
Knowns and Unknowns about CAR-T Cell Dysfunction

 ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Dysfunctional States of T Cells
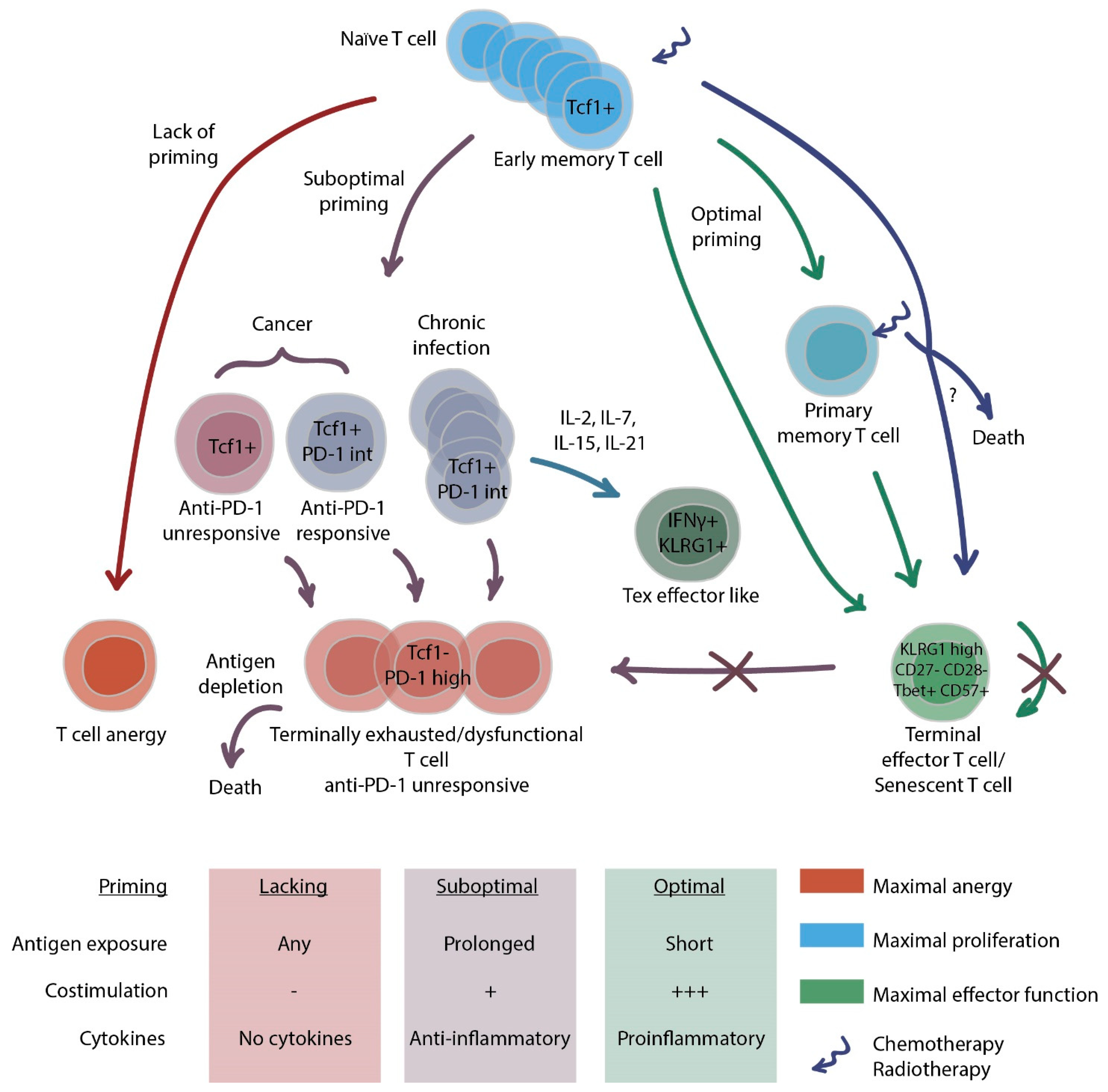
2.1. CD8+ T Cell Exhaustion
2.2. CD4+ T Cell Exhaustion
2.3. How to Combat Exhaustion?
2.4. T cell Senescence
2.5. How to Identify Exhausted and Senescent T Cells
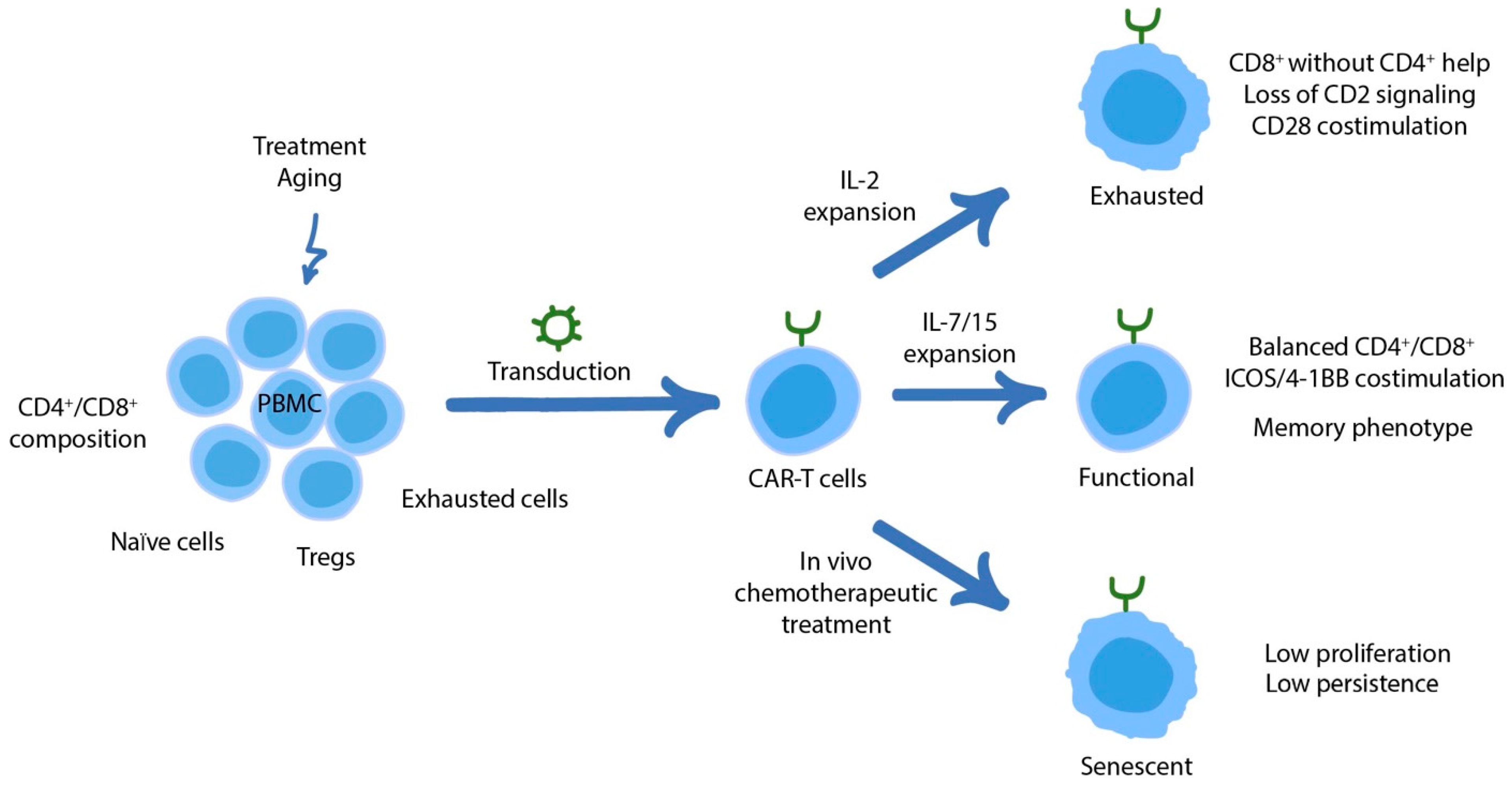
3. Dysfunctional States of CAR-T Cells
3.1. CAR Signaling as a Driver of Dysfunction and the Road to Its Prevention
3.2. Adjusting Regulatory Networks to Counteract T Cell Dysfunction
3.3. The Role of CD4+ CAR-T Cells in Counteracting Exhaustion and Overall Therapeutic Efficacy
3.4. Senescence in CAR-T Cells
3.5. Clinical Correlations of CAR-T Cell Activity/Dysfunction
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
List of Abbreviations
| A2aR | Adenosine Receptor A2 |
| ADAR1 | Adenosine Deaminase RNA Specific |
| AKTi | AKT inhibitor VIII |
| ASS | Argininosuccinate synthase |
| BATF | Basic leucine zipper transcriptional factor ATF-like |
| CAIX | Carbonic anhydrase IX |
| CAR | Chimeric Antigen Receptor |
| CAR-T cell | T cell with Chimeric Antigen Receptor |
| COX | Cyclooxygenase |
| DDR | DNA Damage Response |
| HBV | Hepatitis B Virus |
| HCV | Hepatitis C Virus |
| HIV | Human Immunodeficiency Virus |
| IDO-1 | Indoleamine 2,3-dioxygenase |
| IFN | Interferon |
| iIL | Inducible interleukin |
| IL | Interleukin |
| IR | Inhibitory Receptors |
| IRF4 | Interferon Regulatory Factor 4 |
| KO | Knockout |
| LCMV | Murine Lymphocytic Choriomeningitis |
| LDH | Lactate Dehydrogenase |
| MHC | Main Histocompatibility Complex |
| OTC | Ornithine transcarbamylase |
| PI3Kδ | Phosphatidylinositol-3-kinase p110δ |
| PKA | Protein Kinase A |
| PP2A | Protein Phosphatase 2A |
| PTPN-2 | Protein tyrosine phosphatase non-receptor type 2 |
| ROS | Reactive Oxygen Species |
| SASP | Senescence-Associated Secretory Phenotype |
| shRNA | Small hairpin RNA |
| SLEC | Short-Lived Effector Cells |
| TCR | T Cell Receptor |
| Tex | Exhausted T Cell |
| TIGIT | T cell immunoreceptor with Ig and ITIM domain |
| TIL | Tumor Infiltrating Lymphocyte |
References
- Judge, S.J.; Murphy, W.J.; Canter, R.J. Characterizing the Dysfunctional NK Cell: Assessing the Clinical Relevance of Exhaustion, Anergy, and Senescence. Front. Cell. Infect. Microbiol. 2020, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Beltra, J.-C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental Relationships of Four Exhausted CD8+ T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825–841.e8. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [Green Version]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.-W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utzschneider, D.T.; Gabriel, S.S.; Chisanga, D.; Gloury, R.; Gubser, P.M.; Vasanthakumar, A.; Shi, W.; Kallies, A. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat. Immunol. 2020, 21, 1256–1266. [Google Scholar] [CrossRef]
- Angelosanto, J.M.; Blackburn, S.D.; Crawford, A.; Wherry, E.J. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J. Virol. 2012, 86, 8161–8170. [Google Scholar] [CrossRef] [Green Version]
- West, E.E.; Youngblood, B.; Tan, W.G.; Jin, H.T.; Araki, K.; Alexe, G.; Konieczny, B.T.; Calpe, S.; Freeman, G.J.; Terhorst, C.; et al. Tight Regulation of Memory CD8+ T Cells Limits Their Effectiveness during Sustained High Viral Load. Immunity 2011, 35, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schietinger, A.; Philip, M.; Krisnawan, V.E.; Chiu, E.Y.; Delrow, J.J.; Basom, R.S.; Lauer, P.; Brockstedt, D.G.; Knoblaugh, S.E.; Hämmerling, G.J.; et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 2016, 45, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Mumprecht, S.; Schürch, C.; Schwaller, J.; Solenthaler, M.; Ochsenbein, A.F. Programmed death 1 signaling on chronic myeloid leukemia-specific T cells results in T-cell exhaustion and disease progression. Blood 2009, 114, 1528–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, N.; Madi, A.; Zhang, H.; Klapholz, M.; Escobar, G.; Dulberg, S.; Christian, E.; Ferreira, M.; Dixon, K.O.; Fell, G.; et al. Endogenous Glucocorticoid Signaling Regulates CD8+ T Cell Differentiation and Development of Dysfunction in the Tumor Microenvironment. Immunity 2020, 53, 658–671.e6. [Google Scholar] [CrossRef] [PubMed]
- Horton, B.L.; Morgan, D.M.; Momin, N.; Zagorulya, M.; Torres-Mejia, E.; Bhandarkar, V.; Wittrup, K.D.; Love, J.C.; Spranger, S. Lack of CD8+ T cell effector differentiation during priming mediates checkpoint blockade resistance in non-small cell lung cancer. Sci. Immunol. 2021, 6, eabi8800. [Google Scholar] [CrossRef]
- McKinney, E.F.; Lee, J.C.; Jayne, D.R.W.; Lyons, P.A.; Smith, K.G.C. T cell exhaustion, costimulation and clinical outcome in autoimmunity and infection. Nature 2015, 523, 612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, B.; Liu, X.; Li, X.; Kong, H.; Tian, L.; Chen, Y. T-cell exhaustion in chronic hepatitis B infection: Current knowledge and clinical significance. Cell Death Dis. 2015, 6, e1694. [Google Scholar] [CrossRef] [Green Version]
- Gruener, N.H.; Lechner, F.; Jung, M.C.; Diepolder, H.; Gerlach, T.; Lauer, G.; Walker, B.; Sullivan, J.; Phillips, R.; Pape, G.R.; et al. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J. Virol. 2001, 75, 5550–5558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Kostense, S.; Ogg, G.S.; Manting, E.H.; Gillespie, G.; Joling, J.; Vandenberghe, K.; Veenhof, E.Z.; van Baarle, D.; Jurriaans, S.; Klein, M.R.; et al. High viral burden in the presence of major HIV-specific CD8+ T cell expansions: Evidence for impaired CTL effector function. Eur. J. Immunol. 2001, 31, 677–686. [Google Scholar] [CrossRef]
- Shankar, P.; Russo, M.; Harnisch, B.; Patterson, M.; Skolnik, P.; Lieberman, J. Impaired function of circulating HIV-specific CD8+ T cells in chronic human immunodeficiency virus infection. Blood 2000, 96, 3094–3101. [Google Scholar] [CrossRef]
- Goepfert, P.A.; Bansal, A.; Edwards, B.H.; Ritter, G.D.J.; Tellez, I.; McPherson, S.A.; Sabbaj, S.; Mulligan, M.J. A significant number of human immunodeficiency virus epitope-specific cytotoxic T lymphocytes detected by tetramer binding do not produce gamma interferon. J. Virol. 2000, 74, 10249–10255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.G.; Jang, M.; Kim, Y.; Leem, G.; Kim, K.H.; Lee, H.; Kim, T.-S.; Choi, S.J.; Kim, H.-D.; Han, J.W.; et al. VEGF-A drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci. Immunol. 2019, 4, eaay0555. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, S.; Gong, D.; Qin, Y.; Shen, Q. Programmed death-1 upregulation is correlated with dysfunction of tumor-infiltrating CD8+ T lymphocytes in human non-small cell lung cancer. Cell. Mol. Immunol. 2010, 7, 389–395. [Google Scholar] [CrossRef]
