
Anticancer Effects and Molecular Mechanisms of Apigenin in Cervical Cancer Cells


 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Assay of Cell Viability
2.3. Assay of Cell Cycle Progression
2.4. Wound-Healing Migration Assay
2.5. Western Blot Analysis
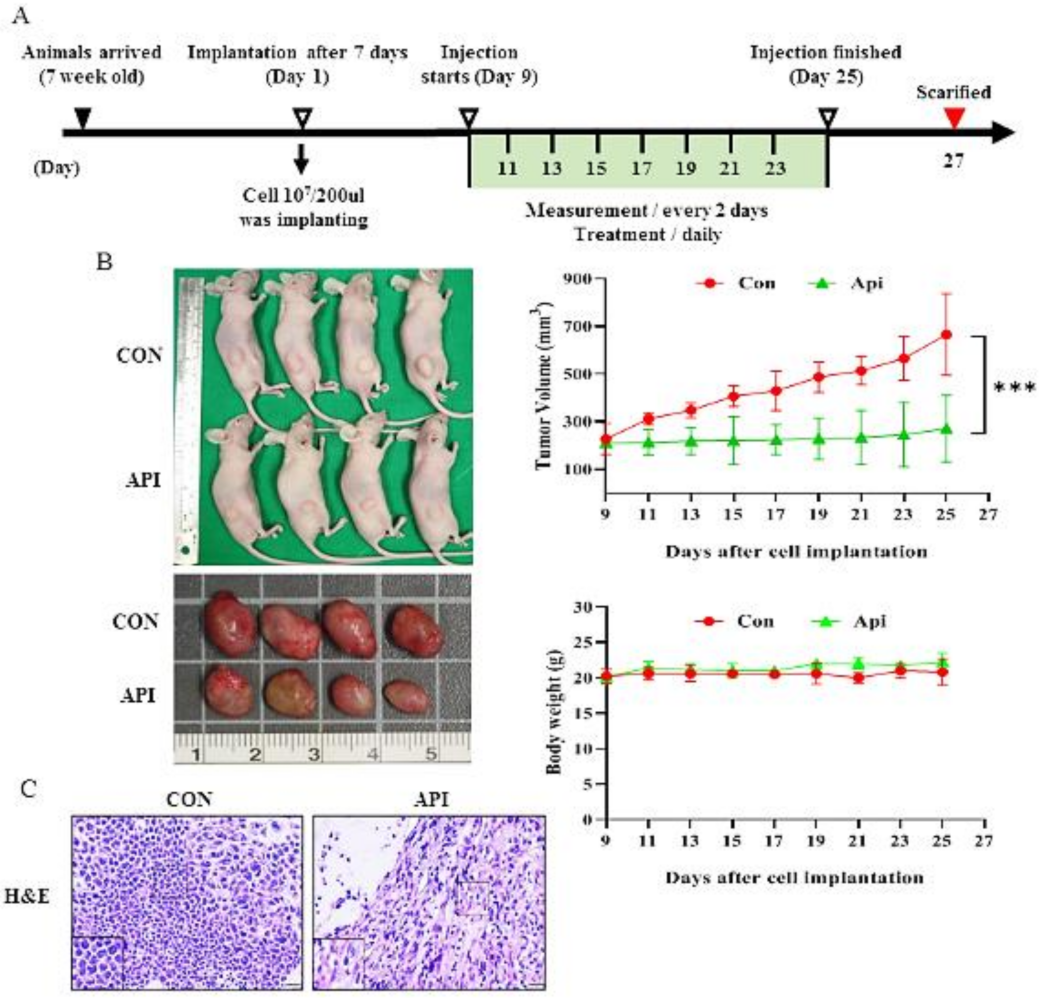
2.6. Human Cervical Tumor Xenograft Mouse Model
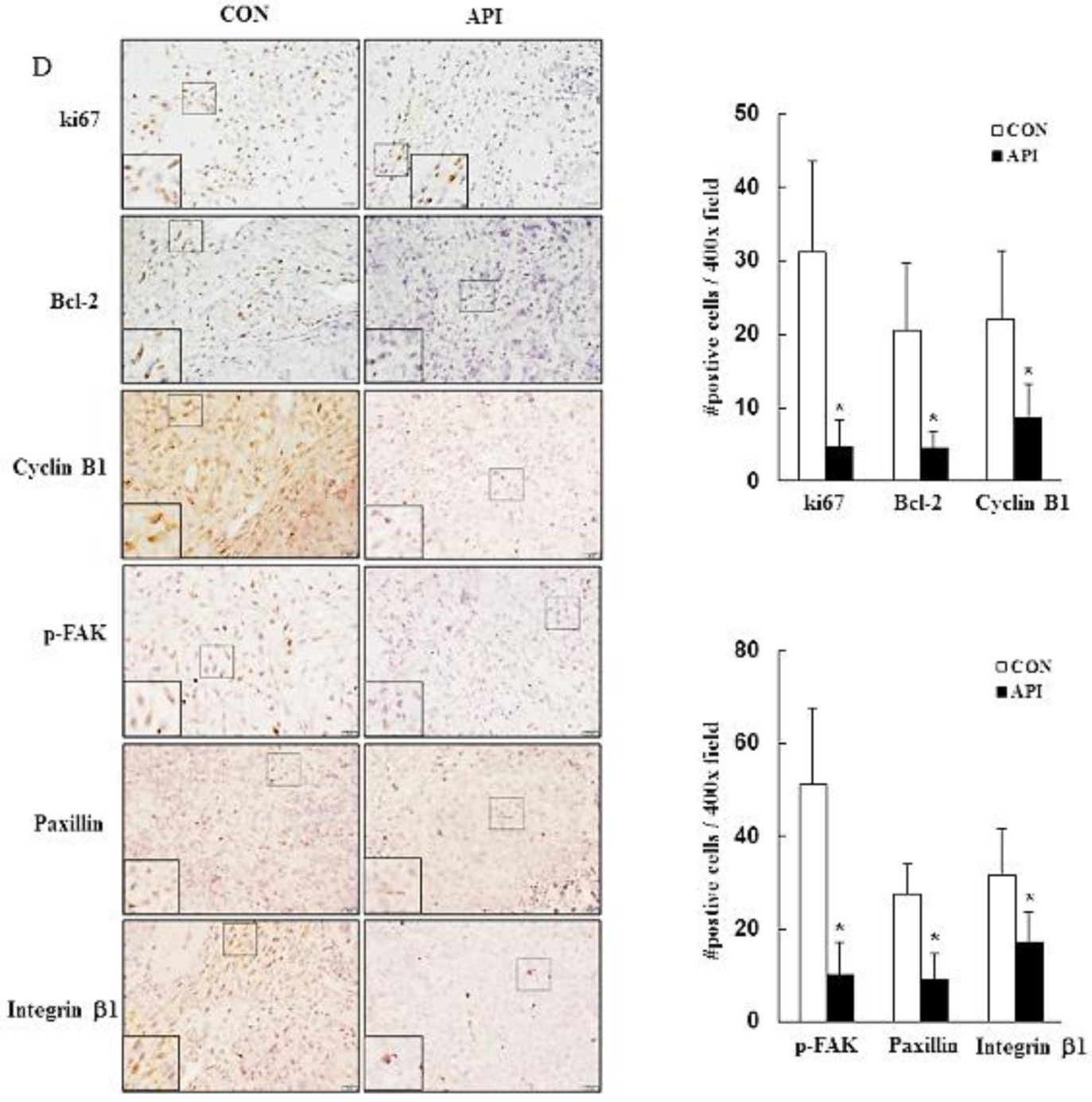
2.7. Histology and Immunohistochemical Analysis
2.8. Statistical Analysis
3. Results
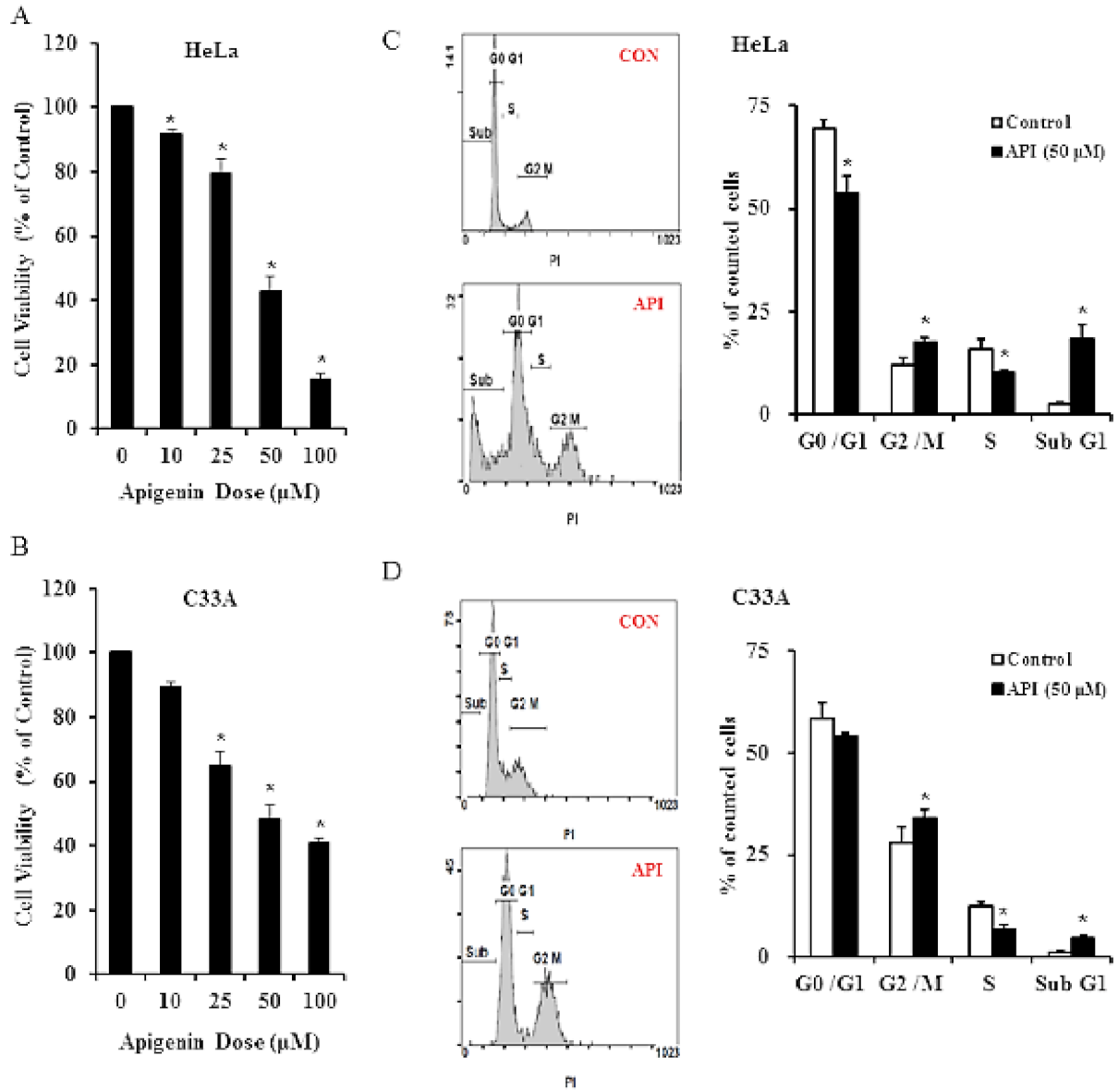
3.1. Apigenin Inhibits Human Cervical Cancer Cell Viability and Induces Cell Cycle Arrest
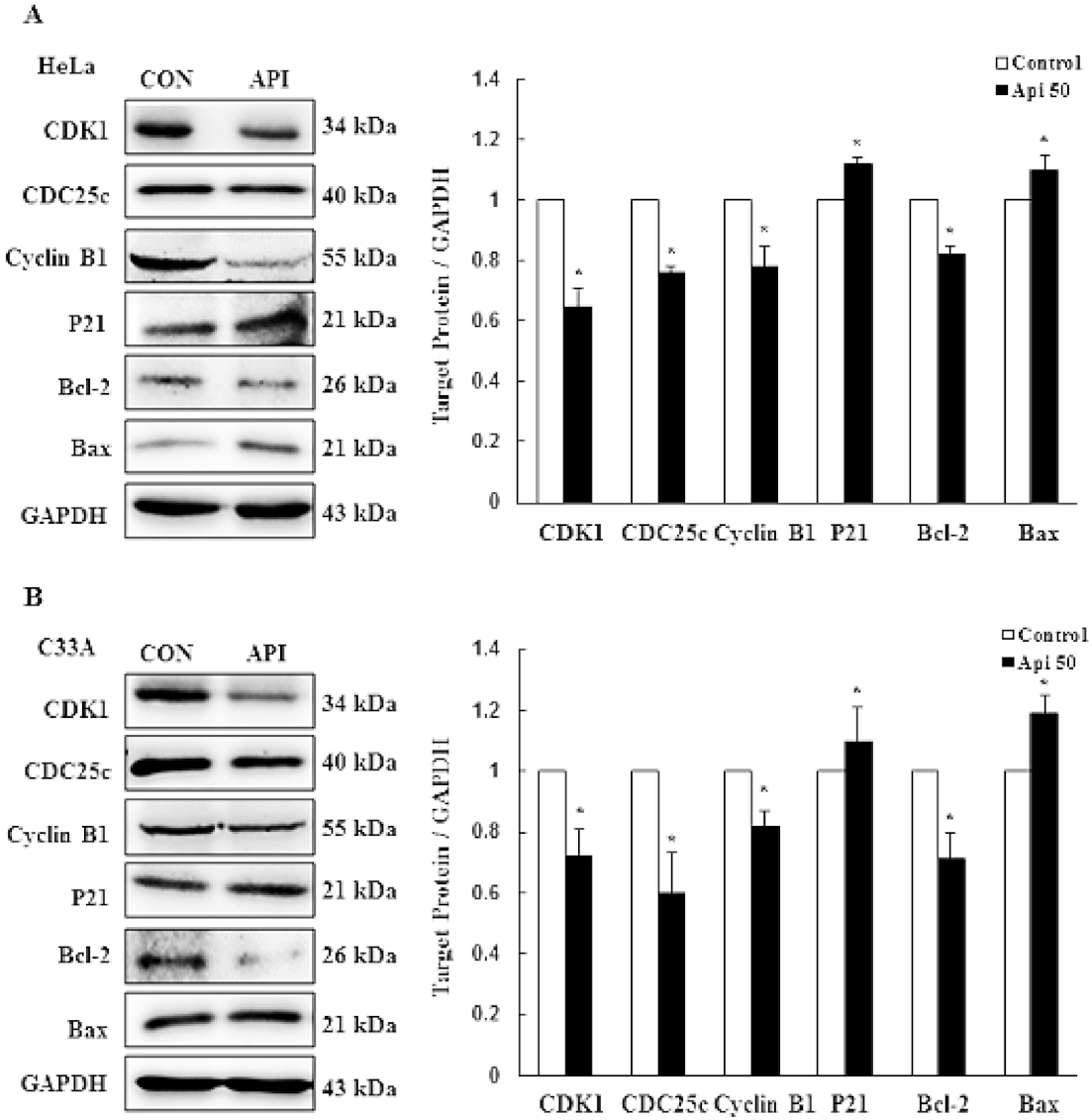
3.2. Apigenin Causes G2/M Phase Arrest by Modulating Cyclin B1/CDK1 and p21cip1 as Well as Activating Mitochondrial-Mediated Apoptosis
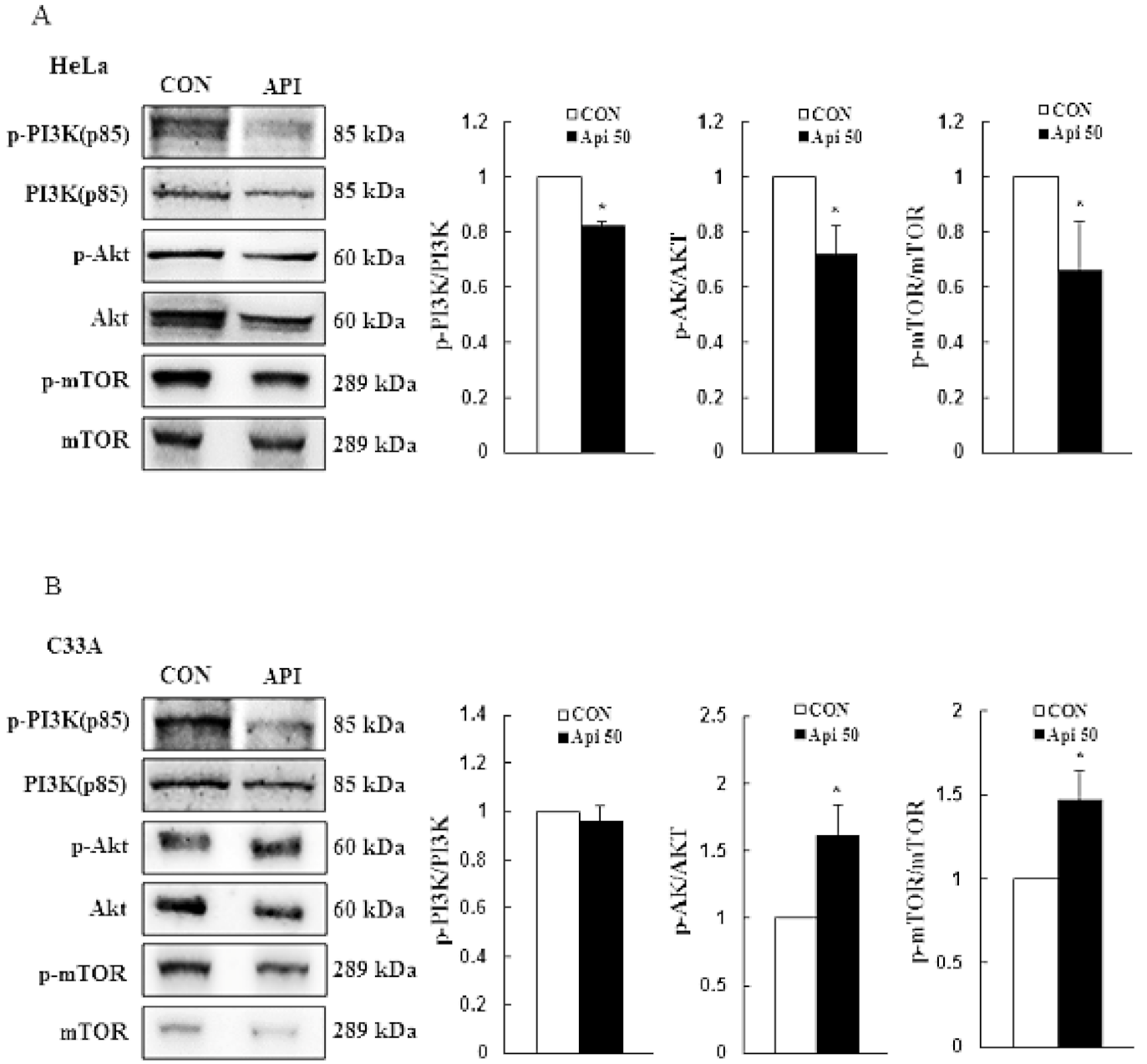
3.3. Apigenin Induces Cytotoxicity and Apoptosis via the PI3K/AKT/mTOR Pathway
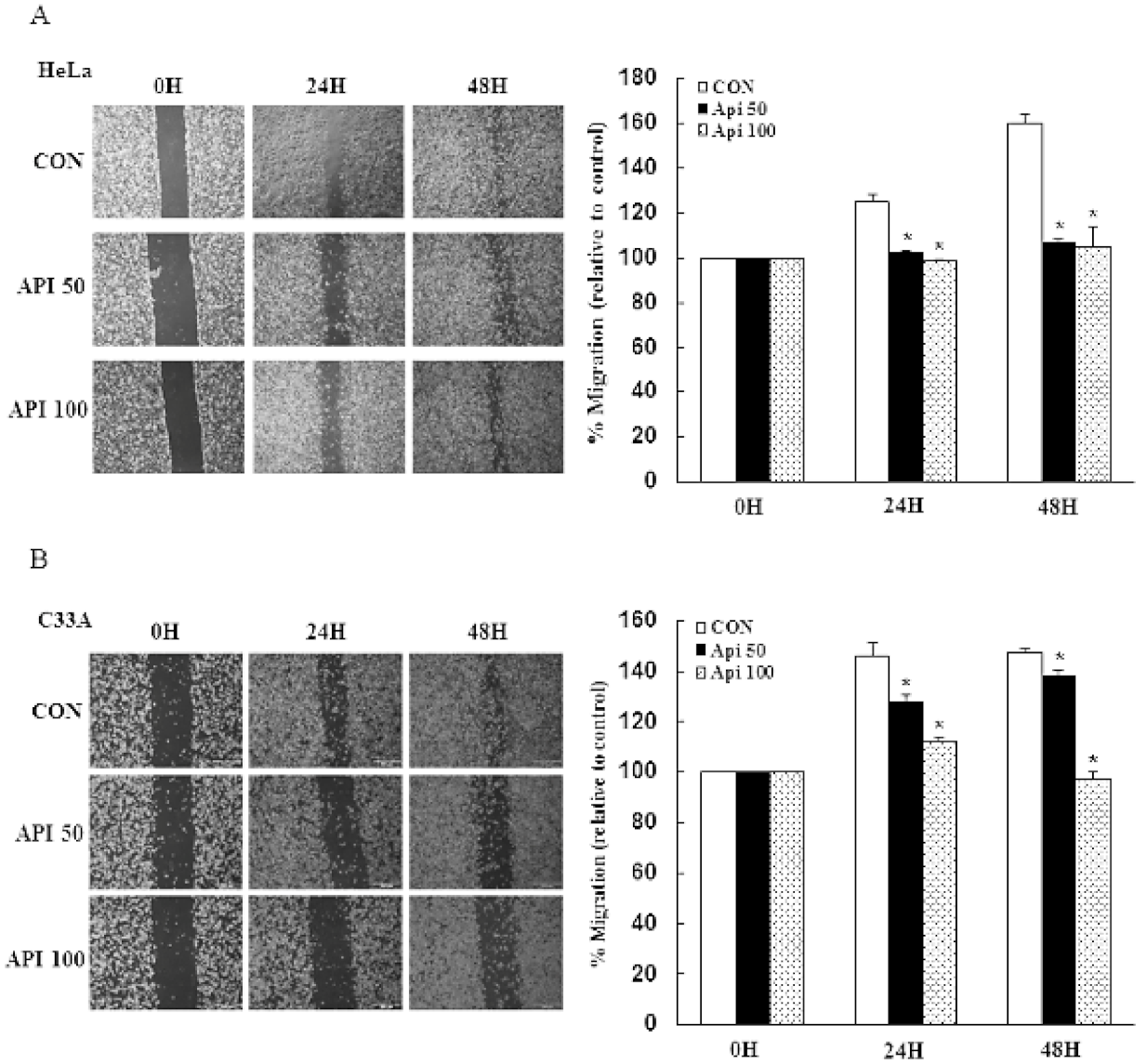
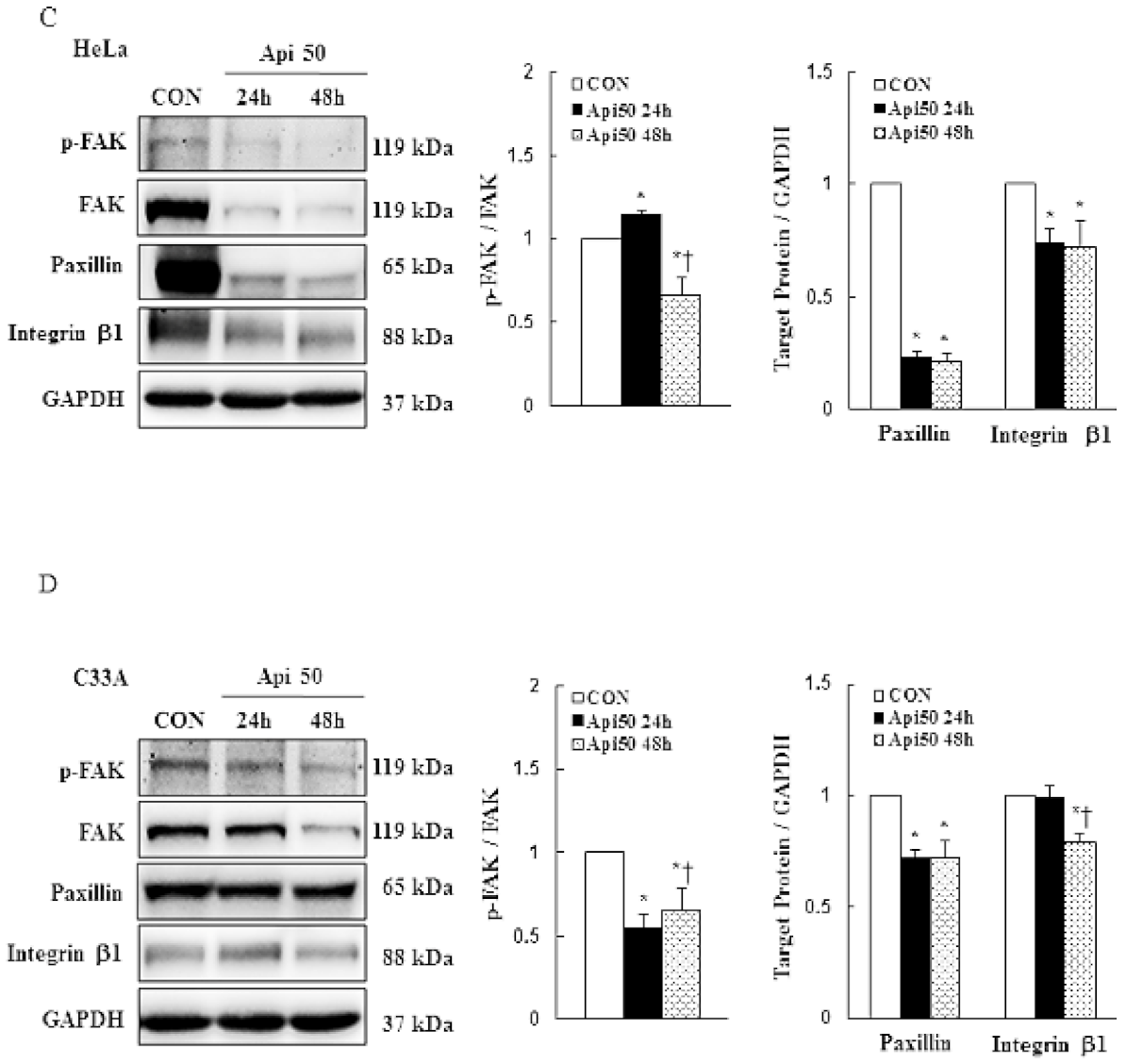
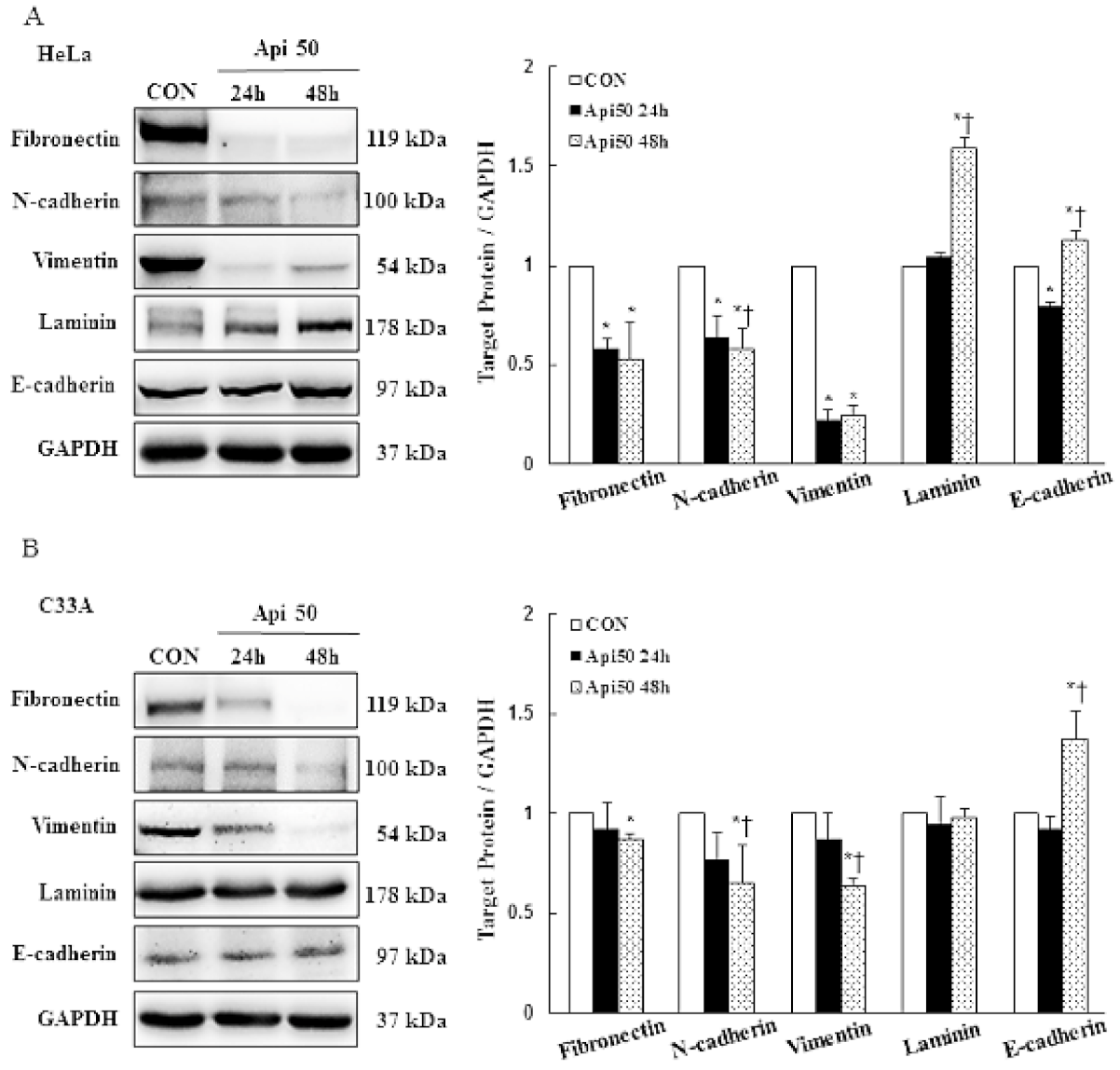
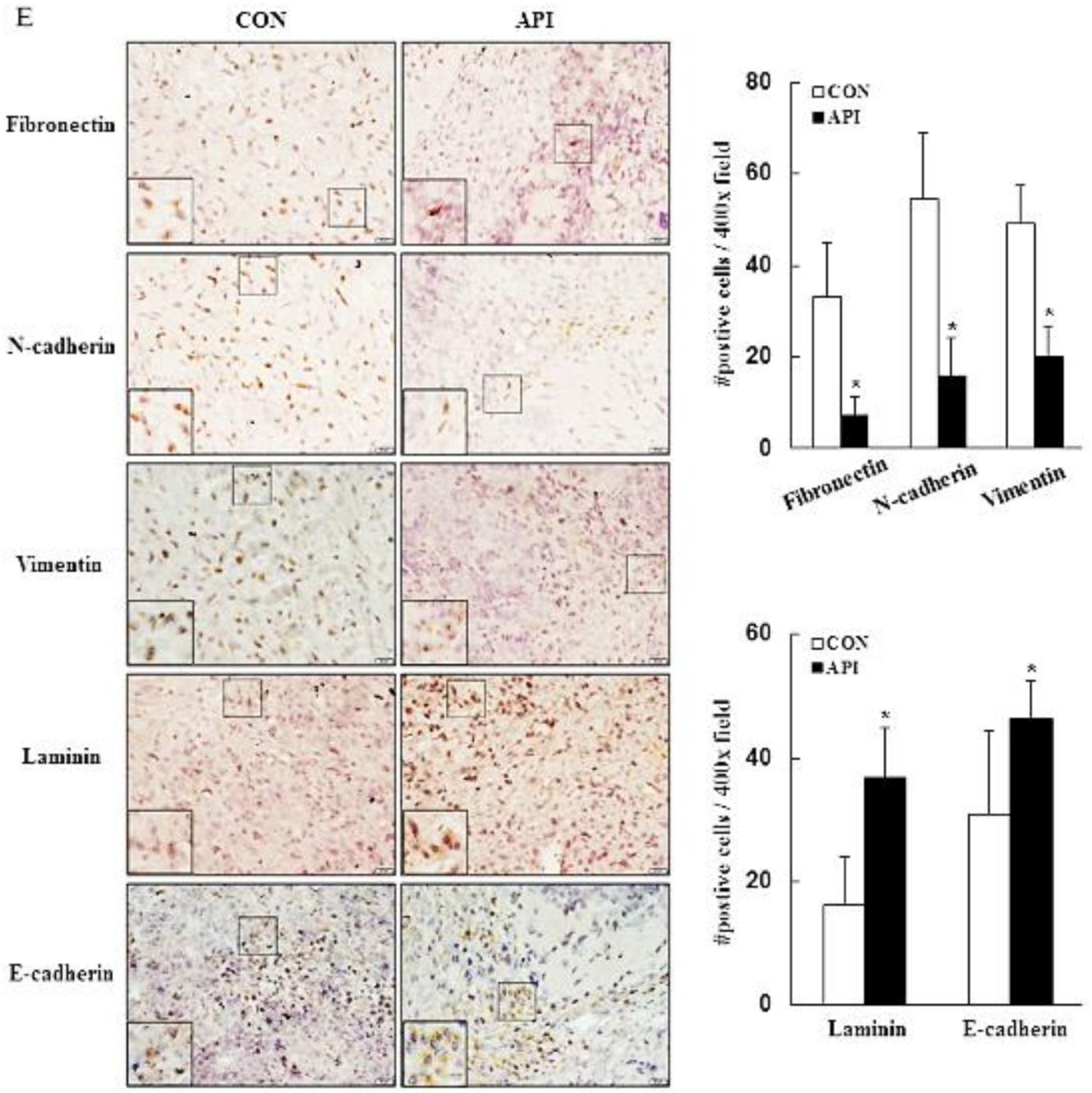
3.4. Apigenin Inhibits Cancer Cell Migration and Epithelial-to-Mesenchymal Transition (EMT) of Human Cervical Cancer
3.5. Apigenin Suppresses the Growth of C33A Xenograft Tumors
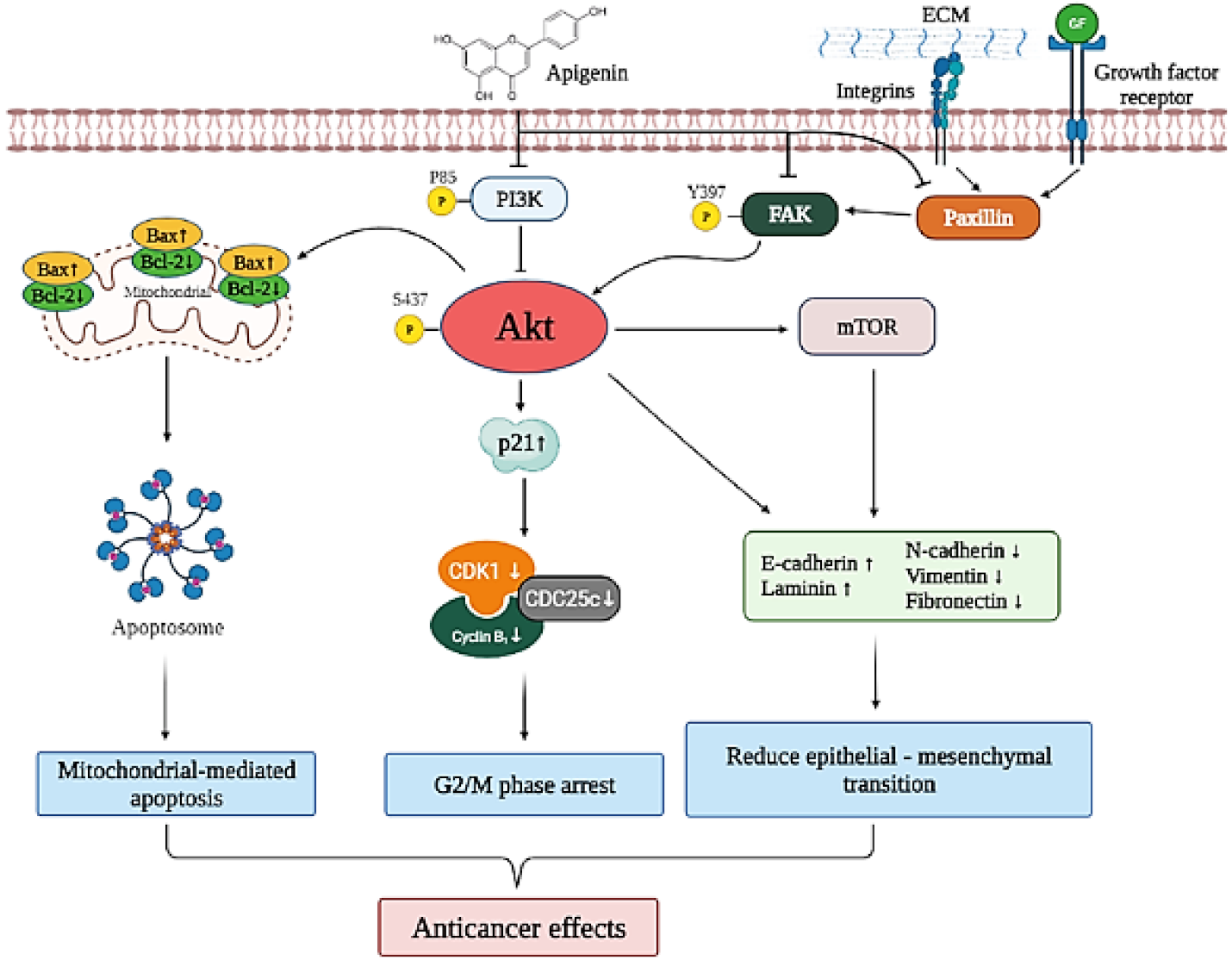
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef] [Green Version]
- Colombo, N.; Carinelli, S.; Marini, C.; Rollo, D.; Sessa, C. Cervical cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23, vii27–vii32. [Google Scholar] [CrossRef] [PubMed]
- Kagabu, M.; Nagasawa, T.; Fukagawa, D.; Tomabechi, H.; Sato, S.; Shoji, T.; Baba, T. Immunotherapy for Uterine Cervical Cancer. Healthcare 2019, 7, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, D.-W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin Resistance: A Cellular Self-Defense Mechanism Resulting from Multiple Epigenetic and Genetic Changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Gupta, S. Apigenin: A Promising Molecule for Cancer Prevention. Pharm. Res. 2010, 27, 962–978. [Google Scholar] [CrossRef]
- Miccadei, S.; Di Venere, D.; Cardinali, A.; Romano, F.; Durazzo, A.; Foddai, M.S.; Fraioli, R.; Mobarhan, S.; Maiani, G. Antioxidative and Apoptotic Properties of Polyphenolic Extracts from Edible Part of Artichoke (Cynara scolymus L.) on Cultured Rat Hepatocytes and on Human Hepatoma Cells. Nutr. Cancer 2008, 60, 276–283. [Google Scholar] [CrossRef]
