
Converged DNA Damage Response Renders Human Hepatocellular Carcinoma Sensitive to CDK7 Inhibition


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Assay
2.3. Western Blot Analysis
2.4. Flow Cytometric Analysis of Cell Cycle
2.5. Flow Cytometry Apoptosis Assay
2.6. Exogenous Overexpression of MYC in the HepG2 Cells
2.7. Immunohistochemical Analysis
2.8. In Vivo Xenograft Tumor Assays
2.9. Statistical Analysis
3. Results
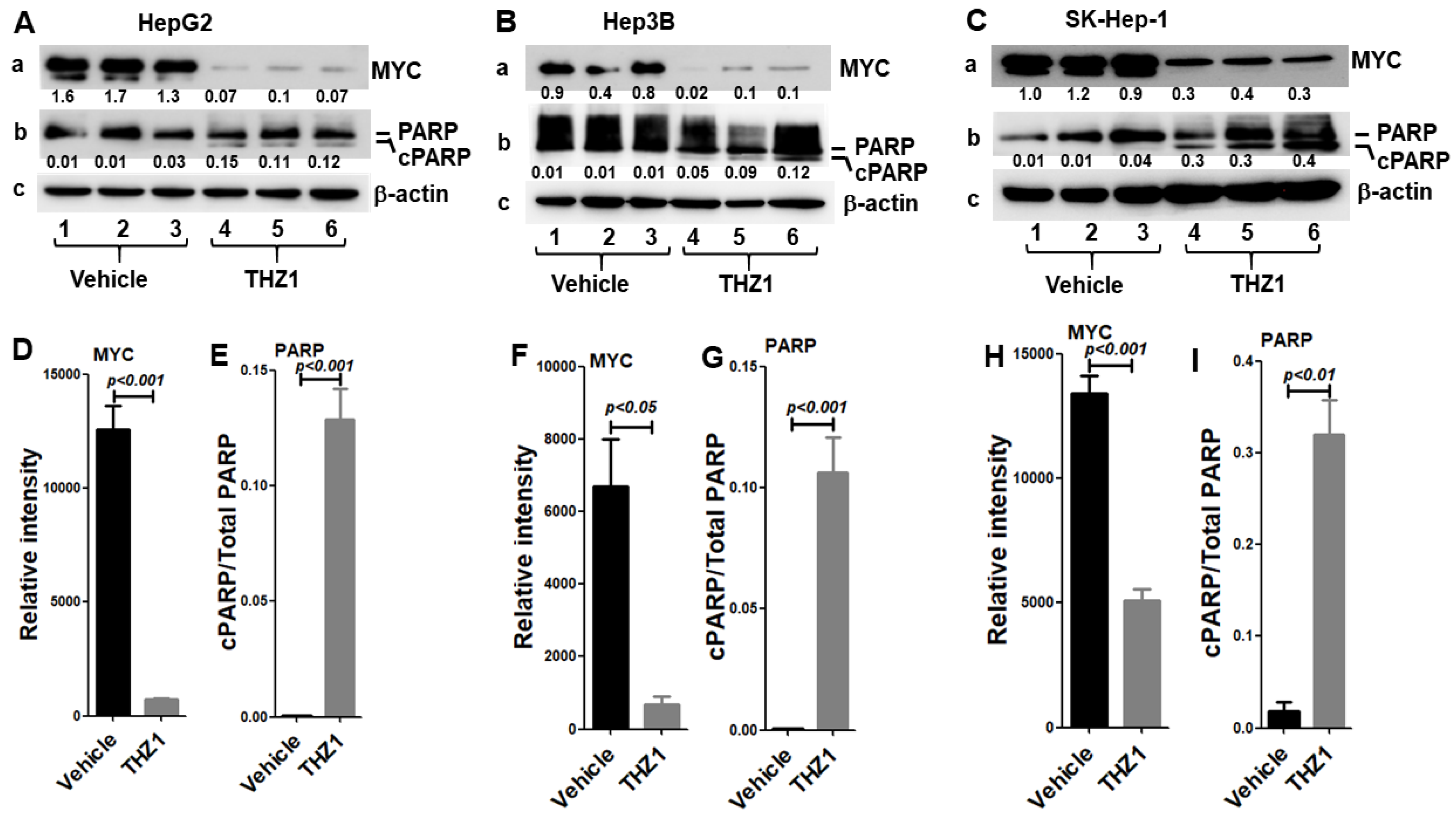
3.1. HCC Cells Are Highly Sensitive to THZ1 Treatment
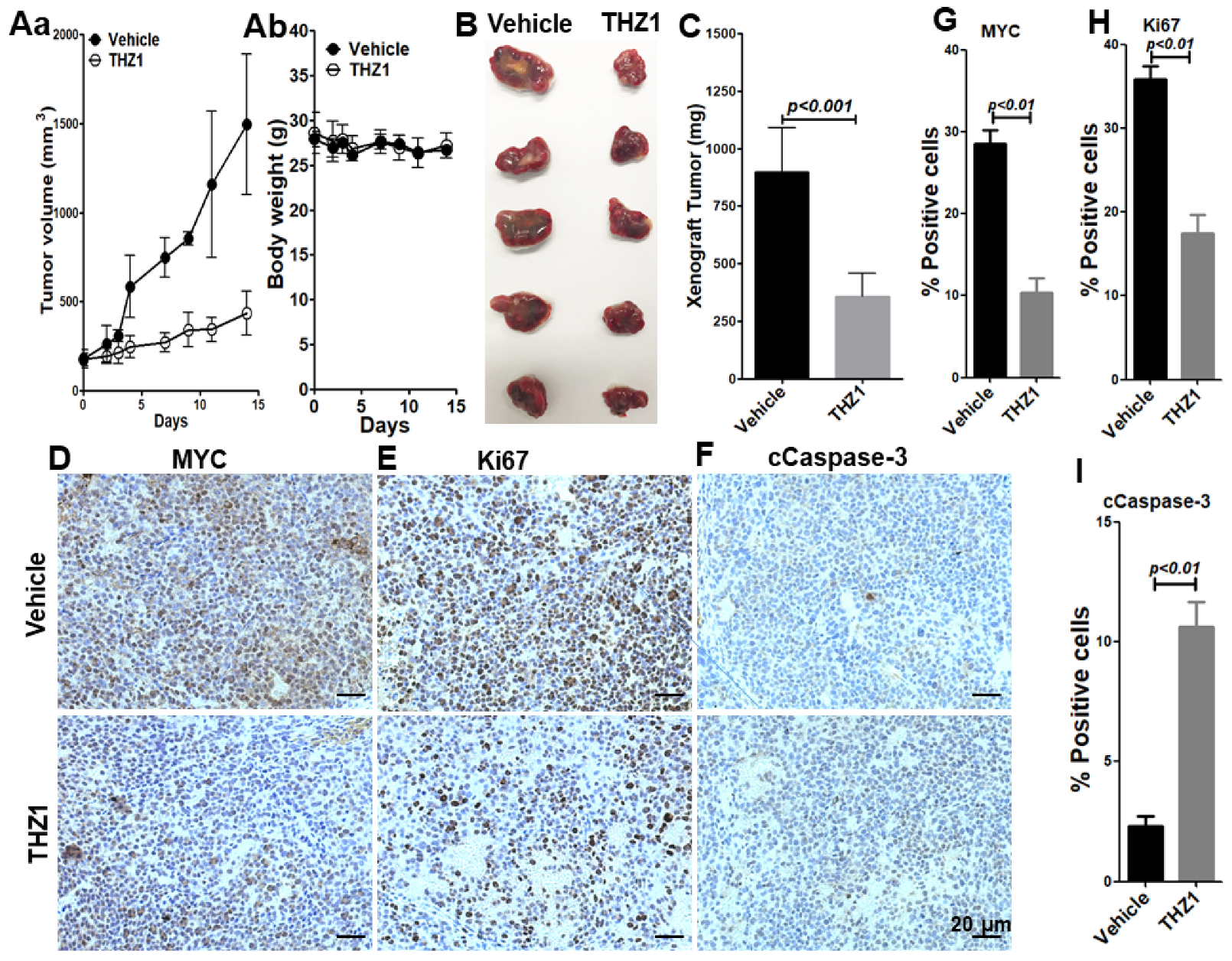
3.2. THZ1 Exhibits Antitumor Activity in Mouse Model
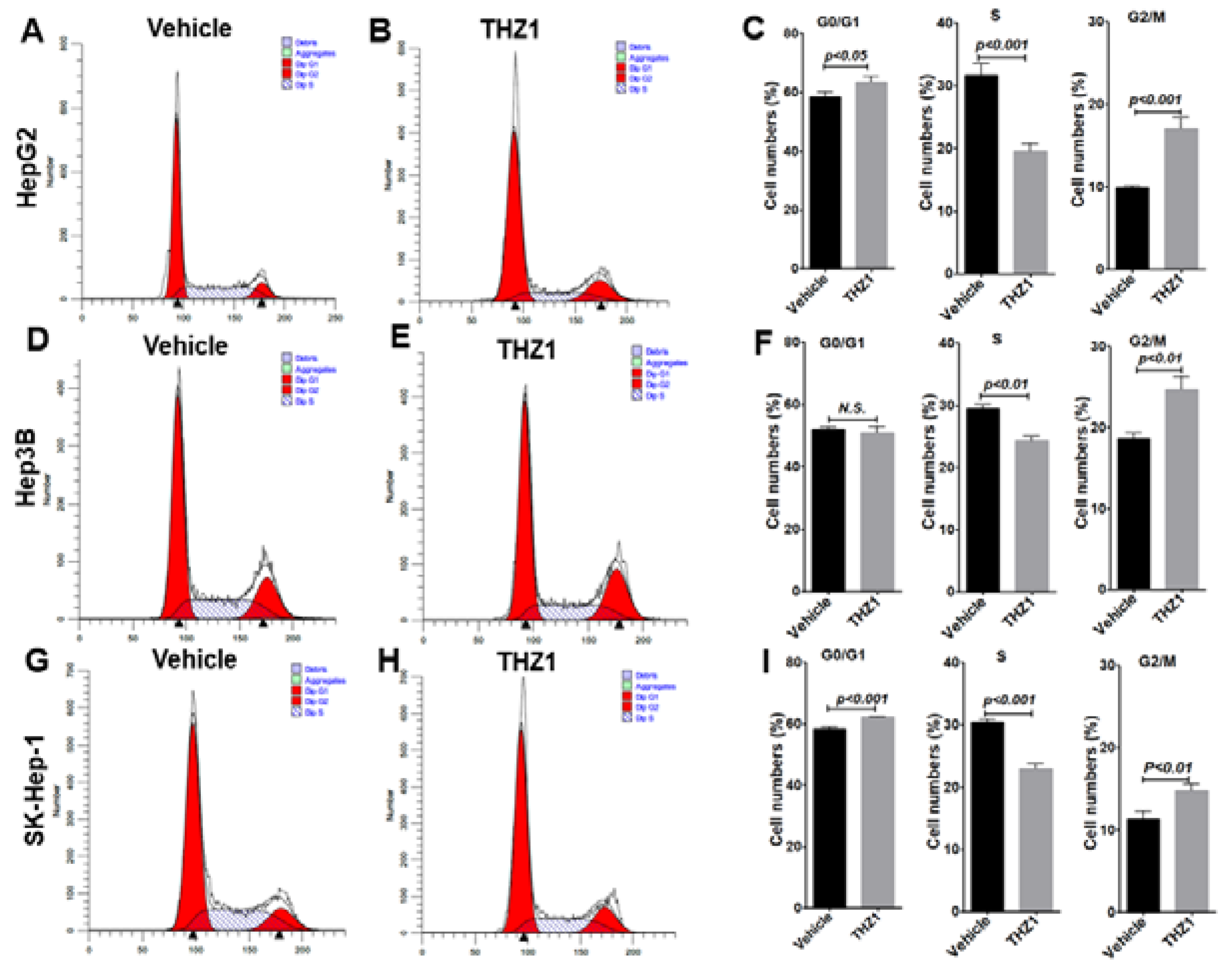
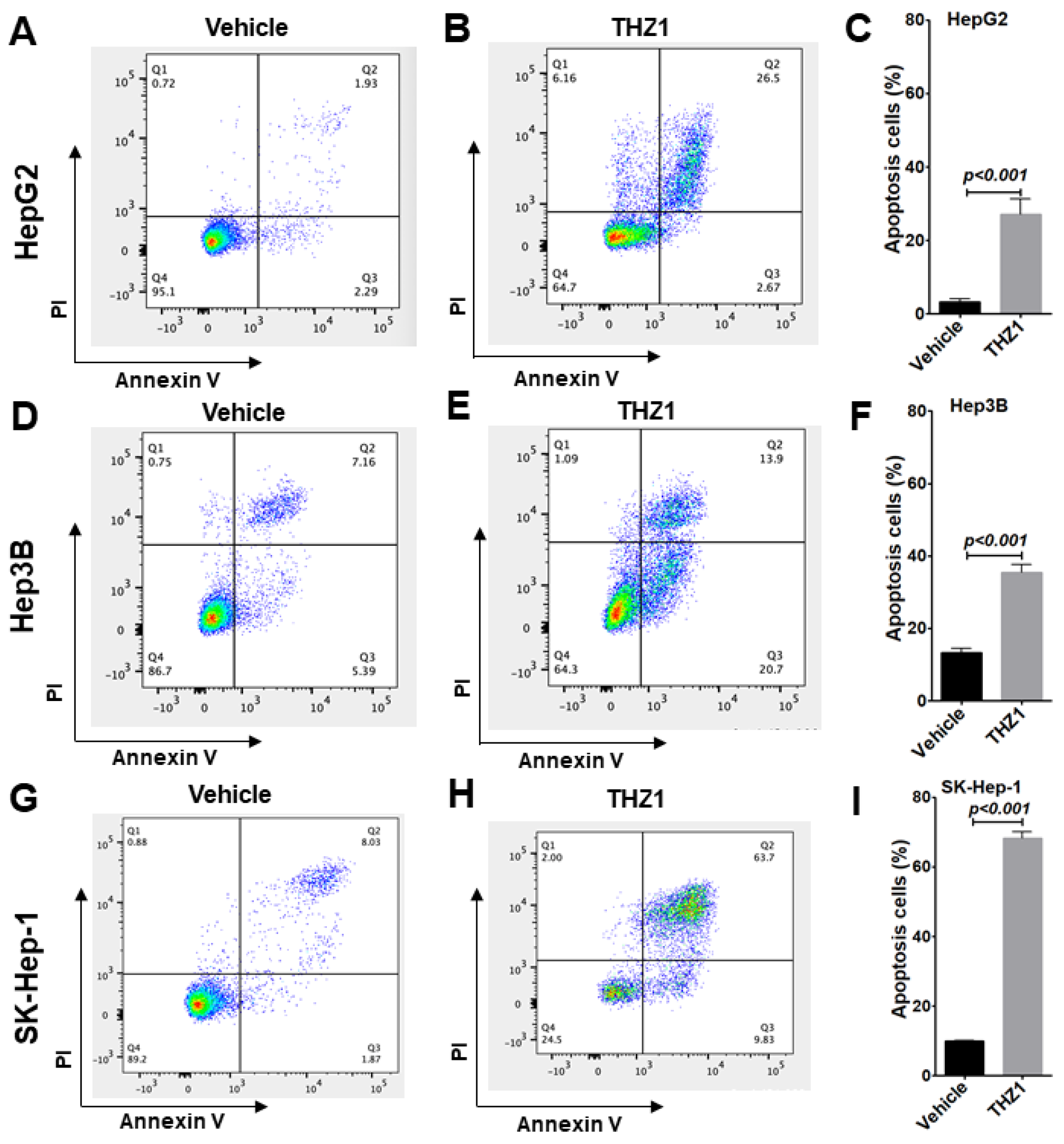
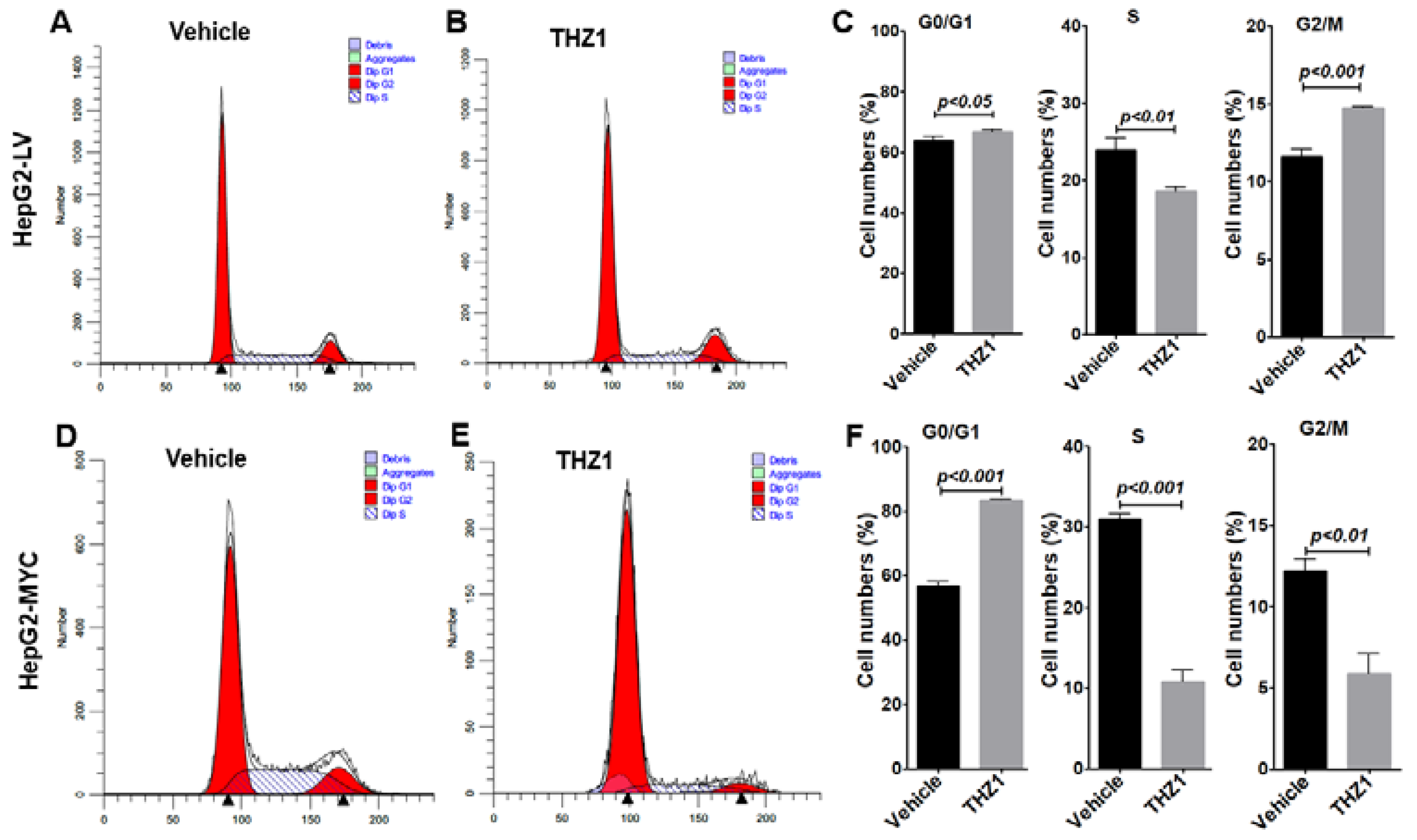
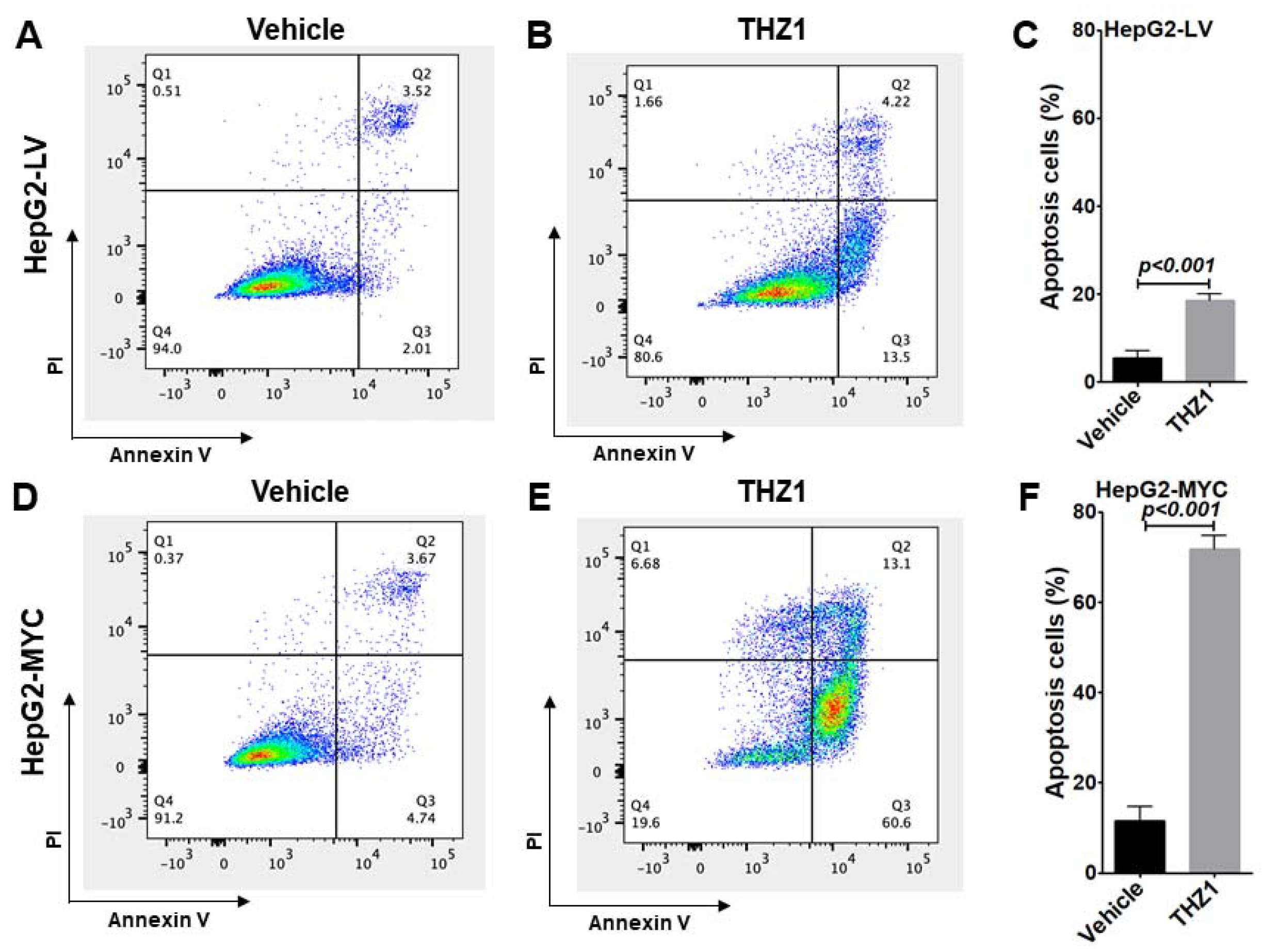
3.3. THZ1 Inhibits Cell Proliferation via Cell Cycle Arrest and Apoptosis Induction
3.4. Overexpressed MYC Promotes Cell Cycle Progression
3.5. Overexpressed MYC Renders Tumor Cells Hypersensitive to THZ1 Treatment
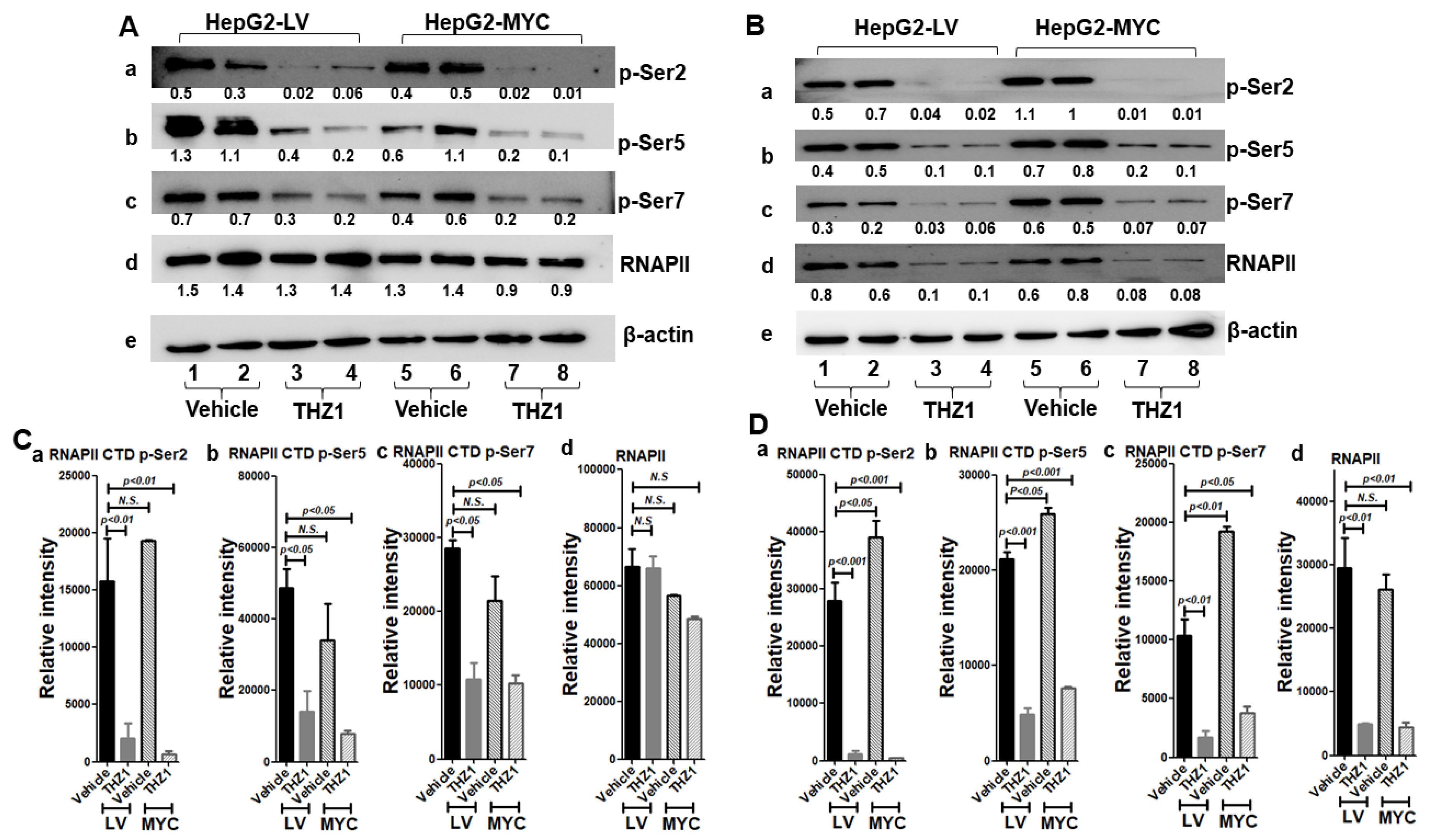
3.6. THZ1 Reduces RNAP II Phosphorylation
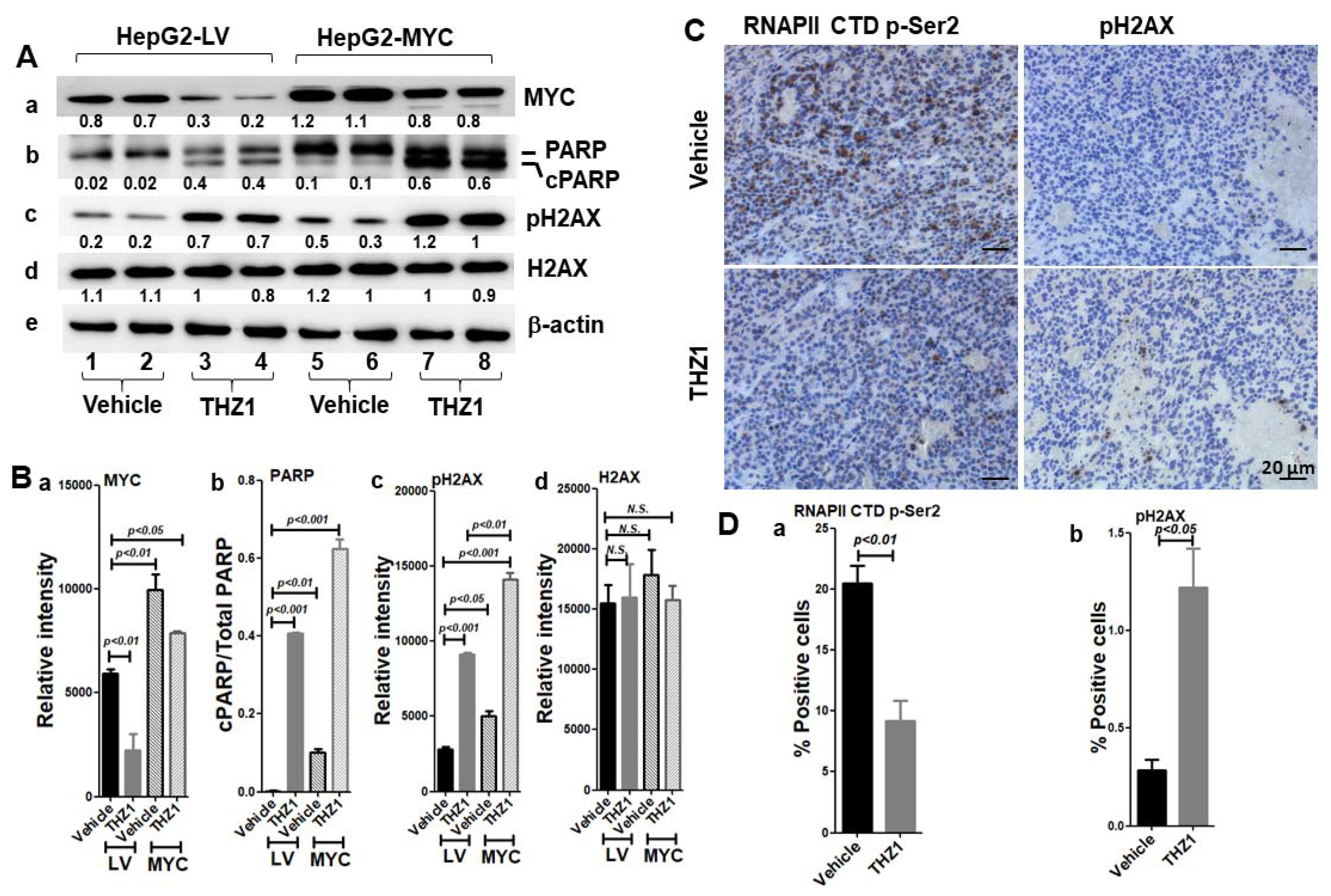
3.7. MYC-Conferred Increased DNA Damage Response to THZ1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; Artaman, A.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Boix, L.; Sala, M.; Llovet, J.M. Focus on hepatocellular carcinoma. Cancer Cell 2004, 5, 215–219. [Google Scholar] [CrossRef] [Green