- Bengsch, B.; Ohtani, T.; Khan, O.; Setty, M.; Manne, S.; O’Brien, S.; Gherardini, P.F.; Herati, R.S.; Huang, A.C.; Chang, K.-M.; et al. Epigenomic-Guided Mass Cytometry Profiling Reveals Disease-Specific Features of Exhausted CD8 T Cells. Immunity 2018, 48, 1029–1045.e5. [Google Scholar] [CrossRef] [Green Version]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef] [Green Version]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2005, 439, 682–687. [Google Scholar] [CrossRef]
- Petrovas, C.; Chaon, B.; Ambrozak, D.R.; Price, D.A.; Melenhorst, J.J.; Hill, B.J.; Geldmacher, C.; Casazza, J.P.; Chattopadhyay, P.K.; Roederer, M.; et al. Differential association of programmed death-1 and CD57 with ex vivo survival of CD8+ T cells in HIV infection. J. Immunol. 2009, 183, 1120–1132. [Google Scholar] [CrossRef] [Green Version]
- Penna, A.; Pilli, M.; Zerbini, A.; Orlandini, A.; Mezzadri, S.; Sacchelli, L.; Missale, G.; Ferrari, C. Dysfunction and functional restoration of HCV-specific CD8 responses in chronic hepatitis C virus infection. Hepatology 2007, 45, 588–601. [Google Scholar] [CrossRef]
- He, R.; Hou, S.; Liu, C.; Zhang, A.; Bai, Q.; Han, M.; Yang, Y.; Wei, G.; Shen, T.; Yang, X.; et al. Follicular CXCR5- expressing CD8+ T cells curtail chronic viral infection. Nature 2016, 537, 412–428. [Google Scholar] [CrossRef] [PubMed]
- Yates, K.B.; Tonnerre, P.; Martin, G.E.; Gerdemann, U.; Al Abosy, R.; Comstock, D.E.; Weiss, S.A.; Wolski, D.; Tully, D.C.; Chung, R.T.; et al. Epigenetic scars of CD8+ T cell exhaustion persist after cure of chronic infection in humans. Nat. Immunol. 2021, 22, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hakeem, M.S.; Manne, S.; Beltra, J.C.; Stelekati, E.; Chen, Z.; Nzingha, K.; Ali, M.A.; Johnson, J.L.; Giles, J.R.; Mathew, D.; et al. Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat. Immunol. 2021, 22, 1008–1019. [Google Scholar] [CrossRef]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749.e18. [Google Scholar] [CrossRef] [Green Version]
- Correa-Rocha, R.; Lopez-Abente, J.; Gutierrez, C.; Pérez-Fernández, V.A.; Prieto-Sánchez, A.; Moreno-Guillen, S.; Muñoz-Fernández, M.-Á.; Pion, M. CD72/CD100 and PD-1/PD-L1 markers are increased on T and B cells in HIV-1+ viremic individuals, and CD72/CD100 axis is correlated with T-cell exhaustion. PLoS ONE 2018, 13, e0203419. [Google Scholar] [CrossRef]
- Brooks, D.G.; McGavern, D.B.; Oldstone, M.B.A. Reprogramming of antiviral T cells prevents inactivation and restores T cell activity during persistent viral infection. J. Clin. Investig. 2006, 116, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef]
- Penaloza-MacMaster, P.; Provine, N.M.; Blass, E.; Barouch, D.H. CD4 T Cell Depletion Substantially Augments the Rescue Potential of PD-L1 Blockade for Deeply Exhausted CD8 T Cells. J. Immunol. 2015, 195, 1054–1063. [Google Scholar] [CrossRef] [Green Version]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Kurtulus, S.; Madi, A.; Escobar, G.; Klapholz, M.; Nyman, J.; Christian, E.; Pawlak, M.; Dionne, D.; Xia, J.; Rozenblatt-Rosen, O.; et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1−CD8+ Tumor-Infiltrating T Cells. Immunity 2019, 50, 181–194.e6. [Google Scholar] [CrossRef] [Green Version]
- Utzschneider, D.T.; Charmoy, M.; Chennupati, V.; Pousse, L.; Ferreira, D.P.; Calderon-Copete, S.; Danilo, M.; Alfei, F.; Hofmann, M.; Wieland, D.; et al. T Cell Factor 1-Expressing Memory-like CD8+ T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 2016, 45, 415–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenkel, J.M.; Herbst, R.H.; Canner, D.; Li, A.; Hillman, M.; Shanahan, S.-L.; Gibbons, G.; Smith, O.C.; Kim, J.Y.; Westcott, P.; et al. Conventional type I dendritic cells maintain a reservoir of proliferative tumor-antigen specific TCF-1+ CD8+ T cells in tumor-draining lymph nodes. Immunity 2021, 54, 2338–2353.e6. [Google Scholar] [CrossRef] [PubMed]
- Rome, K.S.; Stein, S.J.; Kurachi, M.; Petrovic, J.; Schwartz, G.W.; Mack, E.A.; Uljon, S.; Wu, W.W.; DeHart, A.G.; McClory, S.E.; et al. Trib1 regulates T cell differentiation during chronic infection by restraining the effector program. J. Exp. Med. 2020, 217, e20190888. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.A.; Kim, M.; Montalvo, W.; Al-Shamkhani, A.; Allison, J.P. Combination CTLA-4 blockade and 4-1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PLoS ONE 2011, 6, e19499. [Google Scholar] [CrossRef]
- Zander, R.; Schauder, D.; Xin, G.; Nguyen, C.; Wu, X.; Zajac, A.; Cui, W. CD4+ T Cell Help Is Required for the Formation of a Cytolytic CD8+ T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 2019, 51, 1028–1042.e4. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef] [Green Version]
- Backer, R.A.; Hombrink, P.; Helbig, C.; Amsen, D. The Fate Choice Between Effector and Memory T Cell Lineages: Asymmetry, Signal Integration, and Feedback to Create Bistability. Adv. Immunol. 2018, 137, 43–82. [Google Scholar] [CrossRef]
- Pipkin, M.E.; Sacks, J.A.; Cruz-Guilloty, F.; Lichtenheld, M.G.; Bevan, M.J.; Rao, A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 2010, 32, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Xin, A.; Masson, F.; Liao, Y.; Preston, S.; Guan, T.; Gloury, R.; Olshansky, M.; Lin, J.-X.; Li, P.; Speed, T.P.; et al. A molecular threshold for effector CD8+ T cell differentiation controlled by transcription factors Blimp-1 and T-bet. Nat. Immunol. 2016, 17, 422–432. [Google Scholar] [CrossRef]
- Malek, T.R. The biology of interleukin-2. Annu. Rev. Immunol. 2008, 26, 453–479. [Google Scholar] [CrossRef]
- Snell, L.M.; MacLeod, B.L.; Law, J.C.; Osokine, I.; Elsaesser, H.J.; Hezaveh, K.; Dickson, R.J.; Gavin, M.A.; Guidos, C.J.; McGaha, T.L.; et al. CD8+ T Cell Priming in Established Chronic Viral Infection Preferentially Directs Differentiation of Memory-like Cells for Sustained Immunity. Immunity 2018, 49, 678–694.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, K.; Brocker, T.; Oxenius, A. Antigen amount dictates CD8+ T-cell exhaustion during chronic viral infection irrespective of the type of antigen presenting cell. Eur. J. Immunol. 2012, 42, 2290–2304. [Google Scholar] [CrossRef] [PubMed]
- Bénéchet, A.P.; De Simone, G.; Di Lucia, P.; Cilenti, F.; Barbiera, G.; Le Bert, N.; Fumagalli, V.; Lusito, E.; Moalli, F.; Bianchessi, V.; et al. Dynamics and genomic landscape of CD8+ T cells undergoing hepatic priming. Nature 2019, 574, 200–205. [Google Scholar] [CrossRef]
- Mueller, S.N.; Ahmed, R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2009, 106, 8623–8628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef]
- Connolly, K.A.; Kuchroo, M.; Venkat, A.; Khatun, A.; Wang, J.; William, I.; Hornick, N.I.; Fitzgerald, B.L.; Damo, M.; Kasmani, M.Y.; et al. A reservoir of stem-like CD8+ T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci. Immunol. 2021, 6, eabg7836. [Google Scholar] [CrossRef]
- Fransen, M.F.; van Hall, T.; Ossendorp, F. Immune Checkpoint Therapy: Tumor Draining Lymph Nodes in the Spotlights. Int. J. Mol. Sci. 2021, 22, 9401. [Google Scholar] [CrossRef]
