
In Silico Bioinformatics Followed by Molecular Validation Using Archival FFPE Tissue Biopsies Identifies a Panel of Transcripts Associated with Severe Asthma and Lung Cancer

 , ,
, ,  , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.1.1. Microarray Data Selection
2.1.2. Patient Cohort for In Silico Analysis
2.1.3. In Vivo Validation
Ethical Consideration
Formalin-Fixed Paraffin-Embedded Tissue Samples for In Vivo Validation
Blood Samples
Survival Analysis
2.1.4. In Vitro Validation
Cell Culture
2.2. In Silico Analysis
2.2.1. Microarray Data Analysis to Identify Differentially Expressed Genes between Severe Asthmatics and Healthy Controls in Bronchial Epithelium
2.2.2. Gene Set Enrichment Analysis for the Differentially Expressed Pathways among Severe Asthmatics and Healthy Controls
2.2.3. Microarray Data Analysis to Identify Genes Differentially Expressed between NSCLC Patients and Healthy Controls
2.2.4. Gene Set Enrichment Analysis for the Differentially Expressed Pathways among NSCLC Patients and Healthy Controls
2.2.5. In Silico Identification of Intracellular Pathways among Asthmatic and NSCLC Patients in Comparison to Healthy Controls
2.3. Molecular Validation
2.3.1. RNA Extraction
2.3.2. cDNA Synthesis Using Gene-Specific Primer and Random Primer
2.3.3. Quantitative Reverse Transcription PCR (RTq-PCR)
2.4. Statistical Analysis
3. Results
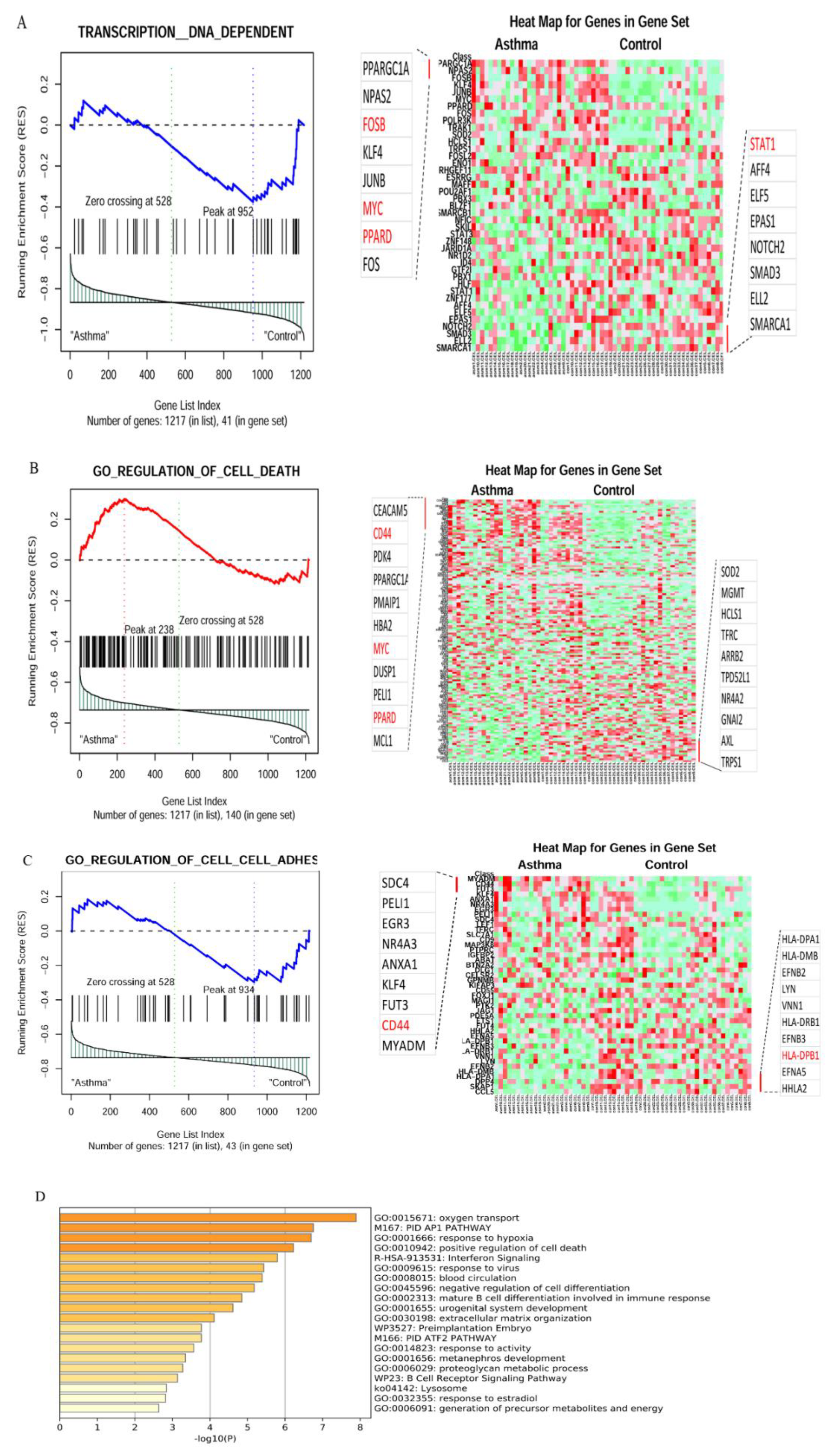
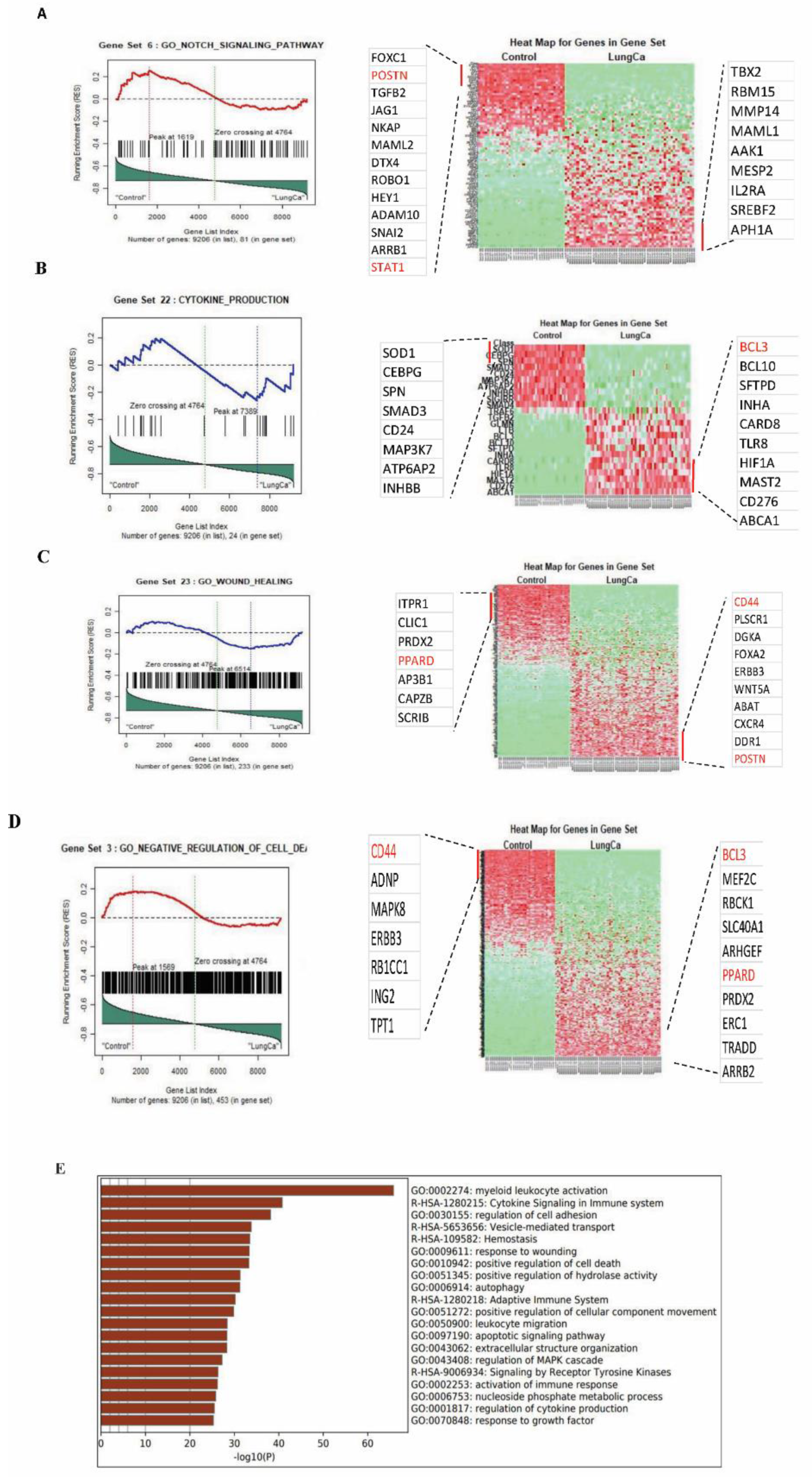
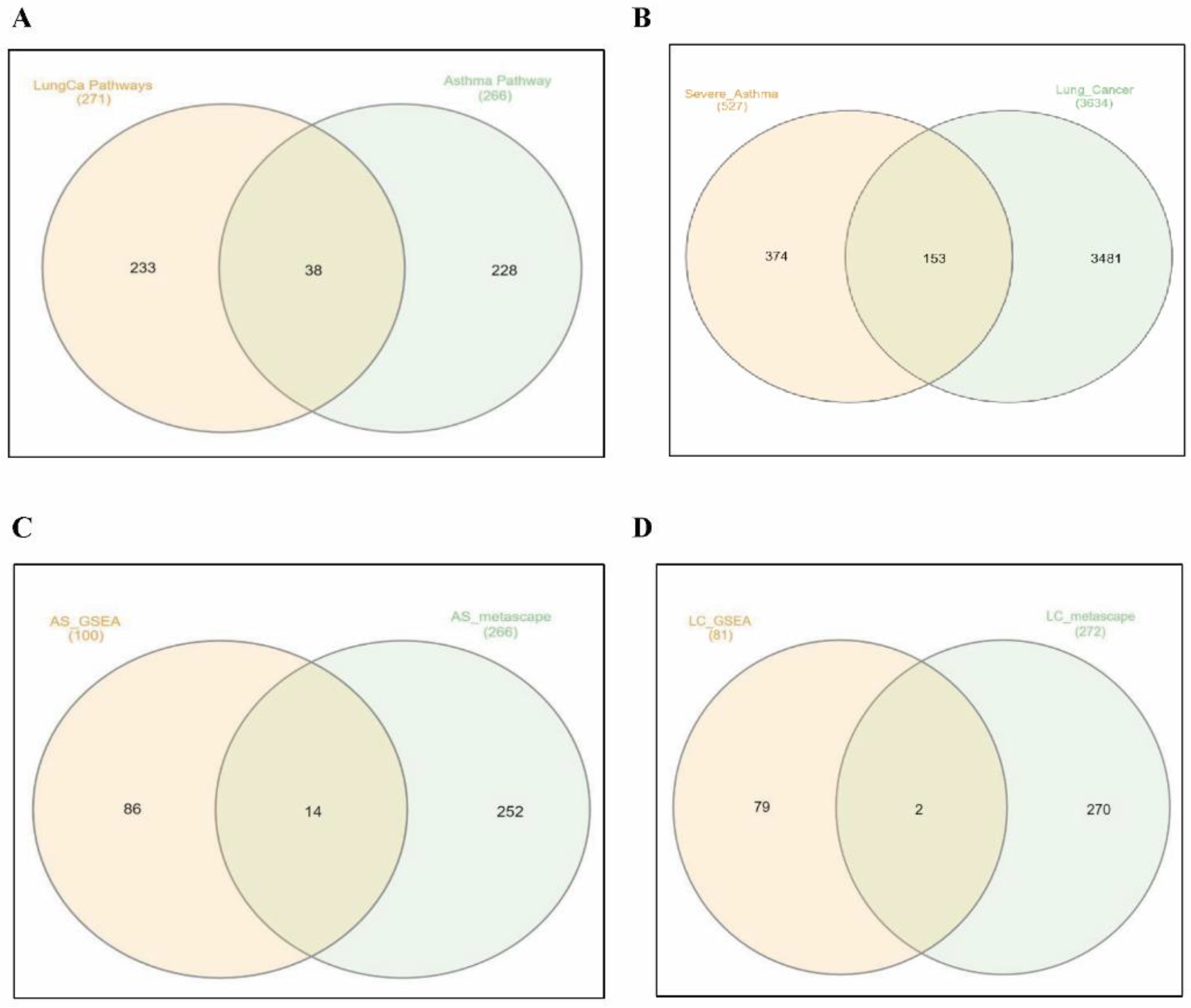
3.1. In Silico Identification of Significant Gene Sets and DEGs between Severe Asthma and Lung Cancer Patients versus Healthy Controls
3.2. In Silico Validation of Differentially Activated Pathways Using Metascape Analysis
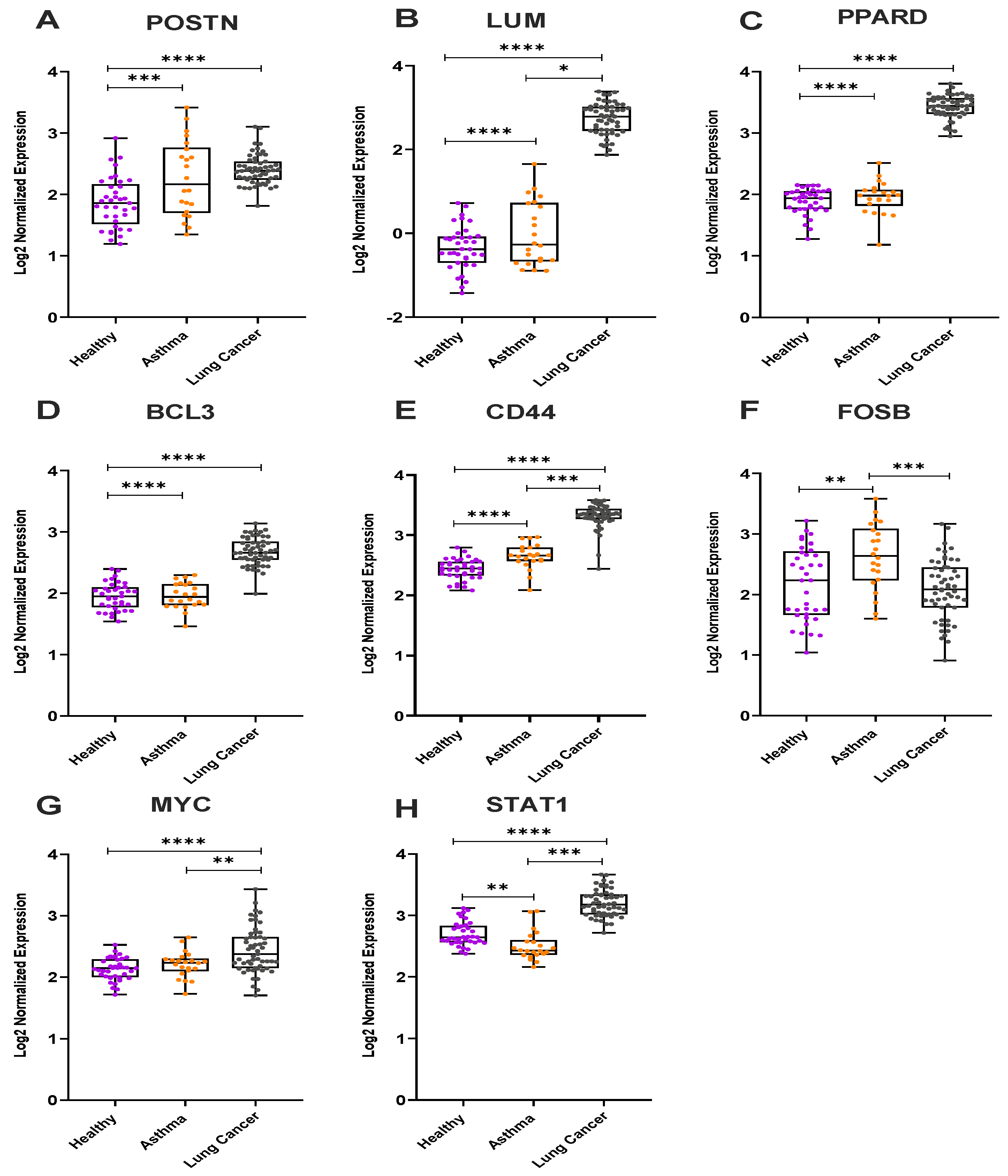
3.3. Gene Expression Analysis from the Microarray Datasets for Severe Asthmatics and Lung Cancer Patients
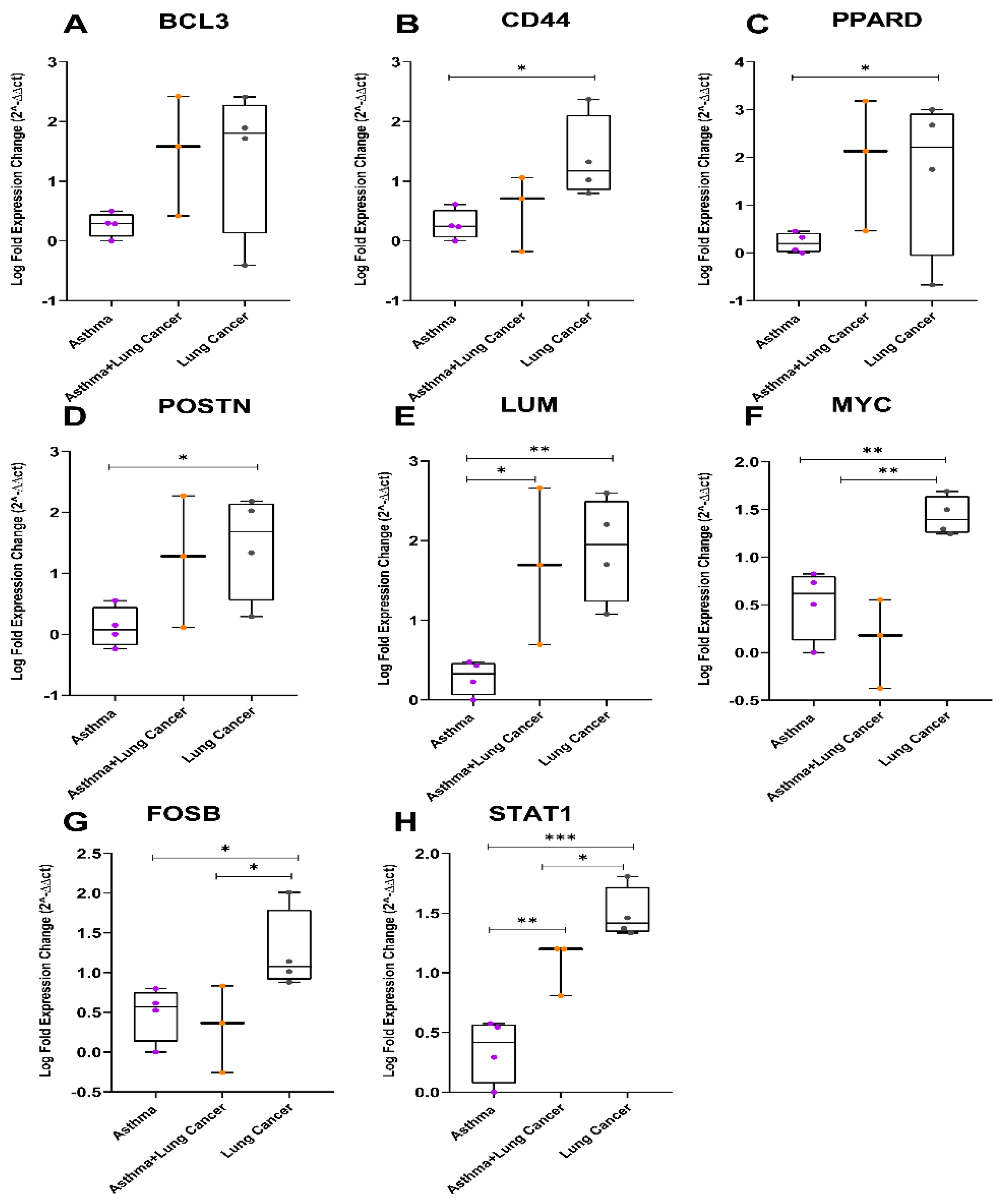
3.4. In Vivo Validation Using Archival Biopsies by RT-qPCR
3.5. Relative Gene Expression of the Eight Genes in Plasma Samples
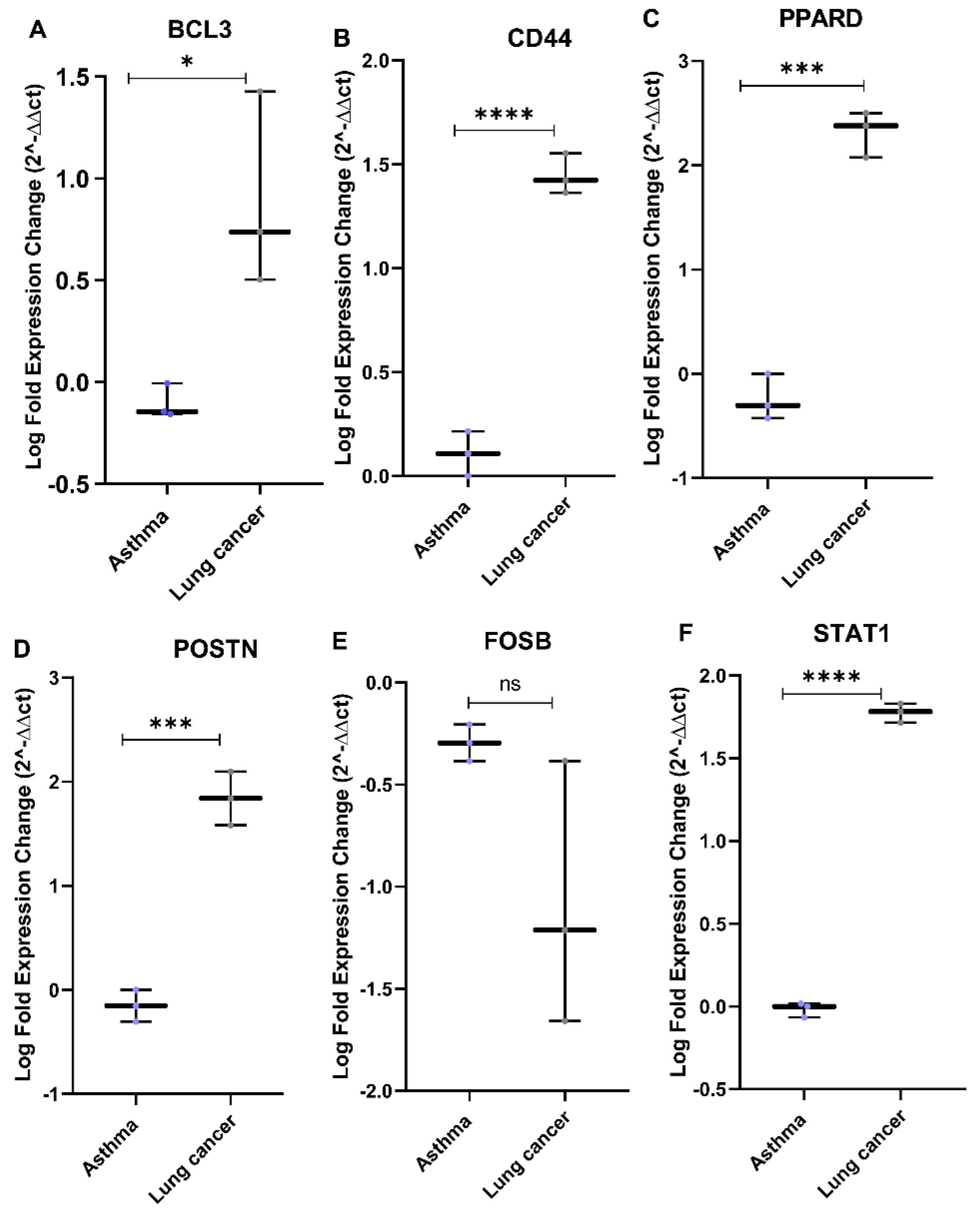
3.6. In Vivo Validation Using Independent NSCLC Patient Cohort
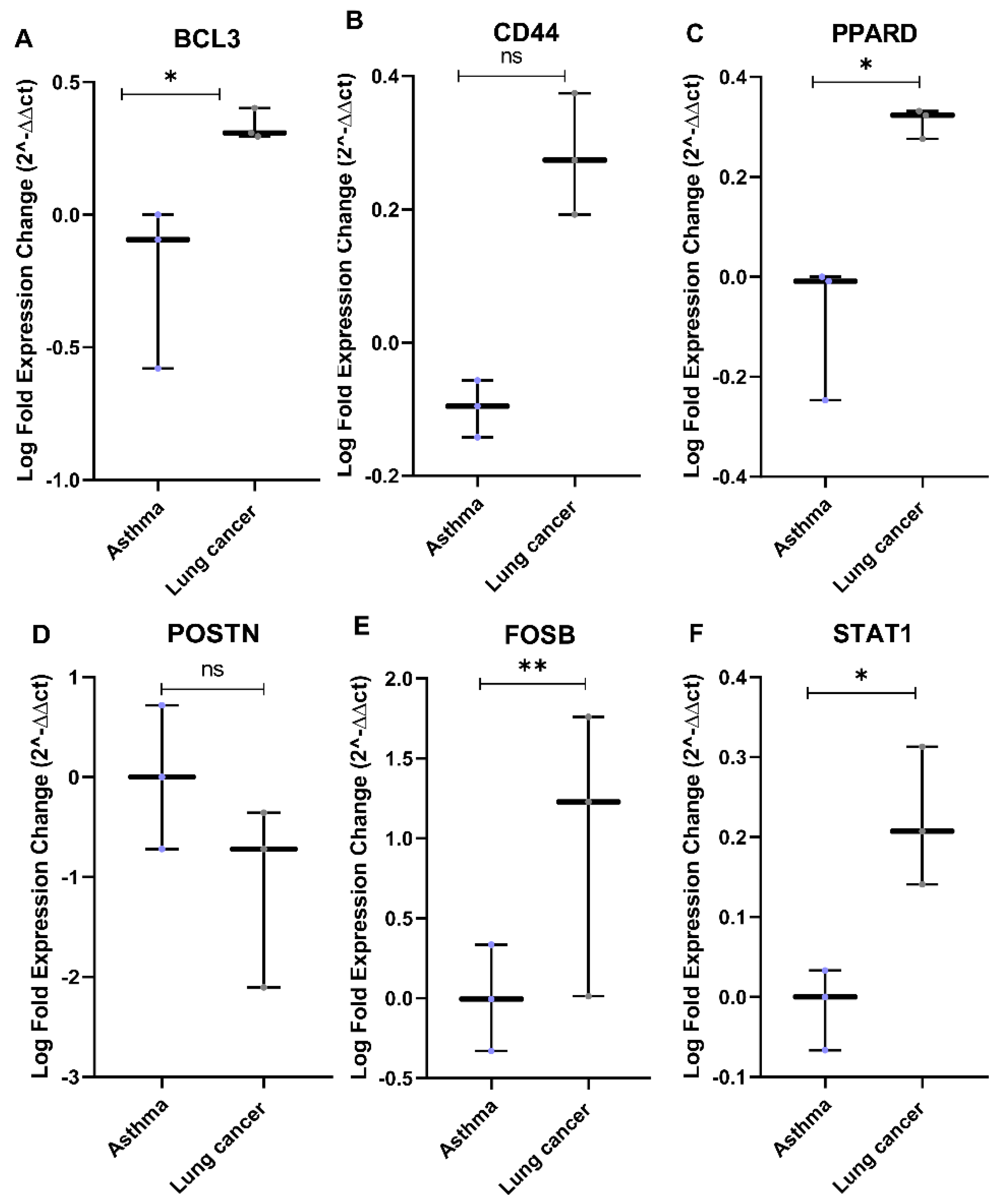
3.7. In Vitro Validation Using Asthmatic and Lung Cancer Cell Lines
4. Discussion
Study Limitations and Justification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FFPE | formalin-fixed paraffin-embedded |
| GINA | The Global Initiative for Asthma |
| GSEA | gene set enrichment analysis |
| NSCLC | non-small-cell lung cancer |
References
- de Sousa, V.M.L.; Carvalho, L. Heterogeneity in Lung Cancer. Pathobiology 2018, 85, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Didkowska, J.; Wojciechowska, U.; Mańczuk, M.; Łobaszewski, J. Lung cancer epidemiology: Contemporary and future challenges worldwide. Ann. Transl. Med. 2016, 4, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagle, P.T.; Allen, T.C.; Olsen, R.J. Lung cancer biomarkers: Present status and future developments. Arch. Pathol. Lab. Med. 2013, 137, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. J. Mol. Diagn. 2018, 20, 129–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindeman, N.I.; Cagle, P.T.; Beasley, M.B.; Chitale, D.A.; Dacic, S.; Giaccone, G.; Jenkins, R.B.; Kwiatkowski, D.J.; Saldivar, J.S.; Squire, J.; et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: Guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J. Mol. Diagn. 2013, 15, 415–453. [Google Scholar] [CrossRef] [Green Version]