Version]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Karaosmanoglu, O.; Banerjee, S.; Sivas, H. Identification of biomarkers associated with partial epithelial to mesenchymal transition in the secretome of slug over-expressing hepatocellular carcinoma cells. Cell Oncol. 2018, 41, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Granito, A.; Forgione, A.; Marinelli, S.; Renzulli, M.; Ielasi, L.; Sansone, V.; Benevento, F.; Piscaglia, F.; Tovoli, F. Experience with regorafenib in the treatment of hepatocellular carcinoma. Ther. Adv. Gastroenterol. 2021, 14, 17562848211016959. [Google Scholar] [CrossRef]
- Granito, A.; Muratori, L.; Lalanne, C.; Quarneti, C.; Ferri, S.; Guidi, M.; Lenzi, M.; Muratori, P. Hepatocellular carcinoma in viral and autoimmune liver diseases: Role of CD4+ CD25+ Foxp3+ regulatory T cells in the immune microenvironment. World J. Gastroenterol. 2021, 27, 2994–3009. [Google Scholar] [CrossRef]
- Rizzo, A.; Brandi, G. Biochemical predictors of response to immune checkpoint inhibitors in unresectable hepatocellular carcinoma. Cancer Treat. Res. Commun. 2021, 27, 100328. [Google Scholar] [CrossRef]
- Rizzo, A.; Dadduzio, V.; Ricci, A.D.; Massari, F.; Di Federico, A.; Gadaleta-Caldarola, G.; Brandi, G. Lenvatinib plus pembrolizumab: The next frontier for the treatment of hepatocellular carcinoma? Expert Opin. Investig. Drugs 2021, 1–8. [Google Scholar] [CrossRef]