- Fransen, M.F.; Schoonderwoerd, M.; Knopf, P.; Camps, M.G.; Hawinkels, L.J.; Kneilling, M.; van Hall, T.; Ossendorp, F. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight 2018, 3, e124507. [Google Scholar] [CrossRef] [Green Version]
- Dammeijer, F.; van Gulijk, M.; Mulder, E.E.; Lukkes, M.; Klaase, L.; van den Bosch, T.; van Nimwegen, M.; Lau, S.P.; Latupeirissa, K.; Schetters, S.; et al. The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell 2020, 38, 685–700.e8. [Google Scholar] [CrossRef] [PubMed]
- Chihara, N.; Madi, A.; Kondo, T.; Zhang, H.; Acharya, N.; Singer, M.; Nyman, J.; Marjanovic, N.D.; Kowalczyk, M.S.; Wang, C.; et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 2018, 558, 454–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakiba, M.; Zumbo, P.; Espinosa-Carrasco, G.; Menocal, L.; Dündar, F.; Carson, S.E.; Bruno, E.M.; Sanchez-Rivera, F.J.; Lowe, S.W.; Camara, S.; et al. TCR signal strength defines distinct mechanisms of T cell dysfunction and cancer evasion. J. Exp. Med. 2021, 219, e20201966. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.; Angelosanto, J.M.; Kao, C.; Doering, T.A.; Odorizzi, P.M.; Barnett, B.E.; Wherry, E.J. Molecular and transcriptional basis of CD4+ T cell dysfunction during chronic infection. Immunity 2014, 40, 289–302. [Google Scholar] [CrossRef] [Green Version]
- Brooks, D.G.; Teyton, L.; Oldstone, M.B.A.; McGavern, D.B. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J. Virol. 2005, 79, 10514–10527. [Google Scholar] [CrossRef] [Green Version]
- Schulze Zur Wiesch, J.; Ciuffreda, D.; Lewis-Ximenez, L.; Kasprowicz, V.; Nolan, B.E.; Streeck, H.; Aneja, J.; Reyor, L.L.; Allen, T.M.; Lohse, A.W.; et al. Broadly directed virus-specific CD4+ T cell responses are primed during acute hepatitis C infection, but rapidly disappear from human blood with viral persistence. J. Exp. Med. 2012, 209, 61–75. [Google Scholar] [CrossRef]
- Morou, A.; Palmer, B.E.; Kaufmann, D.E. Distinctive features of CD4+ T cell dysfunction in chronic viral infections. Curr. Opin. HIV AIDS 2014, 9, 446–451. [Google Scholar] [CrossRef] [Green Version]
- Brooks, D.G.; Trifilo, M.J.; Edelmann, K.H.; Teyton, L.; McGavern, D.B.; Oldstone, M.B.A. Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med. 2006, 12, 1301–1309. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Boettler, T.; Moeckel, F.; Cheng, Y.; Heeg, M.; Salek-Ardakani, S.; Crotty, S.; Croft, M.; von Herrath, M.G. OX40 facilitates control of a persistent virus infection. PLoS Pathog. 2012, 8, e1002913. [Google Scholar] [CrossRef]
- Taraban, V.Y.; Rowley, T.F.; O’Brien, L.; Chan, H.T.C.; Haswell, L.E.; Green, M.H.A.; Tutt, A.L.; Glennie, M.J.; Al-Shamkhani, A. Expression and costimulatory effects of the TNF receptor superfamily members CD134 (OX40) and CD137 (4-1BB), and their role in the generation of anti-tumor immune responses. Eur. J. Immunol. 2002, 32, 3617–3627. [Google Scholar] [CrossRef]
- Melero, I.; Shuford, W.W.; Newby, S.A.; Aruffo, A.; Ledbetter, J.A.; Hellström, K.E.; Mittler, R.S.; Chen, L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat. Med. 1997, 3, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Cormary, C.; Hiver, E.; Mariamé, B.; Favre, G.; Tilkin-Mariamé, A.-F. Coexpression of CD40L and CD70 by semiallogenic tumor cells induces anti-tumor immunity. Cancer Gene Ther. 2005, 12, 963–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, A.M.; Xiao, Y.; Peperzak, V.; Naik, S.H.; Borst, J. Costimulatory ligand CD70 allows induction of CD8+ T-cell immunity by immature dendritic cells in a vaccination setting. Blood 2009, 113, 5167–5175. [Google Scholar] [CrossRef]
- Vezys, V.; Penaloza-MacMaster, P.; Barber, D.L.; Ha, S.-J.; Konieczny, B.; Freeman, G.J.; Mittler, R.S.; Ahmed, R. 4-1BB signaling synergizes with programmed death ligand 1 blockade to augment CD8 T cell responses during chronic viral infection. J. Immunol. 2011, 187, 1634–1642. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, X.; Cheng, D.; Xia, Z.; Luan, M.; Zhang, S. PD-1 blockade and OX40 triggering synergistically protects against tumor growth in a murine model of ovarian cancer. PLoS ONE 2014, 9, e89350. [Google Scholar] [CrossRef]
- Fan, X.; Quezada, S.A.; Sepulveda, M.A.; Sharma, P.; Allison, J.P. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J. Exp. Med. 2014, 211, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Waite., J.; Bei, W.; Lauric, H.; Aynur, H.; Erica, U.; Xuan, Y.; Drew, D.; Rabih, S.; Ajithdoss, D.K.; Godin, S.J.; et al. Tumor-targeted CD28 bispecific antibodies enhance the antitumor efficacy of PD-1 immunotherapy. Sci. Transl. Med. 2020, 12, eaba2325. [Google Scholar] [CrossRef]
- Blattman, J.N.; Grayson, J.M.; Wherry, E.J.; Kaech, S.M.; Smith, K.A.; Ahmed, R. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat. Med. 2003, 9, 540–547. [Google Scholar] [CrossRef]
- Zippelius, A.; Batard, P.; Rubio-Godoy, V.; Bioley, G.; Liénard, D.; Lejeune, F.; Rimoldi, D.; Guillaume, P.; Meidenbauer, N.; Mackensen, A.; et al. Effector Function of Human Tumor-Specific CD8 T Cells in Melanoma Lesions: A State of Local Functional Tolerance. Cancer Res. 2004, 64, 2865–2873. [Google Scholar] [CrossRef] [Green Version]
- Tucker, C.G.; Mitchell, J.S.; Martinov, T.; Burbach, B.J.; Beura, L.K.; Wilson, J.C.; Dwyer, A.J.; Singh, L.M.; Mescher, M.F.; Fife, B.T. Adoptive T Cell Therapy with IL-12-Preconditioned Low-Avidity T Cells Prevents Exhaustion and Results in Enhanced T Cell Activation, Enhanced Tumor Clearance, and Decreased Risk for Autoimmunity. J. Immunol. 2020, 205, 1449–1460. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.P.; Cloud, C.A.; Garrett, T.E.; Moore, C.J.; Schwartz, K.M.; Johnson, C.B.; Craig, D.H.; Salem, M.L.; Paulos, C.M.; Cole, D.J. Ex vivo interleukin-12-priming during CD8+ T cell activation dramatically improves adoptive T cell transfer antitumor efficacy in a lymphodepleted host. J. Am. Coll. Surg. 2012, 214, 700–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.S.; Du, M.; Zajac, A.J. A vital role for interleukin-21 in the control of a chronic viral infection. Science 2009, 324, 1572–1576. [Google Scholar] [CrossRef] [PubMed]
- Micci, L.; Ryan, E.S.; Fromentin, R.; Bosinger, S.E.; Harper, J.L.; He, T.; Paganini, S.; Easley, K.A.; Chahroudi, A.; Benne, C.; et al. Interleukin-21 combined with ART reduces inflammation and viral reservoir in SIV-infected macaques. J. Clin. Investig. 2015, 125, 4497–4513. [Google Scholar] [CrossRef] [Green Version]
- Schurich, A.; Pallett, L.J.; Lubowiecki, M.; Singh, H.D.; Gill, U.S.; Kennedy, P.T.; Nastouli, E.; Tanwar, S.; Rosenberg, W.; Maini, M.K. The third signal cytokine IL-12 rescues the anti-viral function of exhausted HBV-specific CD8 T cells. PLoS Pathog. 2013, 9, e1003208. [Google Scholar] [CrossRef]
- Huang, B.; Luo, L.; Wang, J.; He, B.; Feng, R.; Xian, N.; Zhang, Q.; Chen, L.; Huang, G. B7-H3 specific T cells with chimeric antigen receptor and decoy PD-1 receptors eradicate established solid human tumors in mouse models. Oncoimmunology 2019, 9, 1684127. [Google Scholar] [CrossRef] [Green Version]