- Lim, R.; Barker, G.; Wall, C.A.; Lappas, M. Dietary phytophenols curcumin, naringenin and apigenin reduce infection-induced inflammatory and contractile pathways in human placenta, foetal membranes and myometrium. Mol. Hum. Reprod. 2013, 19, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Villa-Rodriguez, J.A.; Kerimi, A.; Abranko, L.; Tumova, S.; Ford, L.; Blackburn, R.S.; Rayner, C.; Williamson, G. Acute metabolic actions of the major polyphenols in chamomile: An in vitro mechanistic study on their potential to attenuate postprandial hyperglycaemia. Sci. Rep. 2018, 8, 5471. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.S.; Landau, J.M.; Huang, M.-T.; Newmark, H.L. Inhibition of Carcinogenesis by Dietary Polyphenolic Compounds. Annu. Rev. Nutr. 2001, 21, 381–406. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.; Young, J.; Daneshvar, B.; Lauridsen, S.; Knuthsen, P.; Sandström, B.; Dragsted, L.O. Effect of parsley (Petroselinum crispum) intake on urinary apigenin excretion, blood antioxidant enzymes and biomarkers for oxidative stress in human subjects. Br. J. Nutr. 1999, 81, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.-S.; Ku, J.M.; Choi, H.-S.; Woo, J.K.; Jang, B.-H.; Go, H.; Shin, Y.C.; Ko, S.-G. Apigenin induces caspase-dependent apoptosis by inhibiting signal transducer and activator of transcription 3 signaling in HER2-overexpressing SKBR3 breast cancer cells. Mol. Med. Rep. 2015, 12, 2977–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Bhaskaran, N.; Babcook, M.A.; Fu, P.; MacLennan, G.T.; Gupta, S. Apigenin inhibits prostate cancer progression in TRAMP mice via targeting PI3K/Akt/FoxO pathway. Carcinogenesis 2014, 35, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, Y.-A.; Jo, S.-Y.; Lee, H.-Y.; Lee, C. Inhibition of IL-6/STAT3 axis and targeting Axl and Tyro3 receptor tyrosine kinases by apigenin circumvent taxol resistance in ovarian cancer cells. Int. J. Oncol. 2015, 46, 1405–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, H.-Y.; Tang, X.-W. Anti-proliferation and chemo-sensitization effects of apigenin on human lung cancer cells. Zhejiang Da Xue Xue Bao. Yi Xue Ban = J. Zhejiang Univ. Med. Sci. 2011, 40, 508–514. [Google Scholar]
- Zeng, J.; Xie, H.; Zhang, Z.L.; Li, Z.X.; Shi, L.; Wu, K.Y.; Zhou, Y.; Tian, Z.; Zhang, Y.; Zhou, W.; et al. Apigenin regulates the migration, invasion, and autophagy of hepatocellular carcinoma cells by downregulating YAP. Neoplasma 2022, 69, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; de Mejia, E.G. Flavonoid apigenin modified gene expression associated with inflammation and cancer and induced apoptosis in human pancreatic cancer cells through inhibition of GSK-3β/NF-κB signaling cascade. Mol. Nutr. Food Res. 2013, 57, 2112–2127. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, H.; Yu, X.; Wang, X.; Zhu, X.; Xu, X. Apigenin inhibits in vitro and in vivo tumorigenesis in cisplatin-resistant colon cancer cells by inducing autophagy, programmed cell death and targeting m-TOR/PI3K/Akt signalling pathway. J. B. U. ON. Off. J. Balk. Union Oncol. 2019, 24, 488–493. [Google Scholar]
- Zheng, P.-W.; Chiang, L.-C.; Lin, C.-C. Apigenin induced apoptosis through p53-dependent pathway in human cervical carcinoma cells. Life Sci. 2005, 76, 1367–1379. [Google Scholar] [CrossRef]
- Souza, R.P.; Bonfim-Mendonça, P.D.S.; Gimenes, F.; Ratti, B.A.; Kaplum, V.; Bruschi, M.L.; Nakamura, C.V.; Silva, S.O.; Maria-Engler, S.; Consolaro, M.E.L. Oxidative Stress Triggered by Apigenin Induces Apoptosis in a Comprehensive Panel of Human Cervical Cancer-Derived Cell Lines. Oxidative Med. Cell. Longev. 2017, 2017, 1512745. [Google Scholar] [CrossRef]
- Imran, M.; Aslam Gondal, T.; Atif, M.; Shahbaz, M.; Batool Qaisarani, T.; Hanif Mughal, M.; Salehi, B.; Martorell, M.; Sharifi-Rad, J. Apigenin as an anticancer agent. Phytother. Res. 2020, 34, 1812–1828. [Google Scholar] [CrossRef]
- Nicholas, C.; Batra, S.; Vargo, M.A.; Voss, O.H.; Gavrilin, M.A.; Wewers, M.D.; Guttridge, D.C.; Grotewold, E.; Doseff, A.I. Apigenin blocks lipopolysaccharide-induced lethality in vivo and proinflammatory cytokines expression by inactivating NF-κB through the suppression of p65 phosphorylation. J. Immunol. 2007, 179, 7121–7127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strouch, M.J.; Milam, B.M.; Melstrom, L.G.; McGill, J.J.; Salabat, M.R.; Ujiki, M.B.; Ding, X.-Z.; Bentrem, D.J. The Flavonoid Apigenin Potentiates the Growth Inhibitory Effects of Gemcitabine and Abrogates Gemcitabine Resistance in Human Pancreatic Cancer Cells. Pancreas 2009, 38, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.Y.; Liang, J.Y.; Guo, X.J.; Liu, L.; Guo, Y.B. 5-Fluorouracil combined with apigenin enhances anticancer activity through mitochondrial membrane potential (ΔΨm)-mediated apoptosis in hepatocellular carcinoma. Clin. Exp. Pharmacol. Physiol. 2015, 42, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. Akt inhibition enhances the cytotoxic effect of apigenin in combination with PLX4032 in anaplastic thyroid carcinoma cells harboring BRAFV600E. J. Endocrinol. Investig. 2013, 36, 1099–1104. [Google Scholar] [CrossRef]