- Schlaeger, C.; Longerich, T.; Schiller, C.; Bewerunge, P.; Mehrabi, A.; Toedt, G.; Kleeff, J.; Ehemann, V.; Eils, R.; Lichter, P.; et al. Etiology-dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology 2008, 47, 511–520. [Google Scholar] [CrossRef]
- Zondervan, P.E.; Wink, J.; Alers, J.C.; JN, I.J.; Schalm, S.W.; de Man, R.A.; van Dekken, H. Molecular cytogenetic evaluation of virus-associated and non-viral hepatocellular carcinoma: Analysis of 26 carcinomas and 12 concurrent dysplasias. J. Pathol. 2000, 192, 207–215. [Google Scholar] [CrossRef]
- Kaposi-Novak, P.; Libbrecht, L.; Woo, H.G.; Lee, Y.H.; Sears, N.C.; Coulouarn, C.; Conner, E.A.; Factor, V.M.; Roskams, T.; Thorgeirsson, S.S. Central role of c-Myc during malignant conversion in human hepatocarcinogenesis. Cancer Res. 2009, 69, 2775–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, H.L.; Kraus, W.L. No driver behind the wheel? Targeting transcription in cancer. Cell 2015, 163, 28–30. [Google Scholar] [CrossRef] [Green Version]
- Augert, A.; MacPherson, D. Treating transcriptional addiction in small cell lung cancer. Cancer Cell 2014, 26, 783–784. [Google Scholar] [CrossRef] [Green Version]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Geng, J.; Zhang, L.; Wang, Y.; Niu, N.; Fang, Y.; Liu, F.; Shi, J.; Zhang, Z.G.; Sun, Y.W.; et al. THZ1 reveals CDK7-dependent transcriptional addictions in pancreatic cancer. Oncogene 2019, 38, 3932–3945. [Google Scholar] [CrossRef]
- Miller, D.M.; Flaherty, K.T. Cyclin-dependent kinases as therapeutic targets in melanoma. Pigment Cell Melanoma Res. 2014, 27, 351–365. [Google Scholar] [CrossRef] [Green Version]
