
Robust Validation and Comprehensive Analysis of a Novel Signature Derived from Crucial Metabolic Pathways of Pancreatic Ductal Adenocarcinoma

 , ,
, ,  , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. Gene Set Enrichment Analysis
2.3. Consensus Clustering
2.4. Immune Cell Infiltration Evaluation
2.5. Drug Sensitivity Analysis
2.6. Statistical Analysis
3. Results
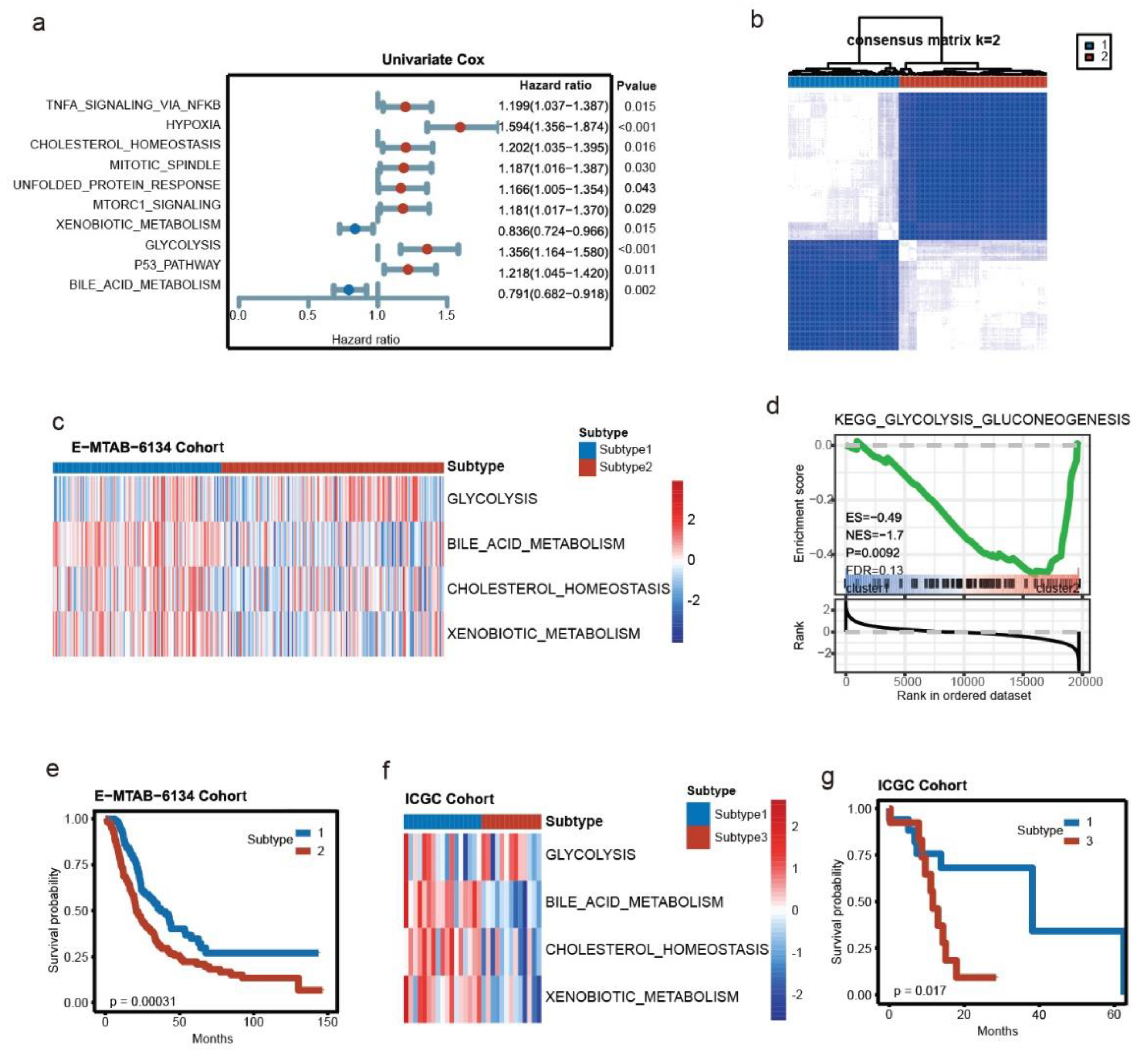
3.1. Key Metabolic Pathways of PDAC and Molecular Subtyping
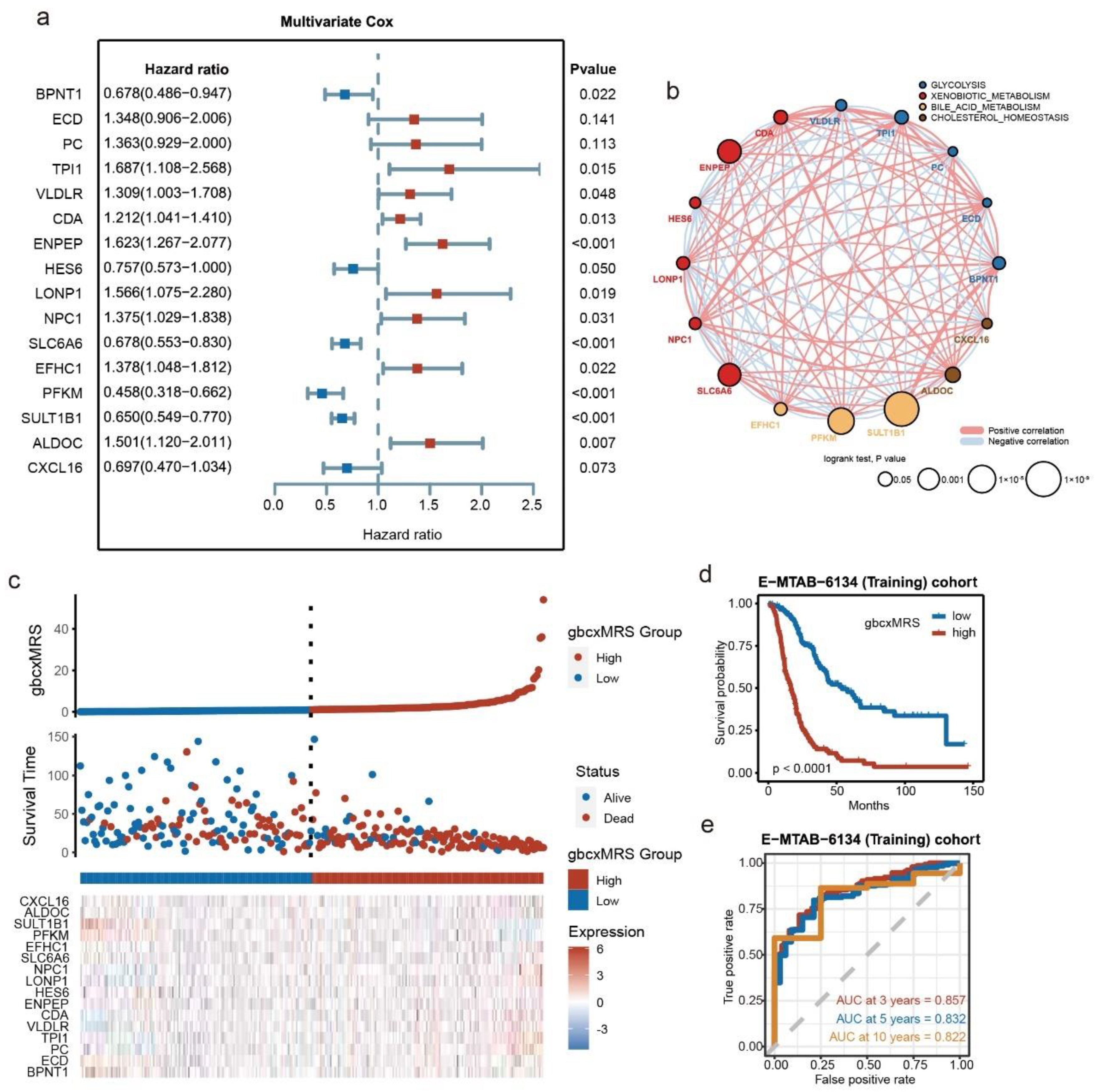