- de Font-Reaulx Rojas, E.; Dorazco-Barragan, G. Clinical stabilisation in neurodegenerative diseases: Clinical study in phase II. Rev. Neurol. 2010, 50, 520–528. [Google Scholar]
- Zick, S.M.; Wright, B.D.; Sen, A.; Arnedt, J.T. Preliminary examination of the efficacy and safety of a standardized chamomile extract for chronic primary insomnia: A randomized placebo-controlled pilot study. BMC Complement. Altern. Med. 2011, 11, 78. [Google Scholar] [CrossRef] [Green Version]
- Amsterdam, J.D.; Shults, J.; Soeller, I.; Mao, J.J.; Rockwell, K.; Newberg, A.B. Chamomile (Matricaria recutita) may provide antidepressant activity in anxious, depressed humans: An exploratory study. Altern. Ther. Health Med. 2012, 18, 44–49. [Google Scholar]
- Shoara, R.; Hashempur, M.H.; Ashraf, A.; Salehi, A.; Dehshahri, S.; Habibagahi, Z. Efficacy and safety of topical Matricaria chamomilla L. (chamomile) oil for knee osteoarthritis: A randomized controlled clinical trial. Complement. Ther. Clin. Pr. 2015, 21, 181–187. [Google Scholar] [CrossRef]
- Mao, J.J.; Xie, S.X.; Keefe, J.R.; Soeller, I.; Li, Q.S.; Amsterdam, J.D. Long-term chamomile (Matricaria chamomilla L.) treatment for generalized anxiety disorder: A randomized clinical trial. Phytomedicine 2016, 23, 1735–1742. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Hugo, H.; Ackland, M.L.; Blick, T.; Lawrence, M.G.; Clements, J.A.; Williams, E.D.; Thompson, E.W. Epithelial—mesenchymal and mesenchymal—epithelial transitions in carcinoma progression. J. Cell. Physiol. 2007, 213, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Ibaragi, S.; Hu, G.-F. Epithelial-Mesenchymal Transition and Cell Cooperativity in Metastasis: Figure 1. Cancer Res. 2009, 69, 7135–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Hiscox, S.; Jiang, W.G.; Obermeier, K.; Taylor, K.; Morgan, L.; Burmi, R.; Barrow, D.; Nicholson, R.I. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of β-catenin phosphorylation. Int. J. Cancer 2006, 118, 290–301. [Google Scholar] [CrossRef]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.I.; Parikh, N.U.; Gallick, G.E. Development and Characterization of Gemcitabine-Resistant Pancreatic Tumor Cells. Ann. Surg. Oncol. 2007, 14, 3629–3637. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-Y.; Chou, C.-Y.; Tang, M.-J.; Shen, M.-R. Epithelial-Mesenchymal Transition in Cervical Cancer: Correlation with Tumor Progression, Epidermal Growth Factor Receptor Overexpression, and Snail Up-Regulation. Clin. Cancer Res. 2008, 14, 4743–4750. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Cui, S.; Wang, J.; Wu, Q.; Qian, J.; Yang, C.; Bo, P. Genistein inhibits the growth and regulates the migration and invasion abilities of melanoma cells via the FAK/paxillin and MAPK pathways. Oncotarget 2017, 8, 21674–21691. [Google Scholar] [CrossRef] [Green Version]
