J. Clin. Oncol. 2006, 24, 3946–3952. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.N.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmaña, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N. Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Kindler, H.L.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Overall Survival Results from the POLO Trial: A Phase III Study of Active Maintenance Olaparib Versus Placebo for Germline BRCA-Mutated Metastatic Pancreatic Cancer. J. Clin. Oncol. 2022, 40, 3929–3939. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.A.; Mick, R.; O’Hara, M.H.; Teitelbaum, U.; Karasic, T.B.; Schneider, C.; Cowden, S.; Southwell, T.; Romeo, J.; Izgur, N.; et al. Phase II Study of Maintenance Rucaparib in Patients with Platinum-Sensitive Advanced Pancreatic Cancer and a Pathogenic Germline or Somatic Variant in BRCA1, BRCA2, or PALB2. J. Clin. Oncol. 2021, 39, 2497–2505. [Google Scholar] [CrossRef]
- Rubinson, D.; Wolpin, B.M.; Warsofsky, I.S.; Ryan, D.P.; Perez, K.; Rahma, O.; Singh, H.; Yurgelun, M.B.; Shapiro, G.I.; Aguirre, A.J.; et al. Durable clinical benefit from PARP inhibition in a platinum-sensitive, BRCA2-mutated pancreatic cancer patient after earlier progression on placebo treatment on the POLO trial: A case report. J. Gastrointest. Oncol. 2021, 12, 3133–3140. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.A.; Mick, R.; Teitelbaum, U.; O’Hara, M.; Schneider, C.; Massa, R.; Karasic, T.; Tondon, R.; Onyiah, C.; Gosselin, M.K.; et al. Niraparib plus nivolumab or niraparib plus ipilimumab in patients with platinum-sensitive advanced pancreatic cancer: A randomised, phase 1b/2 trial. Lancet Oncol. 2022, 23, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Gillson, J.; Ramaswamy, Y.; Singh, G.; Gorfe, A.A.; Pavlakis, N.; Samra, J.; Mittal, A.; Sahni, S. Small Molecule KRAS Inhibitors: The Future for Targeted Pancreatic Cancer Therapy? Cancers 2020, 12, 1341. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.R.; Rubinson, D.A.; Nowak, J.A.; Morales-Oyarvide, V.; Dunne, R.F.; Kozak, M.M.; Welch, M.W.; Brais, L.K.; Da Silva, A.; Li, T.; et al. Association of Alterations in Main Driver Genes with Outcomes of Patients with Resected Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2018, 4, e173420. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.C.; Hannan, L.M.; Zhen, D.B.; Coveler, A.L.; King, G.; Cohen, S.A.; Harris, W.P.; Shankaran, V.; Wong, K.M.; Green, S.; et al. KRAS Mutation Variants and Co-occurring PI3K Pathway Alterations Impact Survival for Patients with Pancreatic Ductal Adenocarcinomas. Oncologist 2022, 27, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Satake, H.; Hollebecque, A.; Sunakawa, Y.; Tomasini, P.; Bajor, D.L.; Schuler, M.H.; Yaeger, R.; George, T.J.; Garrido-Laguna, I.; et al. First data for sotorasib in patients with pancreatic cancer with KRAS p.G12C mutation: A phase I/II study evaluating efficacy and safety. J. Clin. Oncol. 2022, 40, 360490. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.S.; Spira, A.I.; Yaeger, R.; Buchschacher, G.L.; McRee, A.J.; Sabari, J.K.; Johnson, M.L.; Barve, M.A.; Hafez, N.; Velastegui, K.; et al. KRYSTAL-1: Updated activity and safety of adagrasib (MRTX849) in patients (Pts) with unresectable or metastatic pancreatic cancer (PDAC) and other gastrointestinal (GI) tumors harboring a KRASG12C mutation. J. Clin. Oncol. 2022, 40, 519. [Google Scholar] [CrossRef]
- Koltun, E.S.; Rice, M.A.; Gustafson, W.C.; Wilds, D.; Jiang, J.; Lee, B.J.; Wang, Z.; Chang, S.; Flagella, M.; Mu, Y.; et al. Abstract 3597: Direct targeting of KRASG12X mutant cancers with RMC-6236, a first-in-class, RAS-selective, orally bioavailable, tri-complex RASMULTI(ON) inhibitor. Cancer Res. 2022, 82, 3597. [Google Scholar] [CrossRef]
- Hallin, J.; Bowcut, V.; Calinisan, A.; Briere, D.M.; Hargis, L.; Engstrom, L.D.; Laguer, J.; Medwid, J.; Vanderpool, D.; Lifset, E.; et al. Anti-tumor efficacy of a potent and selective non-covalent KRASG12D inhibitor. Nat. Med. 2022, 28, 2171–2182. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019, 156, 2056–2072. [Google Scholar] [CrossRef]