- Tsang, F.H.; Law, C.T.; Tang, T.C.; Cheng, C.L.; Chin, D.W.; Tam, W.V.; Wei, L.; Wong, C.C.; Ng, I.O.; Wong, C.M. Aberrant Super-Enhancer Landscape in Human Hepatocellular Carcinoma. Hepatology 2019, 69, 2502–2517. [Google Scholar] [CrossRef]
- Zhong, L.; Yang, S.; Jia, Y.; Lei, K. Inhibition of cyclin-dependent kinase 7 suppresses human hepatocellular carcinoma by inducing apoptosis. J. Cell Biochem. 2018, 119, 9742–9751. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.P. The CDK Network: Linking Cycles of Cell Division and Gene Expression. Genes Cancer 2012, 3, 731–738. [Google Scholar] [CrossRef] [Green Version]
- Wallenfang, M.R.; Seydoux, G. cdk-7 Is required for mRNA transcription and cell cycle progression in Caenorhabditis elegans embryos. Proc. Natl. Acad. Sci. USA 2002, 99, 5527–5532. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.P.; Morgan, D.O. A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell 1994, 78, 713–724. [Google Scholar] [CrossRef]
- Fisher, R.P.; Jin, P.; Chamberlin, H.M.; Morgan, D.O. Alternative mechanisms of CAK assembly require an assembly factor or an activating kinase. Cell 1995, 83, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Serizawa, H.; Makela, T.P.; Conaway, J.W.; Conaway, R.C.; Weinberg, R.A.; Young, R.A. Association of Cdk-activating kinase subunits with transcription factor TFIIH. Nature 1995, 374, 280–282. [Google Scholar] [CrossRef]