- Garantziotis, S.; Schwartz, D.A. Ecogenomics of respiratory diseases of public health significance. Annu. Rev. Public Health 2010, 31, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Izuhara, K.; Ohta, S.; Ono, J. Using Periostin as a Biomarker in the Treatment of Asthma. Allergy Asthma Immunol. Res. 2016, 8, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Woodruff, P.G.; Modrek, B.; Choy, D.F.; Jia, G.; Abbas, A.R.; Ellwanger, A.; Koth, L.L.; Arron, J.R.; Fahy, J.V. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am. J. Respir. Crit. Care Med. 2009, 180, 388–395. [Google Scholar] [CrossRef]
- Vercelli, D. Discovering susceptibility genes for asthma and allergy. Nat. Rev. Immunol. 2008, 8, 169–182. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.; McGranahan, N.; Birkbak, N.; Watkins, T.; Veeriah, S.; Shafi, S.; Johnson, D.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- Gherasim, A.; Dao, A.; Bernstein, J.A. Confounders of severe asthma: Diagnoses to consider when asthma symptoms persist despite optimal therapy. World Allergy Organ. J. 2018, 11, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Lung Screening Trial Research Team; Aberle, D.R.; Adams, A.M.; Berg, C.D.; Black, W.C.; Clapp, J.D.; Fagerstrom, R.M.; Gareen, I.F.; Gatsonis, C.; Marcus, P.M.; et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N. Engl. J. Med. 2011, 365, 395–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.-L.; Liu, J.; Zhang, L.-X.; Wu, C.-M.; Chu, A.-J.; Wen, B.-L.; Ma, C.; Yan, X.-y.; Zhang, X.; Wang, D.-M.; et al. Asthma and the risk of lung cancer: A meta-analysis. Oncotarget 2017, 8, 11614–11620. [Google Scholar] [CrossRef] [Green Version]
- Salameh, L.; Mahboub, B.; Khamis, A.; Alsharhan, M.; Tirmazy, S.H.; Dairi, Y.; Hamid, Q.; Hamoudi, R.; Al Heialy, S. Asthma severity as a contributing factor to cancer incidence: A cohort study. PLoS ONE 2021, 16, e0250430. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.; Doran, P.; MacMathuna, P.; Moss, A.C. In silico gene expression analysis—An overview. Mol. Cancer 2007, 6, 50. [Google Scholar] [CrossRef] [Green Version]
- Hamoudi, R.A.; Appert, A.; Ye, H.; Ruskone-Fourmestraux, A.; Streubel, B.; Chott, A.; Raderer, M.; Gong, L.; Wlodarska, I.; De Wolf-Peeters, C.; et al. Differential expression of NF-κB target genes in MALT lymphoma with and without chromosome translocation: Insights into molecular mechanism. Leukemia 2010, 24, 1487. [Google Scholar] [CrossRef]
- Singhania, A.; Rupani, H.; Jayasekera, N.; Lumb, S.; Hales, P.; Gozzard, N.; Davies, D.E.; Woelk, C.H.; Howarth, P.H. Altered Epithelial Gene Expression in Peripheral Airways of Severe Asthma. PLoS ONE 2017, 12, e0168680. [Google Scholar] [CrossRef]
- Xie, Y.; Xiao, G.; Coombes, K.R.; Behrens, C.; Solis, L.M.; Raso, G.; Girard, L.; Erickson, H.S.; Roth, J.; Heymach, J.V.; et al. Robust gene expression signature from formalin-fixed paraffin-embedded samples predicts prognosis of non-small-cell lung cancer patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 5705–5714. [Google Scholar] [CrossRef] [Green Version]
- Győrffy, B.; Surowiak, P.; Budczies, J.; Lánczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef] [Green Version]