3.2. Development of the Signature from Key Metabolic Pathways in E-MTAB-6134
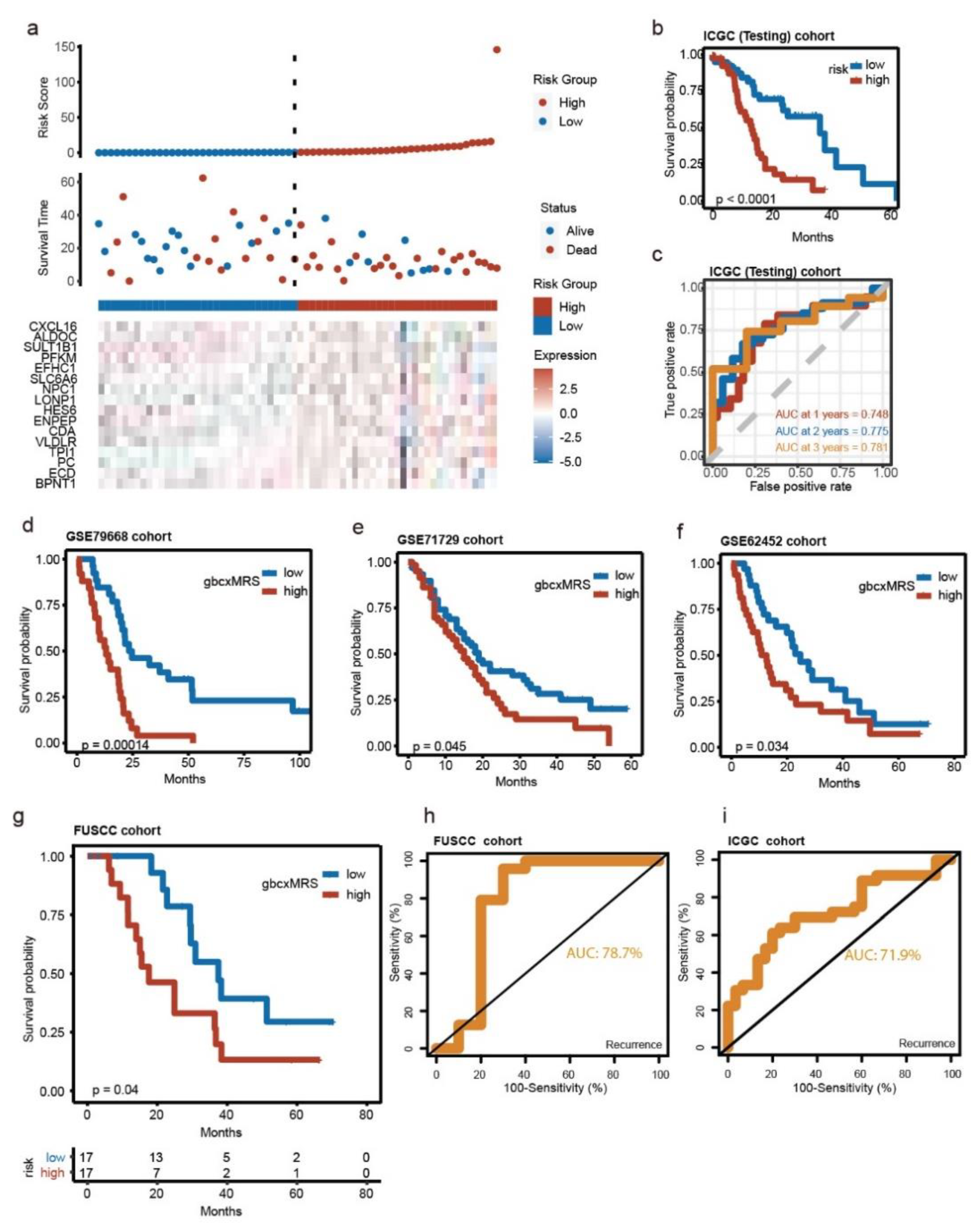
3.3. Robust and Repeated Validation of the 