- Shiekhattar, R.; Mermelstein, F.; Fisher, R.P.; Drapkin, R.; Dynlacht, B.; Wessling, H.C.; Morgan, D.O.; Reinberg, D. Cdk-activating kinase complex is a component of human transcription factor TFIIH. Nature 1995, 374, 283–287. [Google Scholar] [CrossRef]
- Schultz, P.; Fribourg, S.; Poterszman, A.; Mallouh, V.; Moras, D.; Egly, J.M. Molecular structure of human TFIIH. Cell 2000, 102, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Jin, H.; Gao, D.; Wang, L.; Evers, B.; Xue, Z.; Jin, G.; Lieftink, C.; Beijersbergen, R.L.; Qin, W.; et al. A CRISPR screen identifies CDK7 as a therapeutic target in hepatocellular carcinoma. Cell Res. 2018, 28, 690–692. [Google Scholar] [CrossRef]
- Cho, S.J.; Lee, S.S.; Kim, Y.J.; Park, B.D.; Choi, J.S.; Liu, L.; Ham, Y.M.; Moon Kim, B.; Lee, S.K. Xylocydine, a novel Cdk inhibitor, is an effective inducer of apoptosis in hepatocellular carcinoma cells in vitro and in vivo. Cancer Lett. 2010, 287, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Gahr, S.; Peter, G.; Wissniowski, T.T.; Hahn, E.G.; Herold, C.; Ocker, M. The histone-deacetylase inhibitor MS-275 and the CDK-inhibitor CYC-202 promote anti-tumor effects in hepatoma cell lines. Oncol. Rep. 2008, 20, 1249–1256. [Google Scholar] [PubMed]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.Y.; Lin, D.C.; Mayakonda, A.; Hazawa, M.; Ding, L.W.; Chien, W.W.; Xu, L.; Chen, Y.; Xiao, J.F.; Senapedis, W.; et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut 2017, 66, 1358–1368. [Google Scholar] [CrossRef] [Green Version]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 2014, 26, 909–922. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Zhu, A.; Gu, X.; Xie, G. Inhibition of MEK suppresses hepatocellular carcinoma growth through independent MYC and BIM regulation. Cell Oncol. 2019, 42, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gutierrez, L.; Delgado, M.D.; Leon, J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes 2019, 10, 244. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; Cubero, F.J.; Nevzorova, Y.A. c-MYC-Making Liver Sick: Role of c-MYC in Hepatic Cell Function, Homeostasis and Disease. Genes 2017, 8, 123. [Google Scholar] [CrossRef]
- Greenall, S.A.; Lim, Y.C.; Mitchell, C.B.; Ensbey, K.S.; Stringer, B.W.; Wilding, A.L.; O’Neill, G.M.; McDonald, K.L.; Gough, D.J.; Day, B.W.; et al. Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma. Oncogenesis 2017, 6, e336. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryl, T.; Kuchen, E.E.; Bell, E.; Shao, C.; Florez, A.F.; Monke, G.; Gogolin, S.; Friedrich, M.; Lamprecht, F.; Westermann, F.; et al. Cell-Cycle Position of Single MYC-Driven Cancer Cells Dictates Their Susceptibility to a Chemotherapeutic Drug. Cell Syst. 2017, 5, 237–250.e238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salghetti, S.E.; Kim, S.Y.; Tansey, W.P. Destruction of Myc by ubiquitin-mediated proteolysis: Cancer-associated and transforming mutations stabilize Myc. EMBO J. 1999, 18, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, A.; Jiang, C.; Cai, Y.; Kim, J.H.; Tanaka, N.; Ward, J.M.; Shah, Y.M.; Gonzalez, F.J. Role of Myc in hepatocellular proliferation and hepatocarcinogenesis. J. Hepatol. 2014, 60, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Thorgeirsson, S.S.; Grisham, J.W. Molecular pathogenesis of human hepatocellular carcinoma. Nat. Genet. 2002, 31, 339–346. [Google Scholar] [CrossRef]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, W.W.; Zejnullahu, K.; Bradner, J.E.; Varmus, H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 19408–19413. [Google Scholar] [CrossRef] [Green Version]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Ott, C.J.; Kopp, N.; Bird, L.; Paranal, R.M.; Qi, J.; Bowman, T.; Rodig, S.J.; Kung, A.L.; Bradner, J.E.; Weinstock, D.M. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood 2012, 120, 2843–2852. [Google Scholar] [CrossRef] [Green Version]
- Li, G.Q.; Guo, W.Z.; Zhang, Y.; Seng, J.J.; Zhang, H.P.; Ma, X.X.; Zhang, G.; Li, J.; Yan, B.; Tang, H.W.; et al. Suppression of BRD4 inhibits human hepatocellular carcinoma by repressing MYC and enhancing BIM expression. Oncotarget 2016, 7, 2462–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.H.; Eun, J.W.; Choi, S.K.; Shen, Q.; Choi, W.S.; Han, J.W.; Nam, S.W.; You, J.S. Epigenetic reader BRD4 inhibition as a therapeutic strategy to suppress E2F2-cell cycle regulation circuit in liver cancer. Oncotarget 2016, 7, 32628–32640. [Google Scholar] [CrossRef] [PubMed]