- Botling, J.; Edlund, K.; Lohr, M.; Hellwig, B.; Holmberg, L.; Lambe, M.; Berglund, A.; Ekman, S.; Bergqvist, M.; Pontén, F.; et al. Biomarker discovery in non-small cell lung cancer: Integrating gene expression profiling, meta-analysis, and tissue microarray validation. Clin. Cancer Res. 2013, 19, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, M.; Yamaguchi, R.; Nakata, A.; Kohno, T.; Nagasaki, M.; Shimamura, T.; Imoto, S.; Saito, A.; Ueno, K.; Hatanaka, Y. Epidermal growth factor receptor tyrosine kinase defines critical prognostic genes of stage I lung adenocarcinoma. PLoS ONE 2012, 7, e43923. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.-Q.; Ding, K.; Strumpf, D.; Weir, B.A.; Meyerson, M.; Pennell, N.; Thomas, R.K.; Naoki, K.; Ladd-Acosta, C.; Liu, N. Prognostic and predictive gene signature for adjuvant chemotherapy in resected non–small-cell lung cancer. J. Clin. Oncol. 2010, 28, 4417. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Aerts, J.; Den Hamer, B.; Van Ijcken, W.; Den Bakker, M.; Riegman, P.; van der Leest, C.; van der Spek, P.; Foekens, J.A.; Hoogsteden, H.C. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS ONE 2010, 5, e10312. [Google Scholar] [CrossRef]
- Shedden, K.; Taylor, J.M.; Enkemann, S.A.; Tsao, M.S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Venkatraman, S.E.; Meyerson, M. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study: Director’s Challenge Consortium for the molecular classification of lung adenocarcinoma. Nat. Med. 2008, 14, 822. [Google Scholar]
- Lee, E.-S.; Son, D.-S.; Kim, S.-H.; Lee, J.; Jo, J.; Han, J.; Kim, H.; Lee, H.J.; Choi, H.Y.; Jung, Y. Prediction of recurrence-free survival in postoperative non–small cell lung cancer patients by using an integrated model of clinical information and gene expression. Clin. Cancer Res. 2008, 14, 7397–7404. [Google Scholar] [CrossRef] [Green Version]
- Raponi, M.; Zhang, Y.; Yu, J.; Chen, G.; Lee, G.; Taylor, J.M.; MacDonald, J.; Thomas, D.; Moskaluk, C.; Wang, Y. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res. 2006, 66, 7466–7472. [Google Scholar] [CrossRef] [Green Version]
- Bild, A.H.; Yao, G.; Chang, J.T.; Wang, Q.; Potti, A.; Chasse, D.; Joshi, M.-B.; Harpole, D.; Lancaster, J.M.; Berchuck, A. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2006, 439, 353–357. [Google Scholar] [CrossRef]
- Hung, J.-H.; Yang, T.-H.; Hu, Z.; Weng, Z.; DeLisi, C. Gene set enrichment analysis: Performance evaluation and usage guidelines. Brief. Bioinform. 2012, 13, 281–291. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Ye, H.; Dogan, A.; Ranaldi, R.; Hamoudi, R.A.; Bearzi, I.; Isaacson, P.G.; Du, M.-Q. T(11;18)(q21;q21) is associated with advanced mucosa-associated lymphoid tissue lymphoma that expresses nuclear BCL10. Blood 2001, 98, 1182–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.A.D.; AbdelKarem, O.A.; Irlam-Jones, J.J.; Lane, B.; Valentine, H.; Bibby, B.A.S.; Denley, H.; Choudhury, A.; West, C.M.L. Selection of endogenous control genes for normalising gene expression data derived from formalin-fixed paraffin-embedded tumour tissue. Sci. Rep. 2020, 10, 17258. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, A.E.; Liu, H.; Zhang, Y.; Hamoudi, R.; Kocjan, G.; Du, M.Q. Archival cervical smears: A versatile resource for molecular investigations. Cytopathology 2002, 13, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Caminati, M.; Pham, D.L.; Bagnasco, D.; Canonica, G.W. Type 2 immunity in asthma. World Allergy Organ. J. 2018, 11, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schagen, J.; Deane, A.; Andersen, I.; Kelk, D.; Sly, P.; Fantino, E. Impact of atopy, asthma, and asthma treatment on nasal epithelial wound healing. Authorea Prepr. 2020. [Google Scholar] [CrossRef]
