
A Stochastic Binary Model for the Regulation of Gene Expression to Investigate Responses to Gene Therapy

 , , , and
, , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
Rationale
2. Methods and Models
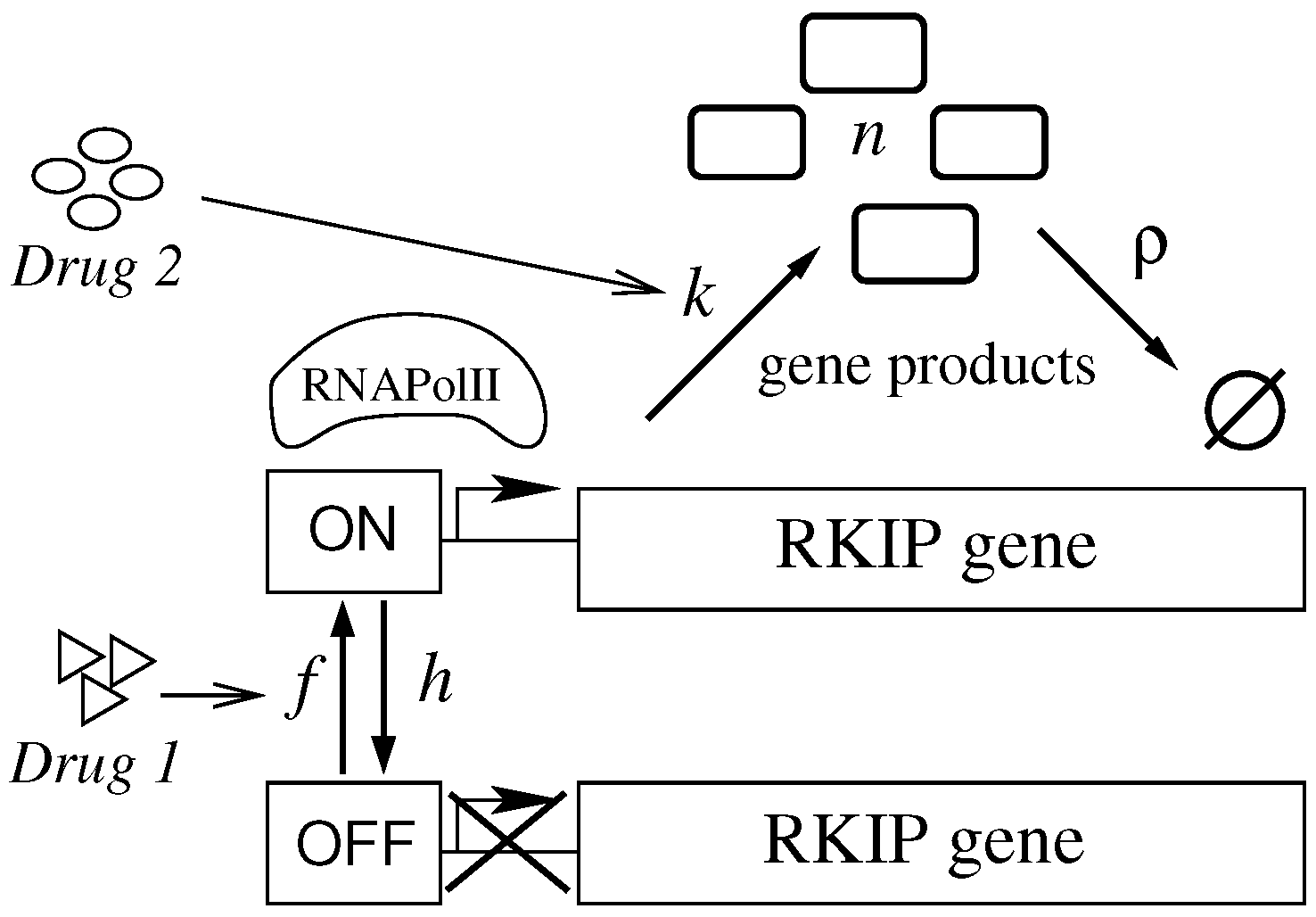
2.1. A Brief Description of the Molecular Role of RKIP
2.2. An Effective Model for the Regulation of Gene Expression
2.3. An Effective Model for Investigating Regulation of Gene Expression Dynamics after Treatment
2.4. An Approach for Investigating Treatment Effects on RKIP Expression Dynamics
2.5. An Approximate Description of the Stochastic Binary Gene Expression Dynamics with Time-Dependent Kinetic Rates
2.6. An Exactly Solvable Model for Benchmarking Cancer Treatment Aiming to Modulate Gene Expression Levels
2.7. Parameter Values and Conditions for Treatment Simulations
3. Results
3.1. Treatment Aiming at the OFF to ON Gene State Switching Rate
3.2. Treatment Aiming at the RKIP mRNA Synthesis Rate
3.3. Treatment with the Two DRUGS Concomitantly
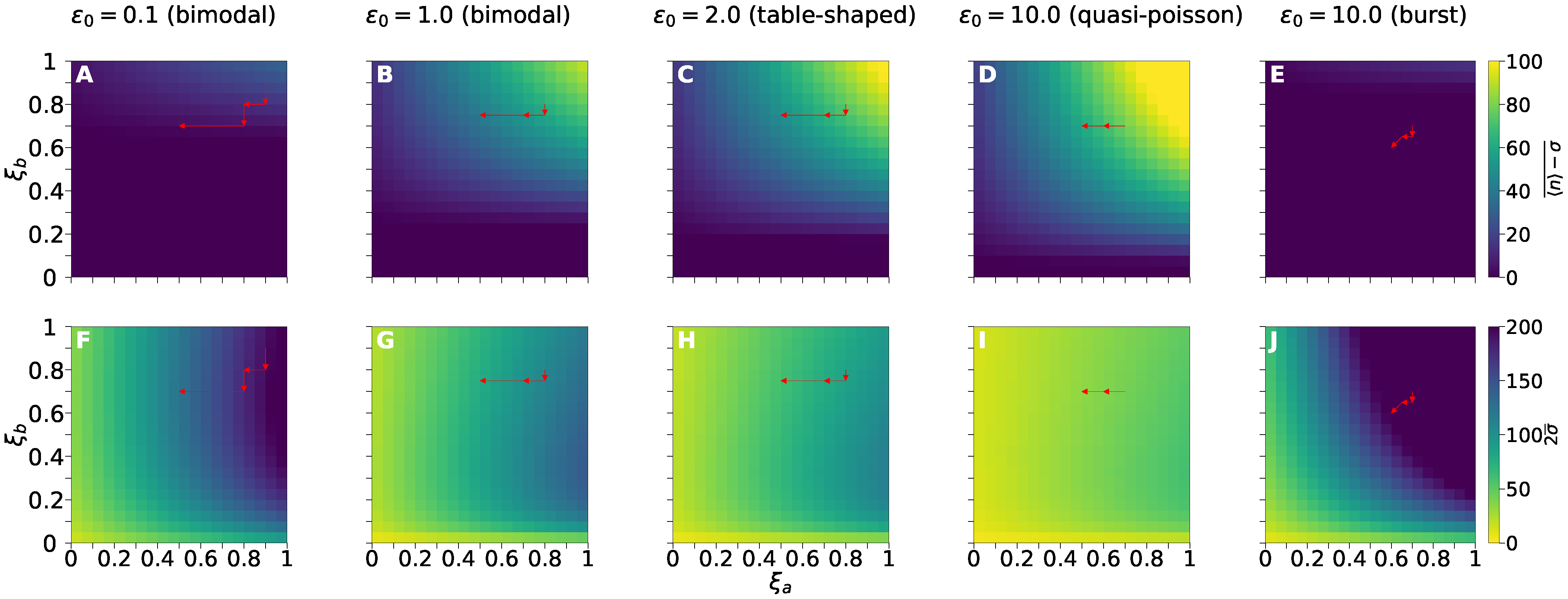
3.4. Treatment Response Mapping and Fractional Effect of Drug Reduction
3.5. Enhancing Ineffective Treatments Aiming at All Kinetic Rates
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Formulae
Appendix A.1. Piecewise Approximation for Time-Dependent Kinetic Parameters
Appendix A.2. System of Coupled ODEs Governing the Moments of the Distribution
Appendix A.3. Steady State Values of the Moments of the Distribution
Appendix A.4. Coefficients of Equations (15) and (16)
Appendix A.5. Exact Formulas for A(t), 〈nα〉(t), and 〈n2〉(t)
Appendix A.6. Steady State Probabilities of Finding n Gene Products
Appendix B. Treatment Parameters and the Agenda of Doses for Enhanced Treatment Designs
| Graph | ||||
| A4 | ||||
| B4 | ||||
| C4/D4 | ||||
| E4 |
References
- Bulaklak, K.; Gersbach, C.A. The once and future gene therapy. Nat. Commun. 2020, 11, 5820. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.I.; Black, J.B.; Hilton, I.B.; Gersbach, C.A. Editing the epigenome: Technologies for programmable transcription and epigenetic modulation. Nat. Methods 2016, 13, 127–137. [Google Scholar] [CrossRef]
- Cai, W.; Zhou, W.; Han, Z.; Lei, J.; Zhuang, J.; Zhu, P.; Wu, X.; Yuan, W. Master regulator genes and their impact on major diseases. PeerJ 2020, 8, e9952. [Google Scholar] [CrossRef] [PubMed]
- Yesilkanal, A.E.; Yang, D.; Valdespino, A.; Tiwari, P.; Sabino, A.U.; Nguyen, L.C.; Lee, J.; Xie, X.H.; Sun, S.; Dann, C.; et al. Limited inhibition of multiple nodes in a driver network blocks metastasis. eLife 2021, 10, e59696. [Google Scholar] [CrossRef]
- Yesilkanal, A.E.; Johnson, G.L.; Ramos, A.F.; Rosner, M.R. New strategies for targeting kinase networks in cancer. J. Biol. Chem. 2021, 297, 101128. [Google Scholar] [CrossRef] [PubMed]
- Delbrück, M. Statistical Fluctuations in Autocatalytic Reactions. J. Chem. Phys. 1940, 8, 120–124. [Google Scholar] [CrossRef]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef]
- Michor, F.; Beal, K. Improving Cancer Treatment via Mathematical Modeling: Surmounting the Challenges Is Worth the Effort. Cell 2015, 163, 1059–1063. [Google Scholar] [CrossRef] [Green Version]