- Antignani, A.; Segal, D.; Simon, N.; Kreitman, R.J.; Huang, D.; FitzGerald, D.J. Essential role for Bim in mediating the apoptotic and antitumor activities of immunotoxins. Oncogene 2017, 36, 4953–4962. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.S.J.; Sharko, A.C.; Munie, J.; Kassler, S.; Melendez, T.; Lim, C.U.; Broude, E.V. CDK7 Inhibition is Effective in all the Subtypes of Breast Cancer: Determinants of Response and Synergy with EGFR Inhibition. Cells 2020, 9, 638. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, G.; Zhu, A.; Gu, X. Converged DNA Damage Response Renders Human Hepatocellular Carcinoma Sensitive to CDK7 Inhibition. Cancers 2022, 14, 1714. https://doi.org/10.3390/cancers14071714
Xie G, Zhu A, Gu X. Converged DNA Damage Response Renders Human Hepatocellular Carcinoma Sensitive to CDK7 Inhibition. Cancers. 2022; 14(7):1714. https://doi.org/10.3390/cancers14071714
Chicago/Turabian StyleXie, Guiqin, Ailin Zhu, and Xinbin Gu. 2022. "Converged DNA Damage Response Renders Human Hepatocellular Carcinoma Sensitive to CDK7 Inhibition" Cancers 14, no. 7: 1714. https://doi.org/10.3390/cancers14071714
APA StyleXie, G., Zhu, A., & Gu, X. (2022). Converged DNA Damage Response Renders Human Hepatocellular Carcinoma Sensitive to CDK7 Inhibition. Cancers, 14(7), 1714. https://doi.org/10.3390/cancers14071714