16-Gene Signature in External Cohorts
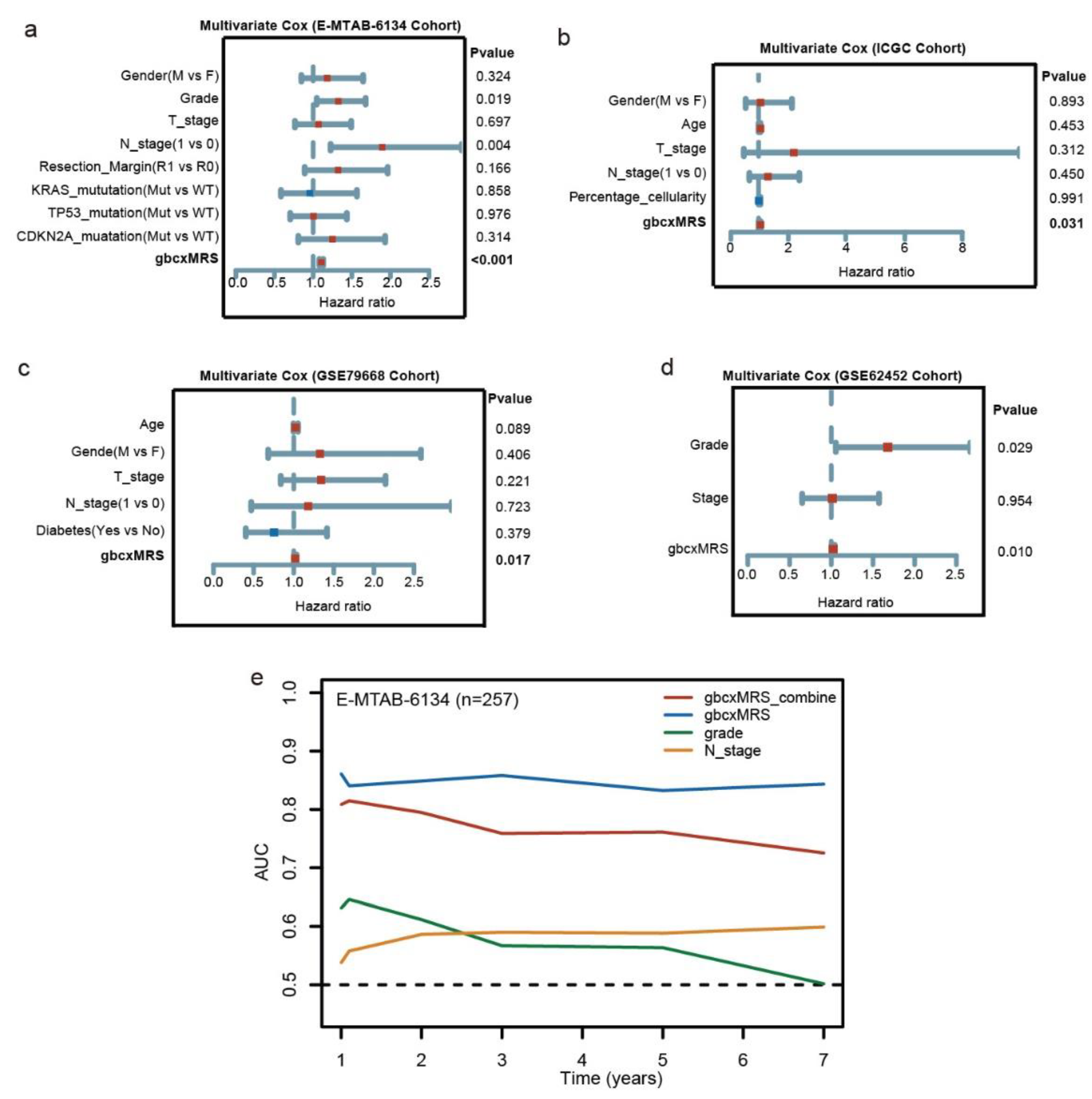
3.4. GbcxMRS Was an Independent and Indispensable Prognostic Factor in PDAC