- Alizadeh, A.A.; Aranda, V.; Bardelli, A.; Blanpain, C.; Bock, C.; Borowski, C.; Caldas, C.; Califano, A.; Doherty, M.; Elsner, M.; et al. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 2015, 21, 846–853. [Google Scholar] [CrossRef]
- Brock, A.; Krause, S.; Ingber, D.E. Control of cancer formation by intrinsic genetic noise and microenvironmental cues. Nat. Rev. Cancer 2015, 15, 499–509. [Google Scholar] [CrossRef]
- Welch, D.R. Tumor Heterogeneity—A ‘Contemporary Concept’ Founded on Historical Insights and Predictions. Cancer Res. 2016, 76, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Guinn, M.T.; Wan, Y.; Levovitz, S.; Yang, D.; Rosner, M.R.; Balázsi, G. Observation and Control of Gene Expression Noise: Barrier Crossing Analogies Between Drug Resistance and Metastasis. Front. Genet. 2020, 11, 586726. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, U.S.; Iyengar, R. Emergent Properties of Networks of Biological Signaling Pathways. Science 1999, 283, 381–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balázsi, G.; van Oudenaarden, A.; Collins, J.J. Cellular decision making and biological noise: From microbes to mammals. Cell 2011, 144, 910–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrell, J.E. Perfect and Near-Perfect Adaptation in Cell Signaling. Cell Syst. 2016, 2, 62–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gérard, C.; Gonze, D.; Goldbeter, A. Revisiting a skeleton model for the mammalian cell cycle: From bistability to Cdk oscillations and cellular heterogeneity. J. Theor. Biol. 2019, 461, 276–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purvis, J.E.; Lahav, G. Encoding and Decoding Cellular Information through Signaling Dynamics. Cell 2013, 152, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Levine, J.H.; Lin, Y.; Elowitz, M.B. Functional Roles of Pulsing in Genetic Circuits. Science 2013, 342, 1193–1200. [Google Scholar] [CrossRef] [Green Version]
- Ramos, A.F.; Innocentini, G.C.P.; Forger, F.M.; Hornos, J.E.M. Symmetry in biology: From genetic code to stochastic gene regulation. IET Syst. Biol. 2010, 4, 311–329. [Google Scholar] [CrossRef] [Green Version]
- Peccoud, J.; Ycart, B. Markovian modelling of gene product synthesis. Theor. Popul. Biol. 1995, 48, 222–234. [Google Scholar] [CrossRef]
- Iyer-Biswas, S.; Hayot, F.; Jayaprakash, C. Stochasticity of gene products from transcriptional pulsing. Phys. Rev. E 2009, 79, 031911. [Google Scholar] [CrossRef] [Green Version]
- Ramos, A.F.; Gama, L.R.; Morais, M.C.C.; Martins, P.C.M. Stochastic modeling for investigation of the regulation of transcription of the RKIP gene. In Prognostic and Therapeutic Applications of RKIP in Cancer; Bonavida, B., Baritaki, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 257–276. [Google Scholar] [CrossRef]
- Giovanini, G.; Sabino, A.U.; Barros, L.R.C.; Ramos, A.F. A comparative analysis of noise properties of stochastic binary models for a self-repressing and for an externally regulating gene. Math. Biosci. Eng. 2020, 17, 5477–5503. [Google Scholar] [CrossRef]
- Jarrett, A.M.; Faghihi, D.; Hormuth, D.A.; Lima, E.A.B.F.; Virostko, J.; Biros, G.; Patt, D.; Yankeelov, T.E. Optimal Control Theory for Personalized Therapeutic Regimens in Oncology: Background, History, Challenges, and Opportunities. J. Clin. Med. 2020, 9, 1314. [Google Scholar] [CrossRef]