- Titov, A.; Zmievskaya, E.; Ganeeva, I.; Valiullina, A.; Petukhov, A.; Rakhmatullina, A.; Miftakhova, R.; Fainshtein, M.; Rizvanov, A.; Bulatov, E. Adoptive immunotherapy beyond CAR T-cells. Cancers 2021, 13, 743. [Google Scholar] [CrossRef]
- West, E.E.; Jin, H.-T.; Rasheed, A.-U.; Penaloza-Macmaster, P.; Ha, S.-J.; Tan, W.G.; Youngblood, B.; Freeman, G.J.; Smith, K.A.; Ahmed, R. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J. Clin. Investig. 2013, 123, 2604–2615. [Google Scholar] [CrossRef]
- Said, E.A.; Dupuy, F.P.; Trautmann, L.; Zhang, Y.; Shi, Y.; El-Far, M.; Hill, B.J.; Noto, A.; Ancuta, P.; Peretz, Y.; et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 2010, 16, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Brooks, D.G.; Ha, S.-J.; Elsaesser, H.; Sharpe, A.H.; Freeman, G.J.; Oldstone, M.B.A. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc. Natl. Acad. Sci. USA 2008, 105, 20428–20433. [Google Scholar] [CrossRef] [Green Version]
- Zarour, H.M. Reversing T-cell dysfunction and exhaustion in cancer. Clin. Cancer Res. 2016, 22, 1856–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damo, M.; Joshi, N.S. T(reg) cell IL-10 and IL-35 exhaust CD8+ T cells in tumors. Nat. Immunol. 2019, 20, 674–675. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, R.; Alcalde, V.; Yang, Y.; Sauer, K.; Zuniga, E.I. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity 2009, 31, 145–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.H.; Perry, C.J.; Tsui, Y.-C.; Staron, M.M.; Parish, I.A.; Dominguez, C.X.; Rosenberg, D.W.; Kaech, S.M. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat. Med. 2015, 21, 327–334. [Google Scholar] [CrossRef]
- Ejrnaes, M.; Filippi, C.M.; Martinic, M.M.; Ling, E.M.; Togher, L.M.; Crotty, S.; von Herrath, M.G. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J. Exp. Med. 2006, 203, 2461–2472. [Google Scholar] [CrossRef]
- Budhu, S.; Schaer, D.A.; Li, Y.; Toledo-Crow, R.; Panageas, K.; Yang, X.; Zhong, H.; Houghton, A.N.; Silverstein, S.C.; Merghoub, T.; et al. Blockade of surface-bound TGF-β on regulatory T cells abrogates suppression of effector T cell function in the tumor microenvironment. Sci. Signal. 2017, 10, eaak9702. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Mancera, P.A.; Young, A.R.J.; Narita, M. Inside and out: The activities of senescence in cancer. Nat. Rev. Cancer 2014, 14, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Akbar, A.N.; Henson, S.M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 2011, 11, 289–295. [Google Scholar] [CrossRef]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Peng, G. T Cell Senescence and Tumor Immunotherapy. In Handbook of Immunosenescence; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; pp. 1–24. [Google Scholar]
- Dunne, P.J.; Belaramani, L.; Fletcher, J.M.; de Mattos, S.F.; Lawrenz, M.; Soares, M.V.D.; Rustin, M.H.A.; Lam, E.W.-F.; Salmon, M.; Akbar, A.N. Quiescence and functional reprogramming of Epstein-Barr virus (EBV)—Specific CD8+ T cells during persistent infection. Blood 2005, 106, 558–565. [Google Scholar] [CrossRef]
- Dunne, P.J.; Faint, J.M.; Gudgeon, N.H.; Fletcher, J.M.; Plunkett, F.J.; Soares, M.V.D.; Hislop, A.D.; Annels, N.E.; Rickinson, A.B.; Salmon, M.; et al. Epstein-Barr virus—Specific CD8+ T cells that re-express CD45RA are apoptosis-resistant memory cells that retain replicative potential. Blood 2002, 100, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Mo, W.; Ye, J.; Li, L.; Zhang, Y.; Hsueh, E.C.; Hoft, D.F.; Peng, G. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat. Commun. 2018, 9, 249. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Ma, C.; Hsueh, E.C.; Dou, J.; Mo, W.; Liu, S.; Han, B.; Huang, Y.; Zhang, Y.; Varvares, M.A.; et al. TLR 8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol. Med. 2014, 6, 1294–1311. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G.; Wagner, W.; Adibzadeh, M.; Engel, A. T cell immunosenescence in vitro and in vivo. Exp. Gerontol. 1999, 34, 419–429. [Google Scholar] [CrossRef]
- Akbar, A.N.; Henson, S.M.; Lanna, A. Senescence of T Lymphocytes: Implications for Enhancing Human Immunity. Trends Immunol. 2016, 37, 866–876. [Google Scholar] [CrossRef]
- Henson, S.M.; Macaulay, R.; Riddell, N.E.; Nunn, C.J.; Akbar, A.N. Blockade of PD-1 or p38 MAP kinase signaling enhances senescent human CD8+ T-cell proliferation by distinct pathways. Eur. J. Immunol. 2015, 45, 1441–1451. [Google Scholar] [CrossRef] [Green Version]
- Henson, S.M.; Lanna, A.; Riddel, N.E.; Franzese, O.; Macaulay, R.; Griffiths, S.J.; Puleston, D.J.; Watson, A.S.; Simon, A.K.; Tooze, S.A.; et al. P38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J. Clin. Investig. 2014, 124, 4004–4016. [Google Scholar] [CrossRef]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Lanna, A.; Coutavas, E.; Levati, L.; Seidel, J.; Rustin, M.H.A.; Henson, S.M.; Akbar, A.N.; Franzese, O. IFN-α Inhibits Telomerase in Human CD8+ T Cells by Both hTERT Downregulation and Induction of p38 MAPK Signaling. J. Immunol. 2013, 191, 3744–3752. [Google Scholar] [CrossRef] [Green Version]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [Green Version]
- Lanna, A.; Gomes, D.C.O.; Muller-Durovic, B.; McDonnell, T.; Escors, D.; Gilroy, D.W.; Lee, J.H.; Karin, M.; Akbar, A.N. A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging. Nat. Immunol. 2017, 18, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, B.I.; De Maeyer, R.P.H.; Covre, L.P.; Nehar-Belaid, D.; Lanna, A.; Ward, S.; Marches, R.; Chambers, E.S.; Gomes, D.C.O.; Riddell, N.E.; et al. Sestrins induce natural killer function in senescent-like CD8+ T cells. Nat. Immunol. 2020, 21, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Callender, L.A.; Carroll, E.C.; Beal, R.W.J.; Chambers, E.S.; Nourshargh, S.; Akbar, A.N.; Henson, S.M. Human CD8+ EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell 2018, 17, e12675. [Google Scholar] [CrossRef]
- Ye, J.; Ma, C.; Hsueh, E.C.; Eickhoff, C.S.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Tumor-Derived γδ Regulatory T Cells Suppress Innate and Adaptive Immunity through the Induction of Immunosenescence. J. Immunol. 2013, 190, 2403–2414. [Google Scholar] [CrossRef]
- Montes, C.L.; Chapoval, A.I.; Nelson, J.; Orhue, V.; Zhang, X.; Schulze, D.H.; Strome, S.E.; Gastman, B.R. Tumor-induced senescent T cells with suppressor function: A potential form of tumor immune evasion. Cancer Res. 2008, 68, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Appay, V.; van Lier, R.A.W.; Sallusto, F.; Roederer, M. Phenotype and function of human T lymphocyte subsets: Consensus and issues. Cytometry. A 2008, 73, 975–983. [Google Scholar] [CrossRef]
- Koch, S.; Larbi, A.; Derhovanessian, E.; Ozcelik, D.; Naumova, E.; Pawelec, G. Multiparameter flow cytometric analysis of CD4 and CD8 T cell subsets in young and old people. Immun. Ageing 2008, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Callender, L.A.; Carroll, E.C.; Bober, E.A.; Akbar, A.N.; Solito, E.; Henson, S.M. Mitochondrial mass governs the extent of human T cell senescence. Aging Cell 2020, 19, e13067. [Google Scholar] [CrossRef] [Green Version]