- Rich, H.E.; Antos, D.; Melton, N.R.; Alcorn, J.F.; Manni, M.L. Insights Into Type I and III Interferons in Asthma and Exacerbations. Front. Immunol. 2020, 11, 574027. [Google Scholar] [CrossRef]
- Burgess, J.T.; Rose, M.; Boucher, D.; Plowman, J.; Molloy, C.; Fisher, M.; O’Leary, C.; Richard, D.J.; O’Byrne, K.J.; Bolderson, E. The Therapeutic Potential of DNA Damage Repair Pathways and Genomic Stability in Lung Cancer. Front. Oncol. 2020, 10, 1256. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Parry, E.M.; Gable, D.L.; Stanley, S.E.; Khalil, S.E.; Antonescu, V.; Florea, L.; Armanios, M. Germline mutations in DNA repair genes in lung adenocarcinoma. J. Thorac. Oncol. 2017, 12, 1673–1678. [Google Scholar] [CrossRef] [Green Version]
- Boffetta, P.; Ye, W.; Boman, G.; Nyrén, O. Lung cancer risk in a population-based cohort of patients hospitalized for asthma in Sweden. Eur. Respir. J. 2002, 19, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Hua, X.; Chen, J.; Wu, Y.; Sha, J.; Han, S.; Zhu, X. Prognostic role of the advanced lung cancer inflammation index in cancer patients: A meta-analysis. World J. Surg. Oncol. 2019, 17, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrakopoulos, F.-I.D.; Antonacopoulou, A.G.; Kottorou, A.E.; Panagopoulos, N.; Kalofonou, F.; Sampsonas, F.; Scopa, C.; Kalofonou, M.; Koutras, A.; Makatsoris, T.; et al. Expression Of Intracellular Components of the NF-κB Alternative Pathway (NF-κB2, RelB, NIK and Bcl3) is Associated With Clinical Outcome of NSCLC Patients. Sci. Rep. 2019, 9, 14299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caramori, G.; Casolari, P.; Adcock, I. Role of Transcription Factors in the Pathogenesis of Asthma and COPD. Cell Commun. Adhes. 2013, 20, 21–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Lu, J.; Ji, X.; Ji, Y.; Zhang, Z.; Peng, H.; Sun, F.; Zhang, C. IL-27 suppresses airway inflammation, hyperresponsiveness and remodeling via the STAT1 and STAT3 pathways in mice with allergic asthma. Int. J. Mol. Med. 2020, 46, 641–652. [Google Scholar] [CrossRef]
- Fulkerson, P.C.; Zimmermann, N.; Hassman, L.M.; Finkelman, F.D.; Rothenberg, M.E. Pulmonary chemokine expression is coordinately regulated by STAT1, STAT6, and IFN-gamma. J. Immunol. 2004, 173, 7565–7574. [Google Scholar] [CrossRef]
- Yang, M.; Chen, H.; Zhou, L.; Chen, K.; Su, F. Expression profile and prognostic values of STAT family members in non-small cell lung cancer. Am. J. Transl. Res. 2019, 11, 4866–4880. [Google Scholar]
- Meissl, K.; Macho-Maschler, S.; Müller, M.; Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017, 89, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Baran-Marszak, F.; Feuillard, J.; Najjar, I.; Le Clorennec, C.; Béchet, J.M.; Dusanter-Fourt, I.; Bornkamm, G.W.; Raphaël, M.; Fagard, R. Differential roles of STAT1alpha and STAT1beta in fludarabine-induced cell cycle arrest and apoptosis in human B cells. Blood 2004, 104, 2475–2483. [Google Scholar] [CrossRef]