- Bonavida, B.; Baritaki, S. (Eds.) Prognostic and Therapeutic Applications of RKIP in Cancer; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar] [CrossRef]
- Martinho, O.; Granja, S.; Jaraquemada, T.; Caeiro, C.; Miranda-Gonçalves, V.; Honavar, M.; Costa, P.; Damasceno, M.; Rosner, M.R.; Lopes, J.M.; et al. Downregulation of RKIP Is Associated with Poor Outcome and Malignant Progression in Gliomas. PLoS ONE 2012, 7, e30769. [Google Scholar] [CrossRef] [Green Version]
- Martinho, O.; Pinto, F.; Granja, S.; Miranda-Gonçalves, V.; Moreira, M.A.R.; Ribeiro, L.F.J.; di Loreto, C.; Rosner, M.R.; Longatto-Filho, A.; Reis, R.M. RKIP Inhibition in Cervical Cancer Is Associated with Higher Tumor Aggressive Behavior and Resistance to Cisplatin Therapy. PLoS ONE 2013, 8, e59104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamiman, K.; Keller, J.M.; Mizokami, A.; Zhang, J.; Keller, E.T. Survey of Raf Kinase Inhibitor Protein (RKIP) in Multiple Cancer Types. Crit. Rev. Oncog. 2014, 19, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Yesilkanal, A.; Rosner, M. Targeting Raf Kinase Inhibitory Protein Regulation and Function. Cancers 2018, 10, 306. [Google Scholar] [CrossRef] [Green Version]
- Dangi-Garimella, S.; Yun, J.; Eves, E.M.; Newman, M.; Erkeland, S.J.; Hammond, S.M.; Minn, A.J.; Rosner, M.R. Raf kinase inhibitory protein suppresses a metastasis signalling cascade involving LIN28 and let-7. EMBO J. 2009, 28, 347–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Smith, P.C.; Zhang, L.; Rubin, M.A.; Dunn, R.L.; Yao, Z.; Keller, E.T. Effects of Raf Kinase Inhibitor Protein Expression on Suppression of Prostate Cancer Metastasis. J. Natl. Cancer Inst. 2003, 95, 878–889. [Google Scholar] [CrossRef]
- Zaravinos, A.; Bonavida, B.; Chatzaki, E.; Baritaki, S. RKIP: A Key Regulator in Tumor Metastasis Initiation and Resistance to Apoptosis: Therapeutic Targeting and Impact. Cancers 2018, 10, 287. [Google Scholar] [CrossRef] [Green Version]
- Ramos, A.F.; Reinitz, J. Physical implications of so(2, 1) symmetry in exact solutions for a self-repressing gene. J. Chem. Phys. 2019, 151, 041101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gama, L.R.; Giovanini, G.; Balázsi, G.; Ramos, A.F. Binary Expression Enhances Reliability of Messaging in Gene Networks. Entropy 2020, 22, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, K.; Seitz, T.; Li, S.; Janosch, P.; McFerran, B.; Kaiser, C.; Fee, F.; Katsanakis, K.D.; Rose, D.W.; Mischak, H.; et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature 1999, 401, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Trakul, N.; Menard, R.E.; Schade, G.R.; Qian, Z.; Rosner, M.R. Raf Kinase Inhibitory Protein Regulates Raf-1 but Not B-Raf Kinase Activation. J. Biol. Chem. 2005, 280, 24931–24940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yesilkanal, A.E.; Rosner, M.R. Raf Kinase Inhibitory Protein (RKIP) as a Metastasis Suppressor: Regulation of Signaling Networks in Cancer. Crit. Rev. Oncog. 2014, 19, 447–454. [Google Scholar] [CrossRef] [Green Version]