- Leidner, R.; Silva, N.S.; Huang, H.; Sprott, D.; Zheng, C.; Shih, Y.-P.; Leung, A.; Payne, R.; Sutcliffe, K.; Cramer, J.; et al. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N. Engl. J. Med. 2022, 386, 2112–2119. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.-Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.-C.; Vlahovic, G.; et al. Durvalumab with or without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Topham, J.T.; Tsang, E.S.; Karasinska, J.M.; Metcalfe, A.; Ali, H.; Kalloger, S.E.; Csizmok, V.; Williamson, L.M.; Titmuss, E.; Nielsen, K.; et al. Integrative analysis of KRAS wildtype metastatic pancreatic ductal adenocarcinoma reveals mutation and expression-based similarities to cholangiocarcinoma. Nat. Commun. 2022, 13, 5941. [Google Scholar] [CrossRef] [PubMed]
- Pishvaian, M.J.; Garrido-Laguna, I.; Liu, S.V.; Multani, P.S.; Chow-Maneval, E.; Rolfo, C. Entrectinib in TRK and ROS1 Fusion-Positive Metastatic Pancreatic Cancer. JCO Precis. Oncol. 2018, 2, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Williamson, L.M.; Topham, J.T.; Lee, M.K.; Goytain, A.; Ho, J.; Denroche, R.E.; Jang, G.-H.; Pleasance, E.D.; Shen, Y.; et al. NRG1 Gene Fusions Are Recurrent, Clinically Actionable Gene Rearrangements in KRAS Wild-Type Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2019, 25, 4674–4681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, W.; O’Connor, C.A.; Bandlamudi, C.; Forman, D.; Chou, J.F.; Umeda, S.; Reyngold, M.; Varghese, A.M.; Keane, F.; Balogun, F.; et al. Clinico-genomic Characterization of ATM and HRD in Pancreas Cancer: Application for Practice. Clin. Cancer Res. 2022, 28, 4782–4792. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Bai, Y.; Wang, Z.; Chen, Z.; Xu, R.; Xu, J.; Zhang, H.; Chen, J.; Yuan, Y.; Liu, T.; et al. Nimotuzumab combined with gemcitabine versus gemcitabine in K-RAS wild-type locally advanced or metastatic pancreatic cancer: A prospective, randomized-controlled, double-blinded, multicenter, and phase III clinical trial. J. Clin. Oncol. 2022, 40, LBA4011. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF -mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Hendifar, A.; Blais, E.M.; Wolpin, B.; Subbiah, V.; Collisson, E.; Singh, I.; Cannon, T.; Shaw, K.; Petricoin, E.F., III; Klempner, S.; et al. Retrospective Case Series Analysis of RAF Family Alterations in Pancreatic Cancer: Real-World Outcomes from Targeted and Standard Therapies. JCO Precis. Oncol. 2021, 1325–1338. [Google Scholar] [CrossRef]
- Fusco, M.J.; Saeed-Vafa, D.; Carballido, E.M.; Boyle, T.A.; Malafa, M.; Blue, K.L.; Teer, J.K.; Walko, C.M.; McLeod, H.L.; Hicks, J.K.; et al. Identification of Targetable Gene Fusions and Structural Rearrangements to Foster Precision Medicine in KRAS Wild-Type Pancreatic Cancer. JCO Precis. Oncol. 2021, 5, 65–74. [Google Scholar] [CrossRef]
- Li, F.; Peiris, M.N.; Donoghue, D.J. Functions of FGFR2 corrupted by translocations in intrahepatic cholangiocarcinoma. Cytokine Growth Factor Rev. 2020, 52, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.M.; Raghavan, S.; Wu, Q.; Li, Y.Y.; Spurr, L.F.; Gupta, H.V.; Rubinson, D.A.; Fetter, I.J.; Hornick, J.L.; Nowak, J.A.; et al. FGFR2 Extracellular Domain In-Frame Deletions Are Therapeutically Targetable Genomic Alterations That Function as Oncogenic Drivers in Cholangiocarcinoma. Cancer Discov. 2021, 11, 2488–2505. [Google Scholar] [CrossRef]
- Subbiah, V.; Iannotti, N.; Gutierrez, M.; Smith, D.; Féliz, L.; Lihou, C.; Tian, C.; Silverman, I.; Ji, T.; Saleh, M. FIGHT-101, a first-in-human study of potent and selective FGFR 1-3 inhibitor pemigatinib in pan-cancer patients with FGF/FGFR alterations and advanced malignancies. Ann. Oncol. 2022, 33, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Helal, C.; Valéry, M.; Ducreux, M.; Hollebecque, A.; Smolenschi, C. FGFR2 fusion in metastatic pancreatic ductal adenocarcinoma: Is there hope? Eur. J. Cancer 2022, 176, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016, 27, iii4–iii15. [Google Scholar] [CrossRef]