- Simon, S.; Labarriere, N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? Oncoimmunology 2018, 7. [Google Scholar] [CrossRef]
- Winkler, F.; Bengsch, B. Use of Mass Cytometry to Profile Human T Cell Exhaustion. Front. Immunol. 2020, 10, 3039. [Google Scholar] [CrossRef] [PubMed]
- Suen, H.; Brown, R.; Yang, S.; Weatherburn, C.; Ho, P.J.; Woodland, N.; Nassif, N.; Barbaro, P.; Bryant, C.; Hart, D.; et al. Multiple myeloma causes clonal T-cell immunosenescence: Identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia 2016, 30, 1716–1724. [Google Scholar] [CrossRef] [PubMed]
- Akondy, R.S.; Monson, N.D.; Miller, J.D.; Edupuganti, S.; Teuwen, D.; Wu, H.; Quyyumi, F.; Garg, S.; Altman, J.D.; Del Rio, C.; et al. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J. Immunol. 2009, 183, 7919–7930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; López-Moyado, I.F.; Seo, H.; Lio, C.-W.J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019, 567, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.J.; Linker, K.E.; Leslie, F.M. THEMIS-SHP1 Recruitment by 4–1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells. Physiol. Behav. 2020, 37, 216–225. [Google Scholar] [CrossRef]
- Kenderian, S.S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Kim, M.; Porter, D.L.; June, C.H.; Gill, S. Identification of PD1 and TIM3 As Checkpoints That Limit Chimeric Antigen Receptor T Cell Efficacy in Leukemia. Biol. Blood Marrow Transpl. 2016, 22, S19–S21. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [Green Version]
- Yazdanifar, M.; Zhou, R.; Grover, P.; Williams, C.; Bose, M.; Moore, L.J.; Wu, S.T.; Maher, J.; Dreau, D.; Mukherjee, A.P. Overcoming Immunological Resistance Enhances the Efficacy of A Novel Anti-tMUC1-CAR T Cell Treatment against Pancreatic Ductal Adenocarcinoma. Cells 2019, 8, 1070. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Deng, Q.; Jiang, Y.Y.; Zhang, R.; Zhu, H.B.; Meng, J.X.; Li, Y.M. CAR-T 19 combined with reduced-dose PD-1 blockade therapy for treatment of refractory follicular lymphoma: A case report. Oncol. Lett. 2019, 18, 4415–4420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, B.T.; Roberts, Z.J.; Xue, A.; Rossi, J.M.; Smith, M.R. Rapid tumor regression from PD-1 inhibition after anti-CD19 chimeric antigen receptor T-cell therapy in refractory diffuse large B-cell lymphoma. Bone Marrow Transpl. 2020, 55, 1184–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. Checkpoint Blockade Reverses Anergy in IL-13Rα2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol. Ther.-Oncolytics 2018, 11, 20–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogi, S.; Eisenberg, V.; Mayer, S.; Shamul, A.; Barliya, T.; Cohen, C.J. A TIGIT-based chimeric co-stimulatory switch receptor improves T-cell anti-tumor function. J. Immunother. Cancer 2019, 7, 243. [Google Scholar] [CrossRef]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, e133977. [Google Scholar] [CrossRef] [PubMed]
- Masoumi, E.; Jafarzadeh, L.; Mirzaei, H.R.; Alishah, K.; Fallah-Mehrjardi, K.; Rostamian, H.; Khakpoor-Koosheh, M.; Meshkani, R.; Noorbakhsh, F.; Hadjati, J. Genetic and pharmacological targeting of A2a receptor improves function of anti-mesothelin CAR T cells. J. Exp. Clin. Cancer Res. 2020, 39, 1–12. [Google Scholar] [CrossRef]
- Beavis, P.A.; Henderson, M.A.; Giuffrida, L.; Mills, J.K.; Sek, K.; Cross, R.S.; Davenport, A.J.; John, L.B.; Mardiana, S.; Slaney, C.Y.; et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J. Clin. Investig. 2017, 127, 929–941. [Google Scholar] [CrossRef] [Green Version]
- Wiede, F.; Lu, K.; Du, X.; Liang, S.; Hochheiser, K.; Dodd, G.T.; Goh, P.K.; Kearney, C.; Meyran, D.; Beavis, P.A.; et al. PTPN 2 phosphatase deletion in T cells promotes anti-tumour immunity and CAR T-cell efficacy in solid tumours. EMBO J. 2020, 39, 1–26. [Google Scholar] [CrossRef]
- Cui, J.; Wang, H.; Medina, R.; Zhang, Q.; Xu, C.; Indig, I.H.; Zhou, J.; Song, Q.; Dmitriev, P.; Sun, M.Y.; et al. Inhibition of PP2A with LB-100 enhances efficacy of CAR-T cell therapy against glioblastoma. Cancers 2020, 12, 139. [Google Scholar] [CrossRef] [Green Version]
- Newick, K.; O’Brien, S.; Sun, J.; Kapoor, V.; Maceyko, S.; Lo, A.; Pure, E.; Moon, E.; Albelda, S.M. Augmentation of CAR T cell trafficking and antitumor efficacy by blocking protein kinase A (PKA) localization. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Bowers, J.S.; Majchrzak, K.; Nelson, M.H.; Aksoy, B.A.; Wyatt, M.M.; Smith, A.S.; Bailey, S.R.; Neal, L.R.; Hammerbacher, J.E.; Paulos, C.M. PI3Kδ inhibition enhances the antitumor fitness of adoptively transferred CD8+ T cells. Front. Immunol. 2017, 8, 1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-Man Clinical Trial of CAR NK-92 Cells: Safety Test of CD33-CAR NK-92 Cells in Patients with Relapsed and Refractory Acute Myeloid Leukemia; e-Century Publishing Corporation: Madison, WI, USA, 2018; Volume 8. [Google Scholar]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 enhances CAR-T-cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Narayan, D. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. J. Comb. Math. Comb. Comput. 2017, 2, e95103. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Kan, S.; Zhou, S.; Wang, Y.; Xu, J.; Cooke, J.P.; Wen, J.; Deng, H. Enhancement of the in vivo persistence and antitumor efficacy of CD19 chimeric antigen receptor T cells through the delivery of modified TERT mRNA. Cell Discov. 2015, 1, 15040. [Google Scholar] [CrossRef]
- Ninomiya, S.; Narala, N.; Huye, L.; Yagyu, S.; Savoldo, B.; Dotti, G.; Heslop, H.E.; Brenner, M.K.; Rooney, C.M.; Ramos, C.A. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 2015, 125, 3905–3916. [Google Scholar] [CrossRef] [Green Version]
- Fultang, L.; Booth, S.; Yogev, O.; da Costa, B.M.; Tubb, V.; Panetti, S.; Stavrou, V.; Scarpa, U.; Jankevics, A.; Lloyd, G.; et al. Metabolic engineering against the arginine microenvironment enhances CAR-T cell proliferation and therapeutic activity. Blood 2020, 136, 1155–1160. [Google Scholar] [CrossRef]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor–Redirected T Cells as well as Bystander Cells from Oxidative Stress–Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bethigh FoxO1low Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Gomes-Silva, D.; Mukherjee, M.; Srinivasan, M.; Krenciute, G.; Dakhova, O.; Zheng, Y.; Cabral, J.M.S.; Rooney, C.M.; Orange, J.S.; Brenner, M.K.; et al. Tonic 4-1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell Rep. 2017, 21, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, E.W.; Parker, K.R.; Sotillo, E.; Lynn, R.C.; Anbunathan, H.; Lattin, J.; Good, Z.; Belk, J.A.; Daniel, B.; Klysz, D.; et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 2021, 372. [Google Scholar] [CrossRef] [PubMed]
- Caulier, B.; Enserink, J.M.; Wälchli, S. Pharmacologic control of car t cells. Int. J. Mol. Sci. 2021, 22, 4320. [Google Scholar] [CrossRef] [PubMed]