- Yeung, K.C.; Rose, D.W.; Dhillon, A.S.; Yaros, D.; Gustafsson, M.; Chatterjee, D.; McFerran, B.; Wyche, J.; Kolch, W.; Sedivy, J.M. Raf Kinase Inhibitor Protein Interacts with NF-κB-Inducing Kinase and TAK1 and Inhibits NF-κB Activation. Mol. Cell. Biol. 2001, 21, 7207–7217. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wenzel, S. Interactions of RKIP with Inflammatory Signaling Pathways. Crit. Rev. Oncog. 2014, 19, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, K.; Lohse, M.J.; Quitterer, U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature 2003, 426, 574–579. [Google Scholar] [CrossRef]
- Corbit, K.C.; Trakul, N.; Eves, E.M.; Diaz, B.; Marshall, M.; Rosner, M.R. Activation of Raf-1 Signaling by Protein Kinase C through a Mechanism Involving Raf Kinase Inhibitory Protein. J. Biol. Chem. 2003, 278, 13061–13068. [Google Scholar] [CrossRef] [Green Version]
- Shvartsur, A.; Givechian, K.B.; Garban, H.; Bonavida, B. Overexpression of RKIP and its cross-talk with several regulatory gene products in multiple myeloma. J. Exp. Clin. Cancer Res. 2017, 36, 62. [Google Scholar] [CrossRef] [Green Version]
- Datar, I.; Tegegne, H.; Qin, K.; Al-Mulla, F.; Bitar, M.S.; Trumbly, R.J.; Yeung, K.C. Genetic and Epigenetic Control of RKIP Transcription. Crit. Rev. Oncog. 2014, 19, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Galal, Y.; Zaravinos, A.; Bonavida, B. Regulation of NKG2D by RKIP: Implications on NK-mediated cytotoxicity and cytokine production. In Successes and Challenges of NK Immunotherapy; Elsevier: Amsterdam, The Netherlands, 2021; pp. 233–265. [Google Scholar] [CrossRef]
- Li, D.X.; Cai, H.Y.; Wang, X.; Feng, Y.L.; Cai, S.W. Promoter methylation of Raf kinase inhibitory protein: A significant prognostic indicator for patients with gastric adenocarcinoma. Exp. Ther. Med. 2014, 8, 844–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Liu, Z.; She, H.; Liu, B.; Gu, J.; Wei, D.; Zhang, X.; Wang, J.; Qi, S.; Ping, F. Promoter methylation and expression of Raf kinase inhibitory protein in esophageal squamous cell carcinoma. Oncol. Lett. 2017, 13, 1866–1872. [Google Scholar] [CrossRef] [Green Version]
- Beach, S.; Tang, H.; Park, S.; Dhillon, A.S.; Keller, E.T.; Kolch, W.; Yeung, K.C. Snail is a repressor of RKIP transcription in metastatic prostate cancer cells. Oncogene 2007, 27, 2243–2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labbozzetta, M.; Poma, P.; Vivona, N.; Gulino, A.; D’Alessandro, N.; Notarbartolo, M. Epigenetic changes and nuclear factor-κB activation, but not microRNA-224, downregulate Raf-1 kinase inhibitor protein in triple-negative breast cancer SUM 159 cells. Oncol. Lett. 2015, 10, 3807–3815. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Baritaki, S.; Marathe, H.; Feng, J.; Park, S.; Beach, S.; Bazeley, P.S.; Beshir, A.B.; Fenteany, G.; Mehra, R.; et al. Polycomb Protein EZH2 Regulates Tumor Invasion via the Transcriptional Repression of the Metastasis Suppressor RKIP in Breast and Prostate Cancer. Cancer Res. 2012, 72, 3091–3104. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Lee, J.; Farquhar, K.S.; Yun, J.; Frankenberger, C.A.; Bevilacqua, E.; Yeung, K.; Kim, E.J.; Balázsi, G.; Rosner, M.R. Network of mutually repressive metastasis regulators can promote cell heterogeneity and metastatic transitions. Proc. Natl. Acad. Sci. USA 2014, 111, E364–E373. [Google Scholar] [CrossRef] [Green Version]