- Cooper, A.J.; Sequist, L.V.; Lin, J.J. Third-generation EGFR and ALK inhibitors: Mechanisms of resistance and management. Nat. Rev. Clin. Oncol. 2022, 19, 499–514. [Google Scholar] [CrossRef]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus crizotinib in patients with ALK -positive non-small-cell lung cancer (J-ALEX): An open-label, randomised phase 3 trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Cleary, J.M.; Rodig, S.; Barr, P.M.; Shinagare, A.B.; Clark, J.W.; Shapiro, G.I.; Armand, P. Crizotinib as salvage and maintenance with allogeneic stem cell transplantation for refractory anaplastic large cell lymphoma. J. Natl. Compr. Cancer Netw. 2014, 12, 323–326. [Google Scholar] [CrossRef] [Green Version]
- Merino, M.; Kasamon, Y.; Li, H.; Ma, L.; Leong, R.; Zhou, J.; Reaman, G.; Chambers, W.; Richardson, N.; Theoret, M.; et al. FDA approval summary: Crizotinib for pediatric and young adult patients with relapsed or refractory systemic anaplastic large cell lymphoma. Pediatr. Blood Cancer 2022, 69, e29602. [Google Scholar] [CrossRef]
- Singh, H.; Li, Y.Y.; Spurr, L.F.; Shinagare, A.B.; Abhyankar, R.; Reilly, E.; Brais, L.K.; Nag, A.; Ducar, M.D.; Thorner, A.R.; et al. Molecular Characterization and Therapeutic Targeting of Colorectal Cancers Harboring Receptor Tyrosine Kinase Fusions. Clin. Cancer Res. 2021, 27, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus Chemotherapy in AdvancedALK-Positive Lung Cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhi, A.D.; Ali, S.M.; Lacy, J.; Hendifar, A.; Nguyen, K.; Koo, J.; Chung, J.H.; Greenbowe, J.; Ross, J.S.; Nikiforova, M.N.; et al. Identification of Targetable ALK Rearrangements in Pancreatic Ductal Adenocarcinoma. J. Natl. Compr. Cancer Netw. 2017, 15, 555–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosini, M.; Del Re, M.; Manca, P.; Hendifar, A.; Drilon, A.; Harada, G.; Ree, A.H.; Klempner, S.; Mælandsmo, G.M.; Flatmark, K.; et al. ALK Inhibitors in Patients with ALK Fusion–Positive GI Cancers: An International Data Set and a Molecular Case Series. JCO Precis. Oncol. 2022, 6, e2200015. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, T.M.; Crinò, L.; Gridelli, C.; Kiura, K.; Liu, G.; Novello, S.; Bearz, A.; Gautschi, O.; Mok, T.; et al. Ceritinib versus chemotherapy in patients with ALK-rearranged non-small-cell lung cancer previously given chemotherapy and crizotinib (ASCEND-5): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 874–886. [Google Scholar] [CrossRef] [PubMed]
- Amatu, A.; Sartore-Bianchi, A.; Bencardino, K.; Pizzutilo, E.; Tosi, F.; Siena, S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann. Oncol. 2019, 30, viii5–viii15. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- O’Reilly, E.; Hechtman, J. Tumour response to TRK inhibition in a patient with pancreatic adenocarcinoma harbouring an NTRK gene fusion. Ann. Oncol. 2019, 30, viii36–viii40. [Google Scholar] [CrossRef] [Green Version]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Werr, L.; Plenker, D.; Dammert, M.A.; Lorenz, C.; Brägelmann, J.; Tumbrink, H.L.; Klein, S.; Schmitt, A.; Büttner, R.; Persigehl, T.; et al. CD74-NRG1 Fusions Are Oncogenic In Vivo and Induce Therapeutically Tractable ERBB2:ERBB3 Heterodimerization. Mol. Cancer Ther. 2022, 21, 821–830. [Google Scholar] [CrossRef]
- Laskin, J.; Liu, S.; Tolba, K.; Heining, C.; Schlenk, R.; Cheema, P.; Cadranel, J.; Jones, M.; Drilon, A.; Cseh, A.; et al. NRG1 fusion-driven tumors: Biology, detection, and the therapeutic role of afatinib and other ErbB-targeting agents. Ann. Oncol. 2020, 31, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J. Oncogenic NRG1 Fusions: A New Hope for Targeted Therapy in Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 4589–4591. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; Goto, K.; Kim, D.-W.; Martin-Romano, P.; Ou, S.-H.I.; O’Kane, G.M.; O’Reilly, E.M.; Umemoto, K.; Duruisseaux, M.; Neuzillet, C.; et al. Efficacy and safety of zenocutuzumab, a HER2 x HER3 bispecific antibody, across advanced NRG1 fusion (NRG1+) cancers. J. Clin. Oncol. 2022, 40, 105. [Google Scholar] [CrossRef]