- Chuang, H.-H.; Zhen, Y.-Y.; Tsai, Y.-C.; Chuang, C.-H.; Hsiao, M.; Huang, M.-S.; Yang, C.-J. FAK in Cancer: From Mechanisms to Therapeutic Strategies. Int. J. Mol. Sci. 2022, 23, 1726. [Google Scholar] [CrossRef]
- Chan, L.-P.; Chou, T.-H.; Ding, H.-Y.; Chen, P.-R.; Chiang, F.-Y.; Kuo, P.-L.; Liang, C.-H. Apigenin induces apoptosis via tumor necrosis factor receptor- and Bcl-2-mediated pathway and enhances susceptibility of head and neck squamous cell carcinoma to 5-fluorouracil and cisplatin. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2012, 1820, 1081–1091. [Google Scholar] [CrossRef]
- Lee, Y.; Sung, B.; Kang, Y.J.; Kim, D.H.; Jang, J.-Y.; Hwang, S.Y.; Kim, M.; Lim, H.S.; Yoon, J.-H.; Chung, H.Y.; et al. Apigenin-induced apoptosis is enhanced by inhibition of autophagy formation in HCT116 human colon cancer cells. Int. J. Oncol. 2014, 44, 1599–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, M.; Kaur, P.; Shukla, S.; Abbas, A.; Fu, P.; Gupta, S. Plant flavone apigenin inhibits HDAC and remodels chromatin to induce growth arrest and apoptosis in human prostate cancer cells: In vitro and in vivo study. Mol. Carcinog. 2012, 51, 952–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggioni, D.; Garavello, W.; Rigolio, R.; Pignataro, L.; Gaini, R.; Nicolini, G. Apigenin impairs oral squamous cell carcinoma growth in vitro inducing cell cycle arrest and apoptosis. Int. J. Oncol. 2013, 43, 1675–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, T.H.; Chien, M.H.; Lin, W.L.; Wen, Y.C.; Chow, J.M.; Chen, C.K.; Kuo, T.C.; Lee, W.J. Inhibition of MDA-MB-231 breast cancer cell proliferation and tumor growth by apigenin through induction of G2/M arrest and histone H3 acetylation-mediated p21WAF1/CIP1 expression. Environ. Toxicol. 2017, 32, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Pelling, J.C. Targeting the PI3K/Akt/mTOR axis by apigenin for cancer prevention. Anti-Cancer Agents Med. Chem. 2013, 13, 971–978. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2018, 20, 69–84. [Google Scholar] [CrossRef]