- Baritaki, S.; Huerta-Yepez, S.; Sahakyan, A.; Karagiannides, I.; Bakirtzi, K.; Jazirehi, A.; Bonavida, B. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer. Cell Cycle 2010, 9, 4931–4940. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Frankenberger, C.A.; Kuo, W.L.; Boelens, M.C.; Eves, E.M.; Cheng, N.; Liang, H.; Li, W.H.; Ishwaran, H.; Minn, A.J.; et al. Signalling pathway for RKIP and Let-7 regulates and predicts metastatic breast cancer. EMBO J. 2011, 30, 4500–4514. [Google Scholar] [CrossRef]
- Du, Y.; Zhu, H.C.; Liu, X.H.; Wang, L.; Ning, J.Z.; Xiao, C.C. MiR-543 Promotes Proliferation and Epithelial-Mesenchymal Transition in Prostate Cancer via Targeting RKIP. Cell. Physiol. Biochem. 2017, 41, 1135–1146. [Google Scholar] [CrossRef]
- de Castro, J.; Odeh, H.N.; Figy, C.; Yeung, M.L.; Trumbly, R.; Yeung, K.C. Regulation of RKIP expression in breast cancer cells by miRNAs. In Prognostic and Therapeutic Applications of RKIP in Cancer; Elsevier: Amsterdam, The Netherlands, 2020; pp. 139–146. [Google Scholar] [CrossRef]
- Du, Y.; Weng, X.D.; Wang, L.; Liu, X.H.; Zhu, H.C.; Guo, J.; Ning, J.Z.; Xiao, C.C. LncRNA XIST acts as a tumor suppressor in prostate cancer through sponging miR-23a to modulate RKIP expression. Oncotarget 2017, 8, 94358–94370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Kitagawa, Y.; Shen, R.; Shah, R.; Mehra, R.; Rhodes, D.; Keller, P.J.; Mizokami, A.; Dunn, R.; Chinnaiyan, A.M.; et al. Metastasis suppressor gene Raf kinase inhibitor protein (RKIP) is a novel prognostic marker in prostate cancer. Prostate 2006, 66, 248–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mulla, F.; Bitar, M.S.; Al-Maghrebi, M.; Behbehani, A.I.; Al-Ali, W.; Rath, O.; Doyle, B.; Tan, K.Y.; Pitt, A.; Kolch, W. Raf Kinase Inhibitor Protein RKIP Enhances Signaling by Glycogen Synthase Kinase-3β. Cancer Res. 2011, 71, 1334–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Dong, Z.; Lin, X.; Zhang, M.; Kuang, G.; Zhu, T. Decreased Expression and Aberrant Methylation of Raf Kinase Inhibitory Protein Gene in Esophageal Squamous Cell Carcinoma. Cancer Investig. 2012, 30, 703–711. [Google Scholar] [CrossRef]
- Kim, G.E.; Kim, N.I.; Lee, J.S.; Park, M.H.; Yoon, J.H. Reduced RKIP Expression is Associated With Breast Neoplastic Progression and is Correlated With Poor Outcomes and Aberrant Methylation in Breast Carcinoma. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 467–474. [Google Scholar] [CrossRef]
- Minoo, P.; Baker, K.; Goswami, R.; Chong, G.; Foulkes, W.D.; Ruszkiewicz, A.R.; Barker, M.; Buchanan, D.; Young, J.; Jass, J.R. Extensive DNA methylation in normal colorectal mucosa in hyperplastic polyposis. Gut 2006, 55, 1467–1474. [Google Scholar] [CrossRef] [Green Version]
- Bonavida, B. Historical Perspectives of the Role of NO/NO Donors in Anti-tumor Activities: Acknowledging Dr. Keefer’s Pioneering Research. Crit. Rev. Oncog. 2021. [Google Scholar] [CrossRef]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef]