- Subbiah, V.; Cassier, P.A.; Siena, S.; Garralda, E.; Paz-Ares, L.; Garrido, P.; Nadal, E.; Vuky, J.; Lopes, G.; Kalemkerian, G.P.; et al. Pan-cancer efficacy of pralsetinib in patients with RET fusion–positive solid tumors from the phase 1/2 ARROW trial. Nat. Med. 2022, 28, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Wolf, J.; Konda, B.; Kang, H.; Spira, A.I.; Weiss, J.; Takeda, M.; Ohe, Y.; Khan, S.A.; Ohashi, K.; et al. Tumor agnostic efficacy of selpercatinib in patients with RET fusion+ solid tumors: A global, multicenter, registrational trial update (LIBRETTO-001). J. Clin. Oncol. 2022, 40, 3094. [Google Scholar] [CrossRef]
- Davies, K.D.; Doebele, R.C. Molecular Pathways: ROS1 Fusion Proteins in Cancer. Clin. Cancer Res. 2013, 19, 4040–4045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad-Nielsen, S.A.; Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Frequency of mismatch repair deficiency in pancreatic ductal adenocarcinoma. Pathol. Res. Pract. 2020, 216, 152985. [Google Scholar] [CrossRef]
- Luchini, C.; Brosens, L.A.A.; Wood, L.D.; Chatterjee, D.; Shin, J.I.; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: Histology, molecular pathology and clinical implications. Gut 2021, 70, 148–156. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Padrón, L.J.; Maurer, D.M.; O’Hara, M.H.; O’Reilly, E.M.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; et al. Sotigalimab and/or nivolumab with chemotherapy in first-line metastatic pancreatic cancer: Clinical and immunologic analyses from the randomized phase 2 PRINCE trial. Nat. Med. 2022, 28, 1167–1177. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Recommended FDA-Approved Targeted Therapies in PDAC | |
|---|---|
| Molecular Target | Drug Name |
| gBRCA1/2 mutations | Olaparib |
| Tumor agnostic FDA-approved targeted therapies | |
| Molecular Target | Drug Name |
| MSI-H/MMRd | Pembrolizumab |
| BRAF V600E | Dabrafenib/Trametinib |
| NTRK fusions | Larotrectinib, Entrectinib |
| RET fusions | Selpercatinib |
| Therapies that are FDA-approved in other tumors | |
| Molecular Target | Drug Name |
| KRAS G12C | Sotorasib, Adagrasib |
| FGFR2 fusions | Pemigatinib |
| ALK fusions | Crizotinib, Alectinib |
| ROS1 fusions | Entrectinib, Crizotinib |
| sBRCA1/2, gPALB2/sPALB2 1 | Niraparib, Olaparib, Rucaparib |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huffman, B.M.; Ellis, H.; Jordan, A.C.; Freed-Pastor, W.A.; Perez, K.; Rubinson, D.A.; Sethi, N.; Singh, H.; Surana, R.; Wolpin, B.M.; et al. Emerging Role of Targeted Therapy in Metastatic Pancreatic Adenocarcinoma. Cancers 2022, 14, 6223. https://doi.org/10.3390/cancers14246223
Huffman BM, Ellis H, Jordan AC, Freed-Pastor WA, Perez K, Rubinson DA, Sethi N, Singh H, Surana R, Wolpin BM, et al. Emerging Role of Targeted Therapy in Metastatic Pancreatic Adenocarcinoma. Cancers. 2022; 14(24):6223. https://doi.org/10.3390/cancers14246223
Chicago/Turabian StyleHuffman, Brandon M., Haley Ellis, Alexander C. Jordan, William A. Freed-Pastor, Kimberly Perez, Douglas A. Rubinson, Nilay Sethi, Harshabad Singh, Rishi Surana, Brian M. Wolpin, and et al. 2022. "Emerging Role of Targeted Therapy in Metastatic Pancreatic Adenocarcinoma" Cancers 14, no. 24: 6223. https://doi.org/10.3390/cancers14246223
APA StyleHuffman, B. M., Ellis, H., Jordan, A. C., Freed-Pastor, W. A., Perez, K., Rubinson, D. A., Sethi, N., Singh, H., Surana, R., Wolpin, B. M., Aguirre, A. J., & Cleary, J. M. (2022). Emerging Role of Targeted Therapy in Metastatic Pancreatic Adenocarcinoma. Cancers, 14(24), 6223. https://doi.org/10.3390/cancers14246223