- Cho, E.S.; Kang, H.E.; Kim, N.H.; Yook, J.I. Therapeutic implications of cancer epithelial-mesenchymal transition (EMT). Arch. Pharmacal Res. 2019, 42, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, M.Q.; Ward, C.; Muller, H.K.; Sohal, S.S.; Walters, E.H. Epithelial mesenchymal transition (EMT) and non-small cell lung cancer (NSCLC): A mutual association with airway disease. Med Oncol. 2017, 34, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-J.; Dai, Z.; Zhou, S.-L.; Hu, Z.-Q.; Chen, Q.; Zhao, Y.-M.; Shi, Y.-H.; Gao, Q.; Wu, W.-Z.; Qiu, S.-J.; et al. HNRNPAB Induces Epithelial–Mesenchymal Transition and Promotes Metastasis of Hepatocellular Carcinoma by Transcriptionally Activating SNAIL. Cancer Res. 2014, 74, 2750–2762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migita, T.; Ueda, A.; Ohishi, T.; Hatano, M.; Seimiya, H.; Horiguchi, S.-I.; Koga, F.; Shibasaki, F. Epithelial–mesenchymal transition promotes SOX2 and NANOG expression in bladder cancer. Lab. Investig. 2017, 97, 567–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Yi, Q.; Tang, L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: A focused review. J. Exp. Clin. Cancer Res. 2019, 38, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, P.; Liu, L.; Liu, B.; Mao, X.-G. Expression of integrin β1 and its significance in squamous cell carcinoma of the cervix. Mol. Med. Rep. 2014, 9, 2473–2478. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, S.; Liu, J.; Tian, Y.; Ma, B.; Xu, S.; Fu, Y.; Luo, Y. Secreted pyruvate kinase M2 promotes lung cancer metastasis through activating the integrin Beta1/FAK signaling pathway. Cell Rep. 2020, 30, 1780–1797.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasnat, A.; Pervin, M.; Lim, J.H.; Lim, B.O. Apigenin Attenuates Melanoma Cell Migration by Inducing Anoikis through Integrin and Focal Adhesion Kinase Inhibition. Molecules 2015, 20, 21157–21166. [Google Scholar] [CrossRef]
- Ruela-De-Sousa, R.R.; Fuhler, G.M.; Blom, N.; Ferreira, C.V.; Aoyama, H.; Peppelenbosch, M.P. Cytotoxicity of apigenin on leukemia cell lines: Implications for prevention and therapy. Cell Death Dis. 2010, 1, e19. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Zhao, X.-L.; Liu, A.-W.; Nian, H.; Zhang, S.-H. Apigenin inhibits hepatoma cell growth through alteration of gene expression patterns. Phytomedicine 2011, 18, 366–373. [Google Scholar] [CrossRef]
- Kuo, C.-H.; Weng, B.-C.; Wu, C.-C.; Yang, S.-F.; Wu, D.-C.; Wang, Y.-C. Apigenin has anti-atrophic gastritis and anti-gastric cancer progression effects in Helicobacter pylori -infected Mongolian gerbils. J. Ethnopharmacol. 2014, 151, 1031–1039. [Google Scholar] [CrossRef]