- Najjar, I.; Schischmanoff, P.; Baran-Marszak, F.; Deglesne, P.-A.; Marfak, I.; Pampin, M.; Feuillard, J.; Bornkamm, G.; Chelbi-Alix, M.; Fagard, R. Novel function of STAT1β in B cells: Induction of cell death by a mechanism different from that of STAT1α. J. Leukoc. Biol. 2008, 84, 1604–1612. [Google Scholar] [CrossRef]
- Tymoszuk, P.; Charoentong, P.; Hackl, H.; Spilka, R.; Müller-Holzner, E.; Trajanoski, Z.; Obrist, P.; Revillion, F.; Peyrat, J.P.; Fiegl, H.; et al. High STAT1 mRNA levels but not its tyrosine phosphorylation are associated with macrophage infiltration and bad prognosis in breast cancer. BMC Cancer 2014, 14, 257. [Google Scholar] [CrossRef] [Green Version]
- Widschwendter, A.; Tonko-Geymayer, S.; Welte, T.; Daxenbichler, G.; Marth, C.; Doppler, W. Prognostic Significance of Signal Transducer and Activator of Transcription 1 Activation in Breast Cancer. Clin. Cancer Res. 2002, 8, 3065–3074. [Google Scholar] [PubMed]
- Brocke-Heidrich, K.; Ge, B.; Cvijic, H.; Pfeifer, G.; Löffler, D.; Henze, C.; McKeithan, T.W.; Horn, F. BCL3 is induced by IL-6 via Stat3 binding to intronic enhancer HS4 and represses its own transcription. Oncogene 2006, 25, 7297–7304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingarelli, B.; Piraino, G.; Hake, P.W.; O’Connor, M.; Denenberg, A.; Fan, H.; Cook, J.A. Peroxisome proliferator-activated receptor {delta} regulates inflammation via NF-{kappa}B signaling in polymicrobial sepsis. Am. J. Pathol. 2010, 177, 1834–1847. [Google Scholar] [CrossRef] [PubMed]
- Kotla, S.; Rao, G.N. Reactive Oxygen Species (ROS) Mediate p300-dependent STAT1 Protein Interaction with Peroxisome Proliferator-activated Receptor (PPAR)-γ in CD36 Protein Expression and Foam Cell Formation. J. Biol. Chem. 2015, 290, 30306–30320. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.-Z.; Wei, X.-W.; Chen, J.-F.; Shi, Y. Overexpression of periostin predicts poor prognosis in non-small cell lung cancer. Oncol. Lett. 2013, 6, 1595–1603. [Google Scholar] [CrossRef]
- González-González, L.; Alonso, J. Periostin: A Matricellular Protein With Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef]
- Morra, L.; Rechsteiner, M.; Casagrande, S.; von Teichman, A.; Schraml, P.; Moch, H.; Soltermann, A. Characterization of periostin isoform pattern in non-small cell lung cancer. Lung Cancer 2012, 76, 183–190. [Google Scholar] [CrossRef]
- Kashyap, M.K.; Marimuthu, A.; Peri, S.; Kumar, G.S.; Jacob, H.K.; Prasad, T.S.; Mahmood, R.; Kumar, K.V.; Kumar, M.V.; Meltzer, S.J.; et al. Overexpression of periostin and lumican in esophageal squamous cell carcinoma. Cancers 2010, 2, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Ballaz, S.; Mulshine, J.L. The Potential Contributions of Chronic Inflammation to Lung Carcinogenesis. Clin. Lung Cancer 2003, 5, 46–62. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Number | GSE64913 | GSE29013 | |
|---|---|---|---|
| Severe Asthmatic (n = 17) | Healthy Control (n = 23) | Lung Cancer (n = 55) | |
| Male | 9 | 14 | 38 |
| Female | 8 | 9 | 17 |
| No. of smokers | 3 | None | 2 |
| Age in years, mean (range) | 41 (20–63) | 26 (19–54) | 63.5 |
| Exacerbations | At least 2 per year | NA | NA |
| NSCLC stage | NA | NA | Stage 1 = 24 Stage 2 = 14 Stage 3 = 17 |
| Clinical Variables | Disease | ||
|---|---|---|---|
| Severe Asthmatic (n = 4) | Asthmatic Patients That Developed Lung Cancer (n = 3) | Lung Cancer (n = 4) | |
| Age in years; mean (range) | 49 (32–61) | 62 (26–83) | 58 (55–91) |