- Wikenheiser, D.J.; Stumhofer, J.S. ICOS Co-Stimulation: Friend or Foe? Front. Immunol. 2016, 7, 304. [Google Scholar] [CrossRef] [Green Version]
- Guedan, S.; Posey, A.D.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [Green Version]
- Guedan, S.; Madar, A.; Casado-Medrano, V.; Shaw, C.; Wing, A.; Liu, F.; Young, R.M.; June, C.H.; Posey, A.D. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J. Clin. Investig. 2020, 130, 3087–3097. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’connor, R.S.; Hwang, W.-T.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Sheih, A.; Voillet, V.; Hanafi, L.A.; DeBerg, H.A.; Yajima, M.; Hawkins, R.; Gersuk, V.; Riddell, S.R.; Maloney, D.G.; Wohlfahrt, M.E.; et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun. 2020, 11, 219. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Frank, M.J.; Mount, C.; Tousley, A.; Kurtz, D.M.; Sworder, B.; Murphy, K.A.; Manousopoulou, A.; Kohler, K.; Rotiroti, M.C.; et al. CD58 Aberrations Limit Durable Responses to CD19 CAR in Large B Cell Lymphoma Patients Treated with Axicabtagene Ciloleucel but Can be Overcome through Novel CAR Engineering. Blood 2020, 136, 53–54. [Google Scholar] [CrossRef]
- Ferraro, B.; Walters, J.N.; Reuschel, E.L.; Balakrishnan, A.; Morrow, M.P.; Yan, J.; Khan, A.S.; Sardesai, N.Y.; Humeau, L.M.; Weiner, D.B.; et al. 211. CD2, the First Identified T cell Co-Stimulator, Demonstrates More Effective Chimeric Antigen Receptor Activity Over CD28 and 4-1BB. Mol. Ther. 2015, 23, S83. [Google Scholar] [CrossRef] [Green Version]
- Odorizzi, P.M.; Pauken, K.E.; Paley, M.A.; Sharpe, A.; John Wherry, E. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 2015, 212, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Bucks, C.M.; Norton, J.A.; Boesteanu, A.C.; Mueller, Y.M.; Katsikis, P.D. Chronic Antigen Stimulation Alone Is Sufficient to Drive CD8+ T Cell Exhaustion. J. Immunol. 2009, 182, 6697–6708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gennert, D.G.; Lynn, R.C.; Granja, J.M.; Weber, E.W.; Mumbach, M.R.; Zhao, Y.; Duren, Z.; Sotillo, E.; Greenleaf, W.J.; Wong, W.H.; et al. Dynamic chromatin regulatory landscape of human CAR T cell exhaustion. Proc. Natl. Acad. Sci. USA 2021, 118, e2104758118. [Google Scholar] [CrossRef] [PubMed]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef]
- Xin, G.; Schauder, D.M.; Lainez, B.; Weinstein, J.S.; Dai, Z.; Chen, Y.; Esplugues, E.; Wen, R.; Wang, D.; Parish, I.A.; et al. A Critical Role of IL-21-Induced BATF in Sustaining CD8-T-Cell-Mediated Chronic Viral Control. Cell Rep. 2015, 13, 1118–1124. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.; González-Avalos, E.; Zhang, W.; Ramchandani, P.; Yang, C.; Lio, C.-W.J.; Rao, A.; Hogan, P.G. BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat. Immunol. 2021, 22, 983–995. [Google Scholar] [CrossRef]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.; Sun, H.-W.; Lacey, N.E.; Ji, Y.; Moseman, E.A.; Shih, H.-Y.; Heuston, E.F.; Kirby, M.; Anderson, S.; Cheng, J.; et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat. Immunol. 2019, 20, 890–901. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef]
- Alfei, F.; Kanev, K.; Hofmann, M.; Wu, M.; Ghoneim, H.E.; Roelli, P.; Utzschneider, D.T.; von Hoesslin, M.; Cullen, J.G.; Fan, Y.; et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 2019, 571, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Naranjo, A.; Brown, C.E.; Bautista, C.; Wong, C.W.; Chang, W.C.; Aguilar, B.; Ostberg, J.R.; Riddell, S.R.; Forman, S.J.; et al. Phenotypic and functional attributes of lentivirus-modified CD19-specific Human CD8+ central memory T cells manufactured at clinical scale. J. Immunother. 2012, 35, 689–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terakura, S.; Yamamoto, T.N.; Gardner, R.A.; Turtle, C.J.; Jensen, M.C.; Riddell, S.R. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood 2012, 119, 72–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, J.; Paczkowski, P.; Shen, Y.-W.; Morse, K.; Flynn, B.; Kaiser, A.; Ng, C.; Gallatin, K.; Cain, T.; Fan, R.; et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 2018, 132, 804–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.; Hanauer, J.D.S.; Frank, A.M.; Riechert, V.; Thalheimer, F.B.; Buchholz, C.J. In Vivo Generation of CAR T Cells Selectively in Human CD4+ Lymphocytes. Mol. Ther. 2020, 28, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Xhangolli, I.; Dura, B.; Lee, G.H.; Kim, D.; Xiao, Y.; Fan, R. Single-cell Analysis of CAR-T Cell Activation Reveals A Mixed TH1/TH2 Response Independent of Differentiation. Genomics. Proteom. Bioinform. 2019, 17, 129–139. [Google Scholar] [CrossRef]
- Wang, D.; Aguilar, B.; Starr, R.; Alizadeh, D.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; Forman, S.J.; Brown, C.E. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight 2018, 3, e99048. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Kohler, M.E.; Chien, C.D.; Sauter, C.T.; Jacoby, E.; Yan, C.; Hu, Y.; Wanhainen, K.; Qin, H.; Fry, T.J. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci. Transl. Med. 2017, 9, eaag1209. [Google Scholar] [CrossRef] [Green Version]
- Liadi, I.; Singh, H.; Romain, G.; Rey-Villamizar, N.; Merouane, A.; Adolacion, J.R.T.; Kebriaei, P.; Huls, H.; Qiu, P.; Roysam, B.; et al. Individual motile CD4+ T cells can participate in efficient multikilling through conjugation to multiple tumor cells. Cancer Immunol. Res. 2015, 3, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [Green Version]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guha, P.; Cunetta, M.; Somasundar, P.; Espat, N.J.; Junghans, R.P.; Katz, S.C. Frontline Science: Functionally impaired geriatric CAR-T cells rescued by increased α5β1 integrin expression. J. Leukoc. Biol. 2017, 102, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Smithey, M.J.; Rudd, B.D.; Nikolich-Žugich, J. Age-associated alterations in CD8α+ dendritic cells impair CD8 T-cell expansion in response to an intracellular bacterium. Aging Cell 2012, 11, 968–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klebanoff, C.A.; Scott, C.D.; Leonardi, A.J.; Yamamoto, T.N.; Cruz, A.C.; Ouyang, C.; Ramaswamy, M.; Roychoudhuri, R.; Ji, Y.; Eil, R.L.; et al. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J. Clin. Investig. 2016, 126, 318–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikuła-Pietrasik, J.; Witucka, A.; Pakuła, M.; Uruski, P.; Begier-Krasińska, B.; Niklas, A.; Tykarski, A.; Książek, K. Comprehensive review on how platinum- and taxane-based chemotherapy of ovarian cancer affects biology of normal cells. Cell. Mol. Life Sci. 2019, 76, 681–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, D.W.; Haider, S.; Robertson, D.; Buus, R.; O’leary, L.; Isacke, C.M. Therapy-induced senescence in normal tissue promotes breast cancer metastasis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Das, R.K.; Storm, J.; Barrett, D.M. Abstract 1631: T cell dysfunction in pediatric cancer patients at diagnosis and after chemotherapy can limit chimeric antigen receptor potential. In Proceedings of the Cancer Research, Chicago, IL, USA, 14–18 April 2018; American Association for Cancer Research (AACR): Philadelphia, PA, US, 2018; Volume 78, p. 1631. [Google Scholar]