- Huerta-Yepez, S.; Vega, M.; Jazirehi, A.; Garban, H.; Hongo, F.; Cheng, G.; Bonavida, B. Nitric oxide sensitizes prostate carcinoma cell lines to TRAIL-mediated apoptosis via inactivation of NF-κB and inhibition of Bcl-xL expression. Oncogene 2004, 23, 4993–5003. [Google Scholar] [CrossRef] [Green Version]
- Bonavida, B. RKIP-Mediated Chemo-Immunosensitization of Resistant Cancer Cells via Disruption of the NF-κB/Snail/YY1/RKIP Resistance-Driver Loop. Crit. Rev. Oncog. 2014, 19, 431–445. [Google Scholar] [CrossRef]
- Rapozzi, V.; Pietra, E.D.; Zorzet, S.; Zacchigna, M.; Bonavida, B.; Xodo, L.E. Nitric oxide-mediated activity in anti-cancer photodynamic therapy. Nitric Oxide 2013, 30, 26–35. [Google Scholar] [CrossRef]
- Bonavida, B.; Baritaki, S. The Novel Role of Yin Yang 1 in the Regulation of Epithelial to Mesenchymal Transition in Cancer Via the Dysregulated NF-κB/Snail/YY1/RKIP/PTEN Circuitry. Crit. Rev. Oncog. 2011, 16, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Pervin, S.; Singh, R.; Hernandez, E.; Wu, G.; Chaudhuri, G. Nitric Oxide in Physiologic Concentrations Targets the Translational Machinery to Increase the Proliferation of Human Breast Cancer Cells: Involvement of Mammalian Target of Rapamycin/eIF4E Pathway. Cancer Res. 2007, 67, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pervin, S.; Tran, L.; Urman, R.; Braga, M.; Parveen, M.; Li, S.A.; Chaudhuri, G.; Singh, R. Oxidative stress specifically downregulates survivin to promote breast tumour formation. Br. J. Cancer 2013, 108, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Rapozzi, V.; Pietra, E.D.; Bonavida, B. Dual roles of nitric oxide in the regulation of tumor cell response and resistance to photodynamic therapy. Redox Biol. 2015, 6, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wink, D.A.; Ridnour, L.A.; Hussain, S.P.; Harris, C.C. The reemergence of nitric oxide and cancer. Nitric Oxide 2008, 19, 65–67. [Google Scholar] [CrossRef] [Green Version]
- Pasqualetti, G.; Ricciardi, S.; Mey, V.; Tacca, M.D.; Danesi, R. Synergistic cytotoxicity, inhibition of signal transduction pathways and pharmacogenetics of sorafenib and gemcitabine in human NSCLC cell lines. Lung Cancer 2011, 74, 197–205. [Google Scholar] [CrossRef]
- Giovannetti, E.; Labots, M.; Dekker, H.; Galvani, E.; Lind, J.; Sciarrillo, R.; Honeywell, R.; Smit, E.; Verheul, H.; Peters, G. Molecular Mechanisms and Modulation of Key Pathways Underlying the Synergistic Interaction of Sorafenib with Erlotinib in Non-Small-Cell-Lung Cancer (NSCLC) Cells. Curr. Pharm. Des. 2012, 19, 927–939. [Google Scholar] [CrossRef]
- Ricciardi, S.; Mey, V.; Nannizzi, S.; Pasqualetti, G.; Crea, F.; Tacca, M.D.; Danesi, R. Synergistic Cytotoxicity and Molecular Interaction on Drug Targets of Sorafenib and Gemcitabine in Human Pancreas Cancer Cells. Chemotherapy 2010, 56, 303–312. [Google Scholar] [CrossRef]
- Chatterjee, D.; Bai, Y.; Wang, Z.; Beach, S.; Mott, S.; Roy, R.; Braastad, C.; Sun, Y.; Mukhopadhyay, A.; Aggarwal, B.B.; et al. RKIP Sensitizes Prostate and Breast Cancer Cells to Drug-induced Apoptosis. J. Biol. Chem. 2004, 279, 17515–17523. [Google Scholar] [CrossRef] [Green Version]