| No. of males; n (%) | 1 (25) | 2 (66.6) | 2 (50) |
| % FEV1; mean (range) | 50.7 (38–61) | 53 (43–64) | NA |
| Reversibility (% FEV1); mean (range) | 16 (12–20) | 21 (18–25) | NA |
| NSCLC Stage | |||
| 1 (%) | NA | 1 (33.3) | |
| 2 (%) | NA | 1 (33.3) | 1 (25) |
| 3 (%) | NA | 1 (33.3) | 1 (25) |
| 4 (%) | NA | 2 (50) | |
| Patient ID | Disease | Gender | Age | FEV1 (/L) |
|---|---|---|---|---|
| AS6 | Asthma | Male | 56 | 1.84 |
| AS14 | Asthma | Female | 57 | 1.33 |
| AS17 | Asthma | Female | 44 | 2.5 |
| LC1 | Lung cancer, stage 3 | Male | 58 | - |
| LC2 | Lung cancer, stage 4 | Male | 63 | - |
| LC3 | Lung cancer, stage 3 | Male | 77 | - |
| Cell ID | Description | Disease | Patient Details Gender, Age, Ethnicity | Catalog Number |
|---|---|---|---|---|
| A549 | Lung epithelial | Lung cancer | Male, 58, Caucasian | C0016002 |
| SK-LU-1 | Lung epithelial | Lung cancer | Female, 46, Caucasian | C0016049 |
| Calu3 | Lung epithelial from metastatic site: pleura | Lung cancer; grade III epidermoid | Male, 25, Caucasian | C0016001 |
| DHBE | Asthmatic epithelial cells | Asthma | Female, 54, Hispanic | 00194911 |
| S13 | Epithelial cells retrieved from severe asthma patient | Severe asthma | Male, 53, East Asian | Isolated from the bronchial biopsy * |
| S14 | Epithelial cells retrieved from severe asthma patient | Severe asthma | Female, 46, East Asian | Isolated from the bronchial biopsy * |
| Gene Sets | Size | Source | ES | NES | NOM p-Value | FDR q-Value | FWER p-Value | Tag % | Gene % | Signal | FDR (Median) | Glob. p-Value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Signal transduction | ||||||||||||
| CELL_CELL_SIGNALING | 22 | GO:0007267 | 0.4642 | 1.5824 | 0.0281 | 0.2633 | 0.3560 | 0.636 | 0.3890 | 0.3960 | 0.0000 | 0.0850 |
| GO_RAS_PROTEIN_SIGNAL_TRANSDUCTION | 17 | GO_RAS_PROTEIN_SIGNAL_TRANSDUCTION | 0.5485 | 1.6490 | 0.0123 | 0.0847 | 0.0670 | 0.529 | 0.3210 | 0.3640 | 0.0000 | 0.0580 |
| GO_GTPASE_REGULATOR_ACTIVITY | 20 | GO_GTPASE_REGULATOR_ACTIVITY | 0.4371 | 1.5238 | 0.0171 | 0.2652 | 0.5940 | 0.65 | 0.4600 | 0.3570 | 0.1522 | 0.0530 |
| POSITIVE_REGULATION_ OF_CELL_ DEATH | 22 | GO Biological Processes | 0.8167 | 1.7937 | 0.0000 | 0.0162 | 0.0239 | 0.682 | 0.1920 | 0.5610 | 0.0000 | 0.0060 |
| GO_NEGATIVE_REGULATION_OF_CELL_ DEATH | 90 | GO_NEGATIVE_REGULATION_OF_CELL_DEATH | 0.3999 | 1.5361 | 0.0381 | 0.3468 | 0.8680 | 0.533 | 0.4350 | 0.3260 | 0.2031 | 0.0730 |
| Regulation of cell-to-cell adhesion | ||||||||||||
| GO_REGULATION_OF_CELL_CELL_ADHESION | 43 | GO_REGULATION_OF_CELL_CELL_ADHESION | 0.4729 | 1.5837 | 0.0236 | 0.1378 | 0.1082 | 0.535 | 0.3820 | 0.3430 | 0.0000 | 0.0860 |
| GO_POSITIVE_REGULATION_OF_CELL_ADHESION | 41 | GO_POSITIVE_REGULATION_OF_CELL_ADHESION | 0.5554 | 1.9064 | 0.0000 | 0.2381 | 0.1430 | 0.439 | 0.2050 | 0.3610 | 0.0000 | 0.0800 |
| GO_REGULATION_OF_CELL_SUBSTRATE_ADHESION | 26 | GO_REGULATION_OF_CELL_SUBSTRATE_ADHESION | 0.5074 | 1.7722 | 0.0021 | 0.2739 | 0.4180 | 0.308 | 0.1310 | 0.2730 | 0.0000 | 0.0700 |
| GO_BIOLOGICAL_ADHESION | 134 | GO_BIOLOGICAL_ADHESION | 0.3695 | 1.6123 | 0.0022 | 0.3383 | 0.7640 | 0.44 | 0.3840 | 0.3050 | 0.1880 | 0.0710 |
| GO_CELL_CELL_ADHESION | 77 | GO_CELL_CELL_ADHESION | 0.4418 | 1.7117 | 0.0067 | 0.3622 | 0.5740 | 0.506 | 0.3820 | 0.3340 | 0.1444 | 0.0840 |
| Transcription and protein modification | ||||||||||||