- Guo, H.; Kong, S.; Chen, W.; Dai, Z.; Lin, T.; Su, J.; Li, S.; Xie, Q.; Su, Z.; Xu, Y.; et al. Apigenin Mediated Protection of OGD-Evoked Neuron-Like Injury in Differentiated PC12 Cells. Neurochem. Res. 2014, 39, 2197–2210. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, X.-C.; Xiao, Q.; Quan, M.-F. Apigenin inhibits HeLa sphere-forming cells through inactivation of casein kinase 2α. Mol. Med. Rep. 2015, 11, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Van Wie, P.G.; Fai, L.Y.; Kim, D.; Wang, L.; Poyil, P.; Luo, J.; Zhang, Z. Downregulation of NEDD9 by apigenin suppresses migration, invasion, and metastasis of colorectal cancer cells. Toxicol. Appl. Pharmacol. 2016, 311, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Wang, S.; Song, Y.; Yao, J.; Huang, K.; Zhu, X. Apigenin suppresses colorectal cancer cell proliferation, migration and invasion via inhibition of the Wnt/β-catenin signaling pathway. Oncol. Lett. 2016, 11, 3075–3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Wei, Y.-X.; Shen, M.-C.; Tu, Y.-H.; Wang, C.-C.; Huang, H.-C. Chrysin, Abundant in Morinda citrifolia Fruit Water–EtOAc Extracts, Combined with Apigenin Synergistically Induced Apoptosis and Inhibited Migration in Human Breast and Liver Cancer Cells. J. Agric. Food Chem. 2016, 64, 4235–4245. [Google Scholar] [CrossRef]
- Erdogan, S.; Turkekul, K.; Serttas, R.; Erdogan, Z. The natural flavonoid apigenin sensitizes human CD44 + prostate cancer stem cells to cisplatin therapy. Biomed. Pharmacother. 2017, 88, 210–217. [Google Scholar] [CrossRef]
- Silvan, S.; Manoharan, S. Apigenin prevents deregulation in the expression pattern of cell-proliferative, apoptotic, inflammatory and angiogenic markers during 7,12-dimethylbenz[a]anthracene-induced hamster buccal pouch carcinogenesis. Arch. Oral Biol. 2013, 58, 94–101. [Google Scholar] [CrossRef]
- Ketkaew, Y.; Osathanon, T.; Pavasant, P.; Sooampon, S. Apigenin inhibited hypoxia induced stem cell marker expression in a head and neck squamous cell carcinoma cell line. Arch. Oral Biol. 2017, 74, 69–74. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Molecular Mechanism and Activity | Refs. |
|---|---|---|
| Leukemia | Apigenin inhibits HL60 cell proliferation via G2/M phase arrest, but TF1 cell was G0/G1 phase arrest | [58] |
| Liver cancer | Apigenin (5–20 μg/mL) inhibits hepatoma Huh7 cell growth via G2/M phase arrest and apoptosis; Apigenin (50 μg/day) significantly suppressed the growth of Huh7 cell-derived xenograft tumor | [59] |