- Das, R.K.; O’Connor, R.S.; Grupp, S.A.; Barrett, D.M. Lingering effects of chemotherapy on mature T cells impair proliferation. Blood Adv. 2020, 4, 4653–4664. [Google Scholar] [CrossRef]
- Fagnoni, F.F.; Lozza, L.; Zibera, C.; Zambelli, A.; Ponchio, L.; Gibelli, N.; Oliviero, B.; Pavesi, L.; Gennari, R.; Vescovini, R.; et al. T-cell dynamics after high-dose chemotherapy in adults: Elucidation of the elusive CD8+ subset reveals multiple homeostatic T-cell compartments with distinct implications for immune competence. Immunology 2002, 106, 27. [Google Scholar] [CrossRef]
- Haining, W.N.; Neuberg, D.S.; Keczkemethy, H.L.; Evans, J.W.; Rivoli, S.; Gelman, R.; Rosenblatt, H.M.; Shearer, W.T.; Guenaga, J.; Douek, D.C.; et al. Antigen-specific T-cell memory is preserved in children treated for acute lymphoblastic leukemia. Blood 2005, 106, 1749. [Google Scholar] [CrossRef]
- Du, M.; Hari, P.; Hu, Y.; Mei, H. Biomarkers in individualized management of chimeric antigen receptor T cell therapy. Biomark. Res. 2020, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Dancy, E.K.; Cohen, A.D.; Hwang, W.T.; Fraietta, J.A.; Davis, M.M.; Levine, B.L.; Siegel, D.L.; Stadtmauer, E.A.; Vogl, D.T.; et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv. 2019, 3, 2812–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramson, J.S.; Lia, P.M.; Gordon, L.I.; Lunning, M.A.; Arnason, J.E.; Wang, M. High Durable CR Rates in Relapsed/Refractory (R/R) Aggressive B-NHL Treated with the CD19-Directed CAR T Cell Product JCAR017 (TRANSCEND NHL 001): Defined Composition Allows for Dose-Finding and Definition of Pivotal Cohort. Blood 2017, 130, 581. [Google Scholar]
- Titov, A.; Petukhov, A.; Staliarova, A.; Motorin, D.; Bulatov, E.; Shuvalov, O.; Soond, S.M.; Piacentini, M.; Melino, G.; Zaritskey, A.; et al. The biological basis and clinical symptoms of CAR-T therapy-associated toxicites. Cell Death Dis. 2018, 9, 897. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Modified Pathway/Molecule | Molecule Type | Upstream Signaling | Downstream Signaling (Activation or Inhibition) | Modification | Tumor Model (Animal, Cell Line) and Observed Effect | CAR Construct, Introduction Method | Reference |
|---|---|---|---|---|---|---|---|
| A2aR (adenosine receptor A2) | surface molecule | adenosine | cAMP | shRNA knockdown A2aR antagonist (SCH-58261) | in vitro, HeLa no significant reduction in cytotoxic function in presence of the adenosine agonist NECA (5′-(N-ethylcarboxamide), higher IL-2 expression in comparison to unmodified cells | mesothelin-BBz, lentiviral | [138] |
| A2aR (adenosine receptor A2) | surface molecule | adenosine | cAMP | shRNA knockdown anti-PD-1 antibody | in vivo, Ly 5.1 mice + 24JK-HER2+ or E0771-HER2+ increased cytokine production of CD8+ and activation of CD8+ and CD4+ CAR-T, particularly with PD-1 blockade; significant survival advantage in mice | HER2-28z, retroviral | [139] |
| ADAR1 (adenosine deaminase RNA specific) | cytoplasmic enzyme | IFN Type I | biogenesis of members of the miR-222 family -> ICAM1 expression -> immune resistance | EHNA drug (ADAR1 inhibitor) | in vitro, HPAFII, CFPAC, MiaPaCa2 combination with EHNA did not result in reduced survival of the target cells | MUC-1-28z, retroviral | [129] |
| AKT | intracellular signal transducer | PI3K | MAPK, FOXO1 | AKT inhibitor VIII (AKTi) | in vitro, NALM6 AKTi repressed glycolysis; in vivo, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice + NALM6 AKTi exposed CAR-T elicit significant increase in animal survival | CD19, retroviral | [146] |
| Argininosuccinate synthase, ornithine transcarbamylase | cancer-associated enzymes | constitutive | arginine, ornithine | Knockin argininosuccinate synthase (ASS) and/or ornithine transcarbamylase (OTC) enzymes | in vivo, arginine-depleted NOG-SCID mice + GD2+ SKNMC, KELLY, and LAN-1; expression of ASS, OTC, or ASS+OTC enhanced proliferation of CAR-T; derived from multiple human donors, regardless of scFv | GD2-BBz CD33-BBz mesothelin-BBz EGFRvIII-BBz | [149] |
| Catalase | intracellular enzyme | constitutive | H2O2 | coexpression of catalase with CAR | in vitro, SkoV3-Her2+ reduced oxidative state, both basal and upon activation, enhanced proliferation and preserved target cell lysis in presence of H2O2 | CEA-28z, HER2-29z, retroviral | [150] |
| CTLA-4 | surface molecule | CD80/CD86 | SHP-2, PP2A | checkpoint blockade by CAR-T-secreted minibodies (reduced checkpoint inhibitors) | in vivo, NSG mice + D270, Hu08-BBz CAR-T cells also showed enhanced tumor reduction when used in combination with anti-CTLA-4, whereas 2173BBz CAR-T cells did not benefit from CTLA-4 checkpoint blockade | see TIM-3 section | [134] |
| Cyclooxygenase (COX-1, COX-2) | cytoplasmic enzymes | constitutive/inflammation (NFkB) | prostaglandin E2 | celecoxib (specific COX-2 inhibitor) and indomethacin (COX1 and 2 inhibitor) | in vitro, HPAFII and CFPAC cells showed a reduction in survival, MiaPaCa2 cells showed no difference in survival; Celecoxib did not change the efficacy of CAR-T cells | MUC-1-28z, retroviral | [129] |
| Telomerase | cancer/memory T-cell-associated enzyme | p38; DDR | Restores telomere length | transient delivery of telomerase mRNA | in vitro, Raji prolonged proliferation and inhibited cell senescence; in vivo, NPG/Vst mice + Raji, improved persistence, proliferation, and long-term antitumor effects | CD19-28z, CD19-BBz, lentiviral | [147] |
| IDO-1 (indoleamine 2,3-dioxygenase) | enzyme with high activity in cancer cells | constitutive | kynurenine | IDO inhibitor (1-methyl-tryptophan) | in vivo, SCID-Beige mice + Raji/Raji-IDO CAR-T inhibited IDO-negative but not IDO-positive tumors growth; IDO inhibitor restored IDO-positive tumor control; tryptophan metabolites inhibited expansion, proliferation, cytotoxicity, cytokine secretion, and increased CAR-T apoptosis; 4-1 BB intracellular domain had no effect on the inhibition | CD19-z/BBz, retroviral | [148] |
| IL-12, IL-18 | cytokines | - | STAT4 | inducible single-chain p45-p30 IL-12 (iIL-12); 18-kD IL-18 (iIL-18); constitutive IL-18 and IL-12 | in vivo, CEA transgenic C57BL/6 mice + Panc02-CEA+ and Rag2/γc/mice + A549 CEA+ iIL-18 CAR-T exhibited superior activity against large pancreatic and lung tumors refractory to CAR-T cells without cytokines. IL-18 induced overall change in the immune microenvironment | CEA-28z, retroviral | [151] |
| mTORC1 | intracellular signal transducer | calcineurin/DAPK1 | mTORC2/T-bet | rapamycin (mTOR inhibitor) expansion with IL-15 | in vivo, NSG mice, Raji IL15-expanded CAR-T mediated superior antitumor activity, longer persistence, and significantly greater survival than IL-2-expanded CAR-T; IL2/rapamycin-cultured CAR-T shared phenotypic features with IL-15-CAR-T, suggesting that IL15- mediated reduction of mTORC1 activity is responsible for preserving less differentiated phenotype | CD19 second generation, IL13Rα2 second generation, lentiviral | [145] |