| TRANSCRIPTION | 46 | GO:0006350 | 0.4410 | 1.5196 | 0.0365 | 0.1963 | 0.4560 | 0.5 | 0.3590 | 0.3330 | 0.0000 | 0.0340 |
| TRANSCRIPTION__DNA_DEPENDENT | 41 | GO:0006351 | 0.4573 | 1.5451 | 0.0340 | 0.1761 | 0.4210 | 0.537 | 0.3590 | 0.3560 | 0.0000 | 0.0270 |
| GO_RNA_SPLICING | 21 | GO_RNA_SPLICING | 0.4809 | 1.5485 | 0.0478 | 0.3423 | 0.8530 | 0.476 | 0.2770 | 0.3500 | 0.2007 | 0.0730 |
| Miscellaneous | ||||||||||||
| GO_HUMORAL_IMMUNE_RESPONSE | 16 | GO_HUMORAL_IMMUNE_RESPONSE | 0.5810 | 1.5893 | 0.0366 | 0.3531 | 0.7910 | 0.5 | 0.3340 | 0.3370 | 0.1959 | 0.0790 |
| GO_HORMONE_TRANSPORT | 23 | GO_HORMONE_TRANSPORT | 0.4065 | 1.4568 | 0.0387 | 0.3800 | 0.9210 | 0.304 | 0.1950 | 0.2500 | 0.2441 | 0.0790 |
| GO_GLYCOSAMINOGLYCAN_BINDING | 18 | GO_GLYCOSAMINOGLYCAN_BINDING | 0.6212 | 1.8600 | 0.0000 | 0.1535 | 0.0500 | 0.333 | 0.0970 | 0.3060 | 0.0000 | 0.0500 |
| Gene Sets | Size | Source | ES | NES | NOM p-Value | FDR q-Value | FWER p-Value | Tag % | Gene % | Signal | FDR (Median) | Glob. p-Value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Signal transduction | ||||||||||||
| GO_NOTCH_SIGNALING_PATHWAY | 81 | GO_NOTCH_SIGNALING_PATHWAY | 0.3330 | 1.5135 | 0.0289 | 0.8450 | 0.2560 | 0.3210 | 0.2590 | 0.2400 | 0.0000 | 0.2540 |
| REGULATION_OF_GENE_EXPRESSION | 351 | GO:0010468 | 0.2664 | 1.4706 | 0.0111 | 0.2621 | 0.8460 | 0.5160 | 0.5030 | 0.2670 | 0.1746 | 0.0270 |
| SECRETORY_PATHWAY | 48 | GO:0045045 | 0.4318 | 1.6364 | 0.0163 | 0.3425 | 0.5770 | 0.3960 | 0.2740 | 0.2890 | 0.1337 | 0.1000 |
| NEGATIVE_REGULATION_OF_APOPTOSIS | 89 | GO:0043066 | 0.3283 | 1.5110 | 0.0323 | 0.2598 | 0.7990 | 0.2920 | 0.2340 | 0.2260 | 0.1580 | 0.0310 |
| NEGATIVE_REGULATION_OF_PROGRAMMED_CELL_DEATH | 90 | GO:0043069 | 0.3255 | 1.5061 | 0.0340 | 0.2575 | 0.8020 | 0.2220 | 0.1370 | 0.1940 | 0.1548 | 0.0290 |
| Tissue and structure morphogenesis | ||||||||||||
| STRUCTURAL_CONSTITUENT_OF_RIBOSOME | 31 | GO:0003735 | 0.5982 | 1.7802 | 0.0119 | 0.4095 | 0.2390 | 0.5810 | 0.1530 | 0.4940 | 0.0000 | 0.1600 |
| ORGAN_MORPHOGENESIS | 54 | GO:0009887 | 0.3809 | 1.4301 | 0.0383 | 0.2742 | 0.8840 | 0.4630 | 0.3450 | 0.3050 | 0.2011 | 0.0210 |
| ORGAN_DEVELOPMENT | 224 | GO:0048513 | 0.3165 | 1.3987 | 0.0383 | 0.2833 | 0.9160 | 0.4380 | 0.3870 | 0.2750 | 0.2173 | 0.0220 |
| Transcription and protein modification | ||||||||||||
| PROTEIN_CATABOLIC_PROCESS | 35 | GO:0030163 | 0.4445 | 1.8389 | 0.0021 | 0.4918 | 0.2090 | 0.5140 | 0.3060 | 0.3580 | 0.0000 | 0.1470 |
| CELLULAR_PROTEIN_CATABOLIC_PROCESS | 32 | GO:0044257 | 0.4190 | 1.7027 | 0.0040 | 0.8381 | 0.4340 | 0.5000 | 0.3060 | 0.3480 | 0.0000 | 0.2620 |
| PROTEIN_RNA_COMPLEX_ASSEMBLY | 35 | GO:0022618 | 0.4196 | 1.6975 | 0.0085 | 0.7046 | 0.4440 | 0.5710 | 0.3360 | 0.3810 | 0.0000 | 0.2330 |
| Miscellaneous | ||||||||||||
| RESPONSE_TO_STRESS | 252 | GO:0006950 | 0.2780 | 1.4447 | 0.0439 | 0.2658 | 0.8740 | 0.4290 | 0.4290 | 0.2520 | 0.1908 | 0.0210 |
| DNA_REPAIR | 70 | GO:0006281 | 0.3601 | 1.5256 | 0.0493 | 0.2525 | 0.7750 | 0.4430 | 0.3640 | 0.2840 | 0.1428 | 0.0320 |
| CYTOKINE_PRODUCTION | 24 | GO:0001816 | 0.4521 | 1.7395 | 0.0103 | 0.6888 | 0.4040 | 0.7920 | 0.4500 | 0.4360 | 0.0000 | 0.2270 |
| Pathway Description | List of the Genes Involved |
|---|---|
| Regulation of cell adhesion | ANXA1, CD44, EGR3, FUT3, CCN1, S100A10, CXCR4, NR4A3, POSTN, MYADM, CCDC80, S100A8, FCGR2A, HBB, JUN |