| Stomach cancer | Apigenin treatment (30–60 mg/kg body weight/day) significantly anti-gastic cancer and anti-atrophic progression in Helicobacter pylori-infected Mongolian gerbils | [60] |
| Brain cancer | PC12 cells were pretreated with apigenin for 6 h, and then apigenin could decreased oxygen and glucose deprivation/reperfusion (OGD/R)-induced neuronal injury through apigenin-triggered antioxidative and antiapoptotic activity | [61] |
| Cervical cancer | Apigenin reduced the HeLa cells viability, the IC50 value was 35.89 μM. Arrested at sub-G1, G1 phase, and the upregulated p21/WAF1, and p53 protein expressions | [62] |
| Colon cancer | Apigenin suppresses colorectal cancer migration and metastasis through inhibition of NEDD9/Src/AKT and Wnt/β-catin signaling pathway | [63,64] |
| Breast cancer | Apigenin combined with chrysin synergistically decreased MDA-AM-231 cell viability, increased apoptosis, and inhibited migration at 72–96 h | [65] |
| Prostate cancer | Apigenin (15 μM) potentiates the anticancer effect of cisplatin to inhibit CD44+PCa cell growth and to significantly rescue suppressed phosphorylation of AKT and PI3K, and increased the cisplatin on the cell migration inhibitory effect | [66] |
| Oral cancer | Apigenin (40 mM) significantly reduced HN-30 cell viability, and apigenin (2.5 mg/kg body weight) deregulated cell proliferation, apoptosis expression, and inflammatory markers in DMBA-induced hamster pouch carcinogenesis | [67,68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-H.; Wu, J.-X.; Yang, S.-F.; Yang, C.-K.; Chen, T.-H.; Hsiao, Y.-H. Anticancer Effects and Molecular Mechanisms of Apigenin in Cervical Cancer Cells. Cancers 2022, 14, 1824. https://doi.org/10.3390/cancers14071824
Chen Y-H, Wu J-X, Yang S-F, Yang C-K, Chen T-H, Hsiao Y-H. Anticancer Effects and Molecular Mechanisms of Apigenin in Cervical Cancer Cells. Cancers. 2022; 14(7):1824. https://doi.org/10.3390/cancers14071824
Chicago/Turabian StyleChen, Ya-Hui, Jyun-Xue Wu, Shun-Fa Yang, Chueh-Ko Yang, Tze-Ho Chen, and Yi-Hsuan Hsiao. 2022. "Anticancer Effects and Molecular Mechanisms of Apigenin in Cervical Cancer Cells" Cancers 14, no. 7: 1824. https://doi.org/10.3390/cancers14071824
APA StyleChen, Y. -H., Wu, J. -X., Yang, S. -F., Yang, C. -K., Chen, T. -H., & Hsiao, Y. -H. (2022). Anticancer Effects and Molecular Mechanisms of Apigenin in Cervical Cancer Cells. Cancers, 14(7), 1824. https://doi.org/10.3390/cancers14071824