- Yousuf, S.; Duan, M.; Moen, E.L.; Cross-Knorr, S.; Brilliant, K.; Bonavida, B.; LaValle, T.; Yeung, K.C.; Al-Mulla, F.; Chin, E.; et al. Raf Kinase Inhibitor Protein (RKIP) Blocks Signal Transducer and Activator of Transcription 3 (STAT3) Activation in Breast and Prostate Cancer. PLoS ONE 2014, 9, e92478. [Google Scholar] [CrossRef]
- Hampl, V.; Tristani-Firouzi, M.; Hutsell, T.C.; Archer, S.L. Nebulized nitric oxide/nucleophile adduct reduces chronic pulmonary hypertension. Cardiovasc. Res. 1996, 31, 55–62. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Walker, E.J.; Rosenberg, S.A.; Wands, J.R.; Kim, M. Role of Raf Kinase Inhibitor Protein in Hepatocellular Carcinoma. For. Immunopathol. Dis. Therap. 2011, 2, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, Y.; Xu, Z.; Li, J.; Yang, B.; Feng, N.; Zhang, Y.; Wang, S. Raf kinase inhibitor protein mediated signaling inhibits invasion and metastasis of hepatocellular carcinoma. Biochim. Biophys. Acta 2016, 1860, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Chang, L.; Ye, W.; Jiang, C.; Zhang, Z. Raf kinase inhibitor protein (RKIP) inhibits the cell migration and invasion in human glioma cell lines in vitro. Int. J. Clin. Exp. Pathol. 2015, 8, 14214–14220. [Google Scholar] [PubMed]
- Zhao, D.; Ma, J.; Shi, J.; Cheng, L.; Li, F.; Jiang, X.; Jiang, H. Raf kinase inhibitor protein inhibits esophageal cancer cell invasion through downregulation of matrix metalloproteinase expression. Oncol. Rep. 2013, 30, 304–312. [Google Scholar] [CrossRef]
- Hao, C.; Wei, S.; Tong, Z.; Li, S.; Shi, Y.; Wang, X.; hua Zhu, Z. The effects of RKIP gene expression on the biological characteristics of human triple-negative breast cancer cells in vitro. Tumour Biol. 2012, 33, 1159–1167. [Google Scholar] [CrossRef]
- Zou, Q.; Wu, H.; Fu, F.; Yi, W.; Pei, L.; Zhou, M. RKIP suppresses the proliferation and metastasis of breast cancer cell lines through up-regulation of miR-185 targeting HMGA2. Arch. Biochem. Biophys. 2016, 610, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Baritaki, S.; Militello, L.; Malaponte, G.; Bevelacqua, Y.; Bonavida, B. The Role of B-RAF Mutations in Melanoma and the Induction of EMT via Dysregulation of the NF- B/Snail/RKIP/PTEN Circuit. Genes Cancer 2010, 1, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Schuierer, M.M.; Bataille, F.; Hagan, S.; Kolch, W.; Bosserhoff, A.K. Reduction in Raf Kinase Inhibitor Protein Expression Is Associated with Increased Ras-Extracellular Signal-Regulated Kinase Signaling in Melanoma Cell Lines. Cancer Res. 2004, 64, 5186–5192. [Google Scholar] [CrossRef] [Green Version]
- Minoo, P.; Zlobec, I.; Baker, K.; Tornillo, L.; Terracciano, L.; Jass, J.R.; Lugli, A. Loss of Raf-1 Kinase Inhibitor Protein Expression Is Associated With Tumor Progression and Metastasis in Colorectal Cancer. Am. J. Clin. Pathol. 2007, 127, 820–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.Y.; Liu, C.L.; Chang, Y.C.; Nieh, S.; Lin, Y.S.; Jao, S.W.; Chen, S.F.; Liu, T.Y. Increased chemoresistance via Snail-Raf kinase inhibitor protein signaling in colorectal cancer in response to a nicotine derivative. Oncotarget 2016, 7, 23512–23520. [Google Scholar] [CrossRef]