| PD-1 | surface molecule | PD-L1/2 | SHP-1, SHP-2 | antibodies | in vitro MOLM-14, primary leukemia improved cytokine production and Ki-67 proliferation marker, especially in combinational treatment with PD-1 and TIM-3 antibodies; in vivo, NSG mice + MOLM14 increased durable complete response rate | CD33-BBz, CD123-BBz, lentiviral | [127] |
| PD-1 | surface molecule | PD-L1/2 | SHP-1, SHP-2 | CRISPR/Cas9 KO | in vivo, NSG mice + CD19+PD-1L+ K562, improved clearance of tumor | CD19-BBz, lentiviral | [128] |
| PD-1 | surface molecule | PD-L1/2 | SHP-1, SHP-2 | checkpoint blockade by CAR-T-secreted minibodies (reduced checkpoint inhibitors) | see TIM-3 section | [144] | |
| PD-1 | surface molecule | PD-L1/2 | SHP-1, SHP-2 | anti-PD-1 Ab | in vitro, no improvement in the killing of the resistant cell lines (HPAFII, CFPAC), while sensitive cell line (MiaPaCa2) killing was enhanced | MUC-1-28z, retroviral | [129] |
| PD-1 | surface molecule | PD-L1/2 | SHP-1, SHP-2 | pembrolisumab (anti-PD-1 Ab) | clinical case report, mediastinal B cell lymphoma; remission continuing 12 months posttherapy | CD19-BBz, lentiviral | [131] |
| PD-1 | surface molecule | PD-L1/2 | SHP-1, SHP-2 | nivolumab (anti-PD-1 Ab) | clinical case report, diffuse large B cell lymphoma short tumor volume reduction, lasting 2 months, followed by progression | CD19-BBz | [132] |
| PD-1 | surface molecule | PD-L1/2 | SHP-1, SHP-2 | nivolumab (anti-PD-1 Ab) | clinical case report, refractory follicular lymphoma remission lasted for >10 months. | CD19-28z, Axicabtagene ciloleucel (Kymriah®), retroviral | [133] |
| PD-1 | surface molecule | PD-L1/2 | SHP-1, SHP-2 | CRISPR-Cas9 KO | in vitro, PC3; in vivo, NSG mice + NALM6/NALM6-PD-L1+; increased cytotoxicity | PSCA-second generation, lentiviral | [130] |
| PI3Kδ (phosphatidylinositol-3-kinase p110δ) | intracellular signal transducer | TCR and costimulatory molecules (CD28, 4-1BB, and ICOS) | AKT | Idelalisib and AKT inhibitor VIII (AKTi) | in vivo, NSG mice + M108; Idelalisib-treated CAR-T cells exerted longer tumor control compared to the AKTi-treated; Idelalisib T cells have improved engraftment, persistence, less differentiated phenotype, and transcriptional signature | mesothelin-BBz, lentiviral | [143] |
| PI3Kδ (phosphatidylinositol-3-kinase p110δ) | intracellular signal transducer | TCR and costimulatory molecules (CD28, 4-1BB, and ICOS) | AKT | PI3K inhibitor LY294002 | in vitro, MOLM-13 increased effector molecules expression, less differentiated phenotype, higher cell number | CD33-BBz, MOLM-13 target cells (CD33+), retroviral | [144] |
| PKA (protein kinase A) | intracellular signal transducer | cAMP | Csk | transduction with regulatory subunit I anchoring disruptor (RIAD) | in vitro, AE17ova, PDA4662 cells, increased cytokine release; in vivo, C57BL/6 (strain CD45.2) + EM-meso+ and NSG mice + AE17-meso+; adenosine or PGE2 did not affect RIAD CAR-T; higher median reduction in tumor volume | mesothelin-BBz, lentiviral (human), retroviral (mice) | [142] |
| PP2A (protein phosphatase 2A) | intracellular signal transducer | constitutive | AKT, mTOR, Erk, CaMKKII/IV | protein phosphatase 2A (PP2A) inhibitor (LB-100) | in vitro, U251-Luc significantly increased cytotoxicity in a dose-dependent manner; in vivo, NSG mice + U251-Luc, significant increase in CD3+ cells within tumor tissue and more frequent tumor regression | CAIX (carbonic anhydrase IX)-BBz | [141] |
| PTPN-2 (protein tyrosine phosphatase nonreceptor type 2) | constitutive | c-Src | PTPN2-inhibitor compound 8 | siRNA duplexes transient knockdown, CRISPR-Cas9 KO | in vivo, female Ly5.1 B6.SJL-Ptprc a Pepc b/BoyJ, human HER-2 transgenic + E0771-HER-2+; improved immune surveillance on spontaneous tumors and after adoptive transfer | murine CAR-T: human HER-2-28z, human CAR-T: human LeY-28z, retroviral | [140] |
| SHP-1/THEMIS complex | intracellular signal transducer | PD-1; CD19-BBz | CD3ζ | knockdown THEMIS or SHP1 in CD19-BBz- CAR-T cells, shRNA | in vitro, BV173-CD19+; increased CAR-CD3ζ; basal phosphorylation | CD19-BBz, CD19-28 | [126] |
| TGF-β receptor II | surface molecule | TGF-β | SMAD2/3/4 | knockin (together with CAR), dominant-negative TGF-βRII | in vivo, NSG mice + PC3; increased proliferation, cytokine secretion, resistance to exhaustion, long-term in vivo persistence, induction tumor eradication in aggressive human prostate cancer | PSMA-BBz, lentiviral | [136] |
| TGF-β receptor II | surface molecule | TGF-β | SMAD2/3/4 | CRISPR/Cas9 KO | in vitro, mesothelin+ CRL5826 and OVCAR-3, improved killing; in vivo, NPG mice + CRL5826; better controlling tumor growth; TGF-β-RII KO outperform PD-1 KO in efficacy; PD-1 KO complement; TGF-β-RII KO | Mesothelin-28z, lentiviral | [137] |
| TIGIT | surface molecule | CD155 and CD112 | SHP-1; b-arrestin/SHIP1-mediated downstream inhibition of NF-kB, PI3K and MAPK pathways | TIGIT-28 chimeric switch receptor (TIGIT exo- + CD28 signaling domain) | in vitro, Raji, JY, 721.221, Nalm6; improved killing and cytokine secretion | CD19-BBz, MSGV1-based, retroviral | [135] |
| TIM-3 | surface molecule | galectin-9 | CD45; CD148 | checkpoint blockade by CAR-T-secreted minibodies (reduced checkpoint inhibitors) | in vivo, NSG mice + D270; significant inhibition of tumor growth with either 2173BBz or Hu08BBz CAR-T cells and anti-PD-1/anti-TIM-3 | anti-IL-13Rα2-BBz (humanized Hu08BBz, murine 2173BBz), lentiviral | [134] |
| TIM-3 | surface molecule | galectin-9 | CD45; CD148 | Gal-9 blocking Ab | in vitro, HPAFII and CFPAC cells—improvement; MiaPaCa2 cells—no improvement | MUC-1-28z, retroviral | [129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Titov, A.; Kaminskiy, Y.; Ganeeva, I.; Zmievskaya, E.; Valiullina, A.; Rakhmatullina, A.; Petukhov, A.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Knowns and Unknowns about CAR-T Cell Dysfunction. Cancers 2022, 14, 1078. https://doi.org/10.3390/cancers14041078
Titov A, Kaminskiy Y, Ganeeva I, Zmievskaya E, Valiullina A, Rakhmatullina A, Petukhov A, Miftakhova R, Rizvanov A, Bulatov E. Knowns and Unknowns about CAR-T Cell Dysfunction. Cancers. 2022; 14(4):1078. https://doi.org/10.3390/cancers14041078
Chicago/Turabian StyleTitov, Aleksei, Yaroslav Kaminskiy, Irina Ganeeva, Ekaterina Zmievskaya, Aygul Valiullina, Aygul Rakhmatullina, Alexey Petukhov, Regina Miftakhova, Albert Rizvanov, and Emil Bulatov. 2022. "Knowns and Unknowns about CAR-T Cell Dysfunction" Cancers 14, no. 4: 1078. https://doi.org/10.3390/cancers14041078
APA StyleTitov, A., Kaminskiy, Y., Ganeeva, I., Zmievskaya, E., Valiullina, A., Rakhmatullina, A., Petukhov, A., Miftakhova, R., Rizvanov, A., & Bulatov, E. (2022). Knowns and Unknowns about CAR-T Cell Dysfunction. Cancers, 14(4), 1078. https://doi.org/10.3390/cancers14041078