| Response to activity | CXCR4, POSTN, PPARGC1A, G0S2, FOSB, PDK4, NR4A3, PMAIP1 |
| Embryonic placenta development | CCN1, KRT8, SOCS3, TM4SF1, PSPH, ANXA1, SERPINB5, NR4A3 |
| Extracellular matrix organization | CD44, CCN1, SERPINB5, POSTN, CCDC80, CXCR4, NR4A3, SOCS3 |
| Cell morphogenesis involved in differentiation | EGR2, S100A10, CXCR4, NR4A3, POSTN, MYADM, DPYSL3, CD44 |
| Interferon signaling | CD44, IFIT2, SOCS3 |
| Epithelial cell development | CXCR4, TFCP2L1, MYADM |
| Pathway Description | Example of Genes Involved |
|---|---|
| Signaling by receptor tyrosine kinases | DUSP6, EGFR, EGR3, ELK1, FGFR1, FN1, FYN, GRB2, GRB10, ID2, IGF1R, IRSA6A, JCAD, SNX6, PLEK |
| Signaling by Rho GTPases | BRCA1, PPP2R2A, PPP6C, TYMS, NSD2, WRN, ALMS1, CDC7, RAE1, CDC23, CCNE2, PTTG1, KIF23, ESPL1, RAB1B, CEP78, NEDD1 |
| Blood-vessel development | STAT1, EGFR, PPARD, PCDC73, RAB33B, EPPK1, FOXP1, ANLN, CORO1B, PLEKHG5, EPB41L5, ARID5B, SYDE1, CYGB, DNMT1, NFATC1 |
| Regulation of cell projection organization | FN1, FYN, GAK, GATA3, MYC, TGFBR1, BCL11A, AMIGO2, ACTA2, NOS1, SERPINF2, MP14, MAP2K5, RPS6KA1, SLC9A1, SPHK1, PPM1F, ADNP2, EXOSC2 |
| Response to growth factor | EGFR, EGR3, FRP4, SHC1, SPHK1, HGS, NREP, USP15, LUM, POSTN, EHD1, FERMT2, TBC1D7, WWOX, ERRFI1, IL17RD, FAM83G |
| Extracellular matrix organization | BCL3, BGN, BMP1, BSG, CAPN1, CAV1, CD36, IGFBP4, MATN2, THBS2, TNFAIP6, SRPX, CILP, EDIL3, SPON2, SPON1, MXRA5, TSKU, CRIM1, CTHRC1, EMID1 |
| Response to growth factor | COL4A2, CREBBP, DAB2, DCN, DTYMK, DUSP6, E2F1, EGFR, EGR3, ERN1, FBN1, FGFR1, LUM, POSTN, EHD1, FERMT2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salameh, L.; Bhamidimarri, P.M.; Saheb Sharif-Askari, N.; Dairi, Y.; Hammoudeh, S.M.; Mahdami, A.; Alsharhan, M.; Tirmazy, S.H.; Rawat, S.S.; Busch, H.; et al. In Silico Bioinformatics Followed by Molecular Validation Using Archival FFPE Tissue Biopsies Identifies a Panel of Transcripts Associated with Severe Asthma and Lung Cancer. Cancers 2022, 14, 1663. https://doi.org/10.3390/cancers14071663
Salameh L, Bhamidimarri PM, Saheb Sharif-Askari N, Dairi Y, Hammoudeh SM, Mahdami A, Alsharhan M, Tirmazy SH, Rawat SS, Busch H, et al. In Silico Bioinformatics Followed by Molecular Validation Using Archival FFPE Tissue Biopsies Identifies a Panel of Transcripts Associated with Severe Asthma and Lung Cancer. Cancers. 2022; 14(7):1663. https://doi.org/10.3390/cancers14071663
Chicago/Turabian StyleSalameh, Laila, Poorna Manasa Bhamidimarri, Narjes Saheb Sharif-Askari, Youssef Dairi, Sarah Musa Hammoudeh, Amena Mahdami, Mouza Alsharhan, Syed Hammad Tirmazy, Surendra Singh Rawat, Hauke Busch, and et al. 2022. "In Silico Bioinformatics Followed by Molecular Validation Using Archival FFPE Tissue Biopsies Identifies a Panel of Transcripts Associated with Severe Asthma and Lung Cancer" Cancers 14, no. 7: 1663. https://doi.org/10.3390/cancers14071663
APA StyleSalameh, L., Bhamidimarri, P. M., Saheb Sharif-Askari, N., Dairi, Y., Hammoudeh, S. M., Mahdami, A., Alsharhan, M., Tirmazy, S. H., Rawat, S. S., Busch, H., Hamid, Q., Al Heialy, S., Hamoudi, R., & Mahboub, B. (2022). In Silico Bioinformatics Followed by Molecular Validation Using Archival FFPE Tissue Biopsies Identifies a Panel of Transcripts Associated with Severe Asthma and Lung Cancer. Cancers, 14(7), 1663. https://doi.org/10.3390/cancers14071663