- Morozova, N.; Zinovyev, A.; Nonne, N.; Pritchard, L.L.; Gorban, A.N.; Harel-Bellan, A. Kinetic signatures of microRNA modes of action. RNA 2012, 18, 1635–1655. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Shan, Y.; Zheng, J.; Lambrecht, R.W.; Donohue, S.E.; Bonkovsky, H.L. Zinc mesoporphyrin induces rapid and marked degradation of the transcription factor bach1 and up-regulates HO-1. Biochim. Biophys. Acta Gene Regul. Mech. 2008, 1779, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Shahrezaei, V.; Swain, P.S. Analytical distributions for stochastic gene expression. Proc. Natl. Acad. Sci. USA 2008, 105, 17256–17261. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Ehnert, S.; Culmes, M.; Bachmann, A.; Seeliger, C.; Schyschka, L.; Wang, Z.; Rahmanian-Schwarz, A.; Stöckle, U.; Sousa, P.A.D.; et al. 5-Azacytidine Improves the Osteogenic Differentiation Potential of Aged Human Adipose-Derived Mesenchymal Stem Cells by DNA Demethylation. PLoS ONE 2014, 9, e90846. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Liu, Z.; Li, X.; Zhou, Y.; Hu, Z. Generation of new hair cells by DNA methyltransferase (Dnmt) inhibitor 5-azacytidine in a chemically-deafened mouse model. Sci. Rep. 2019, 9, 7997. [Google Scholar] [CrossRef] [PubMed]
- Hays, E.; Bonavida, B. Nitric Oxide-Mediated Enhancement and Reversal of Resistance of Anticancer Therapies. Antioxidants 2019, 8, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arfken, G.B.; Weber, H.J. Mathematical Methods for Physicists, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Innocentini, G.C.P.; Hornos, J.E.M. Modeling stochastic gene expression under repression. J. Math. Biol. 2007, 55, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Abramowitz, M.; Stegun, I.A. (Eds.) Handbook of Mathematical Functions: With Formulas, Graphs, and Mathematical Tables, 10th ed.; National Bureau of Standards Applied Mathematics Series; U.S. Department of Commerce, National Bureau of Standards: Washington, DC, USA, 1972; Volume 55.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Graph | Sequence of Number of Doses × Fraction for | Cumulative Reduction in | ||
|---|---|---|---|---|
| F3 | ; ; | ; ; | ||
| G3 | ; ; | ; | ||
| H3 | ; ; | ; | ||
| I3 | ; ; | |||
| J3 | ; ; | ; ; | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giovanini, G.; Barros, L.R.C.; Gama, L.R.; Tortelli, T.C., Jr.; Ramos, A.F. A Stochastic Binary Model for the Regulation of Gene Expression to Investigate Responses to Gene Therapy. Cancers 2022, 14, 633. https://doi.org/10.3390/cancers14030633
Giovanini G, Barros LRC, Gama LR, Tortelli TC Jr., Ramos AF. A Stochastic Binary Model for the Regulation of Gene Expression to Investigate Responses to Gene Therapy. Cancers. 2022; 14(3):633. https://doi.org/10.3390/cancers14030633
Chicago/Turabian StyleGiovanini, Guilherme, Luciana R. C. Barros, Leonardo R. Gama, Tharcisio C. Tortelli, Jr., and Alexandre F. Ramos. 2022. "A Stochastic Binary Model for the Regulation of Gene Expression to Investigate Responses to Gene Therapy" Cancers 14, no. 3: 633. https://doi.org/10.3390/cancers14030633
APA StyleGiovanini, G., Barros, L. R. C., Gama, L. R., Tortelli, T. C., Jr., & Ramos, A. F. (2022). A Stochastic Binary Model for the Regulation of Gene Expression to Investigate Responses to Gene Therapy. Cancers, 14(3), 633. https://doi.org/10.3390/cancers14030633