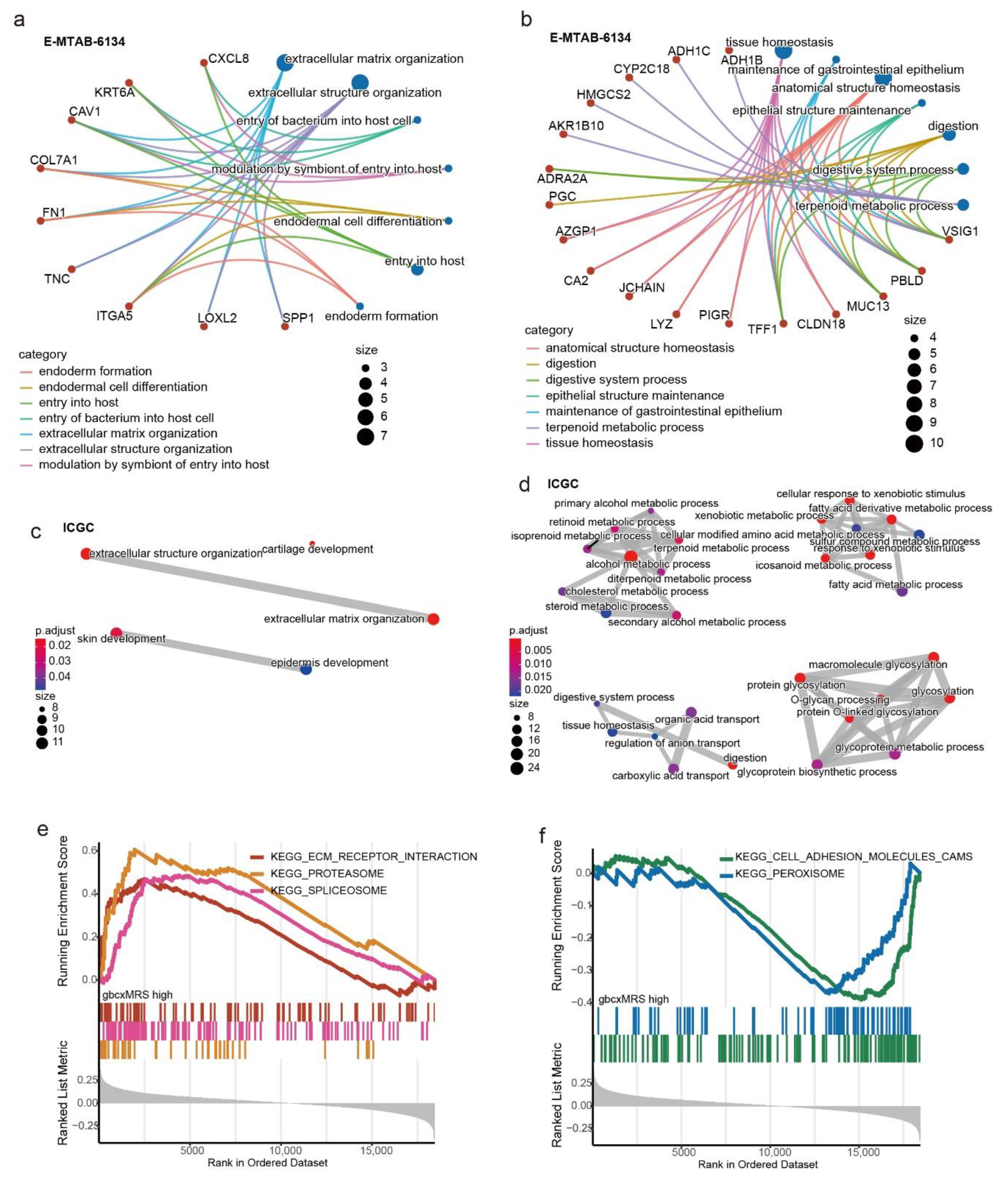
3.5. Functional Enrichment for the gbcxMRS
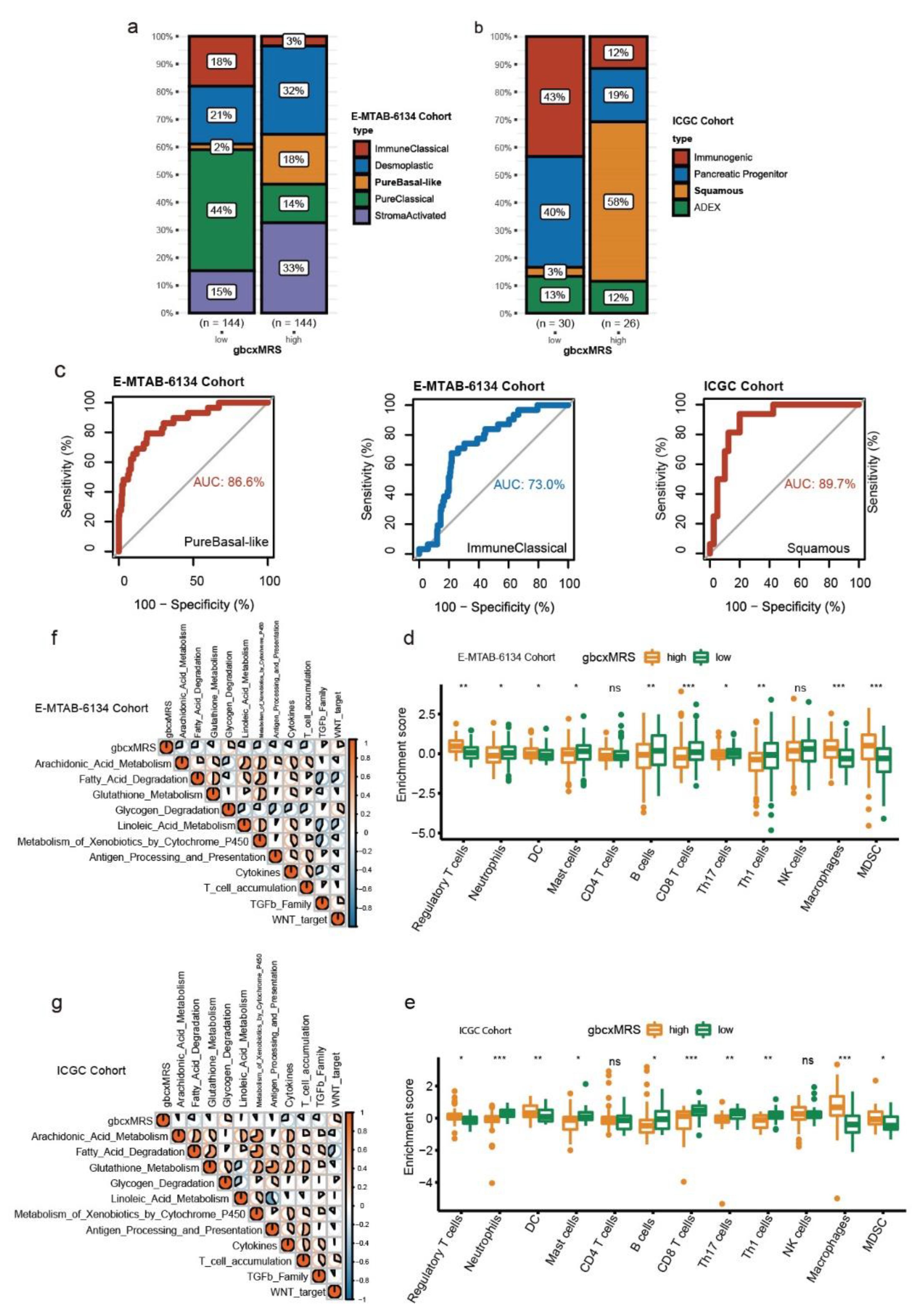
3.6. GbcxMRS Predicted PDAC Subtypes and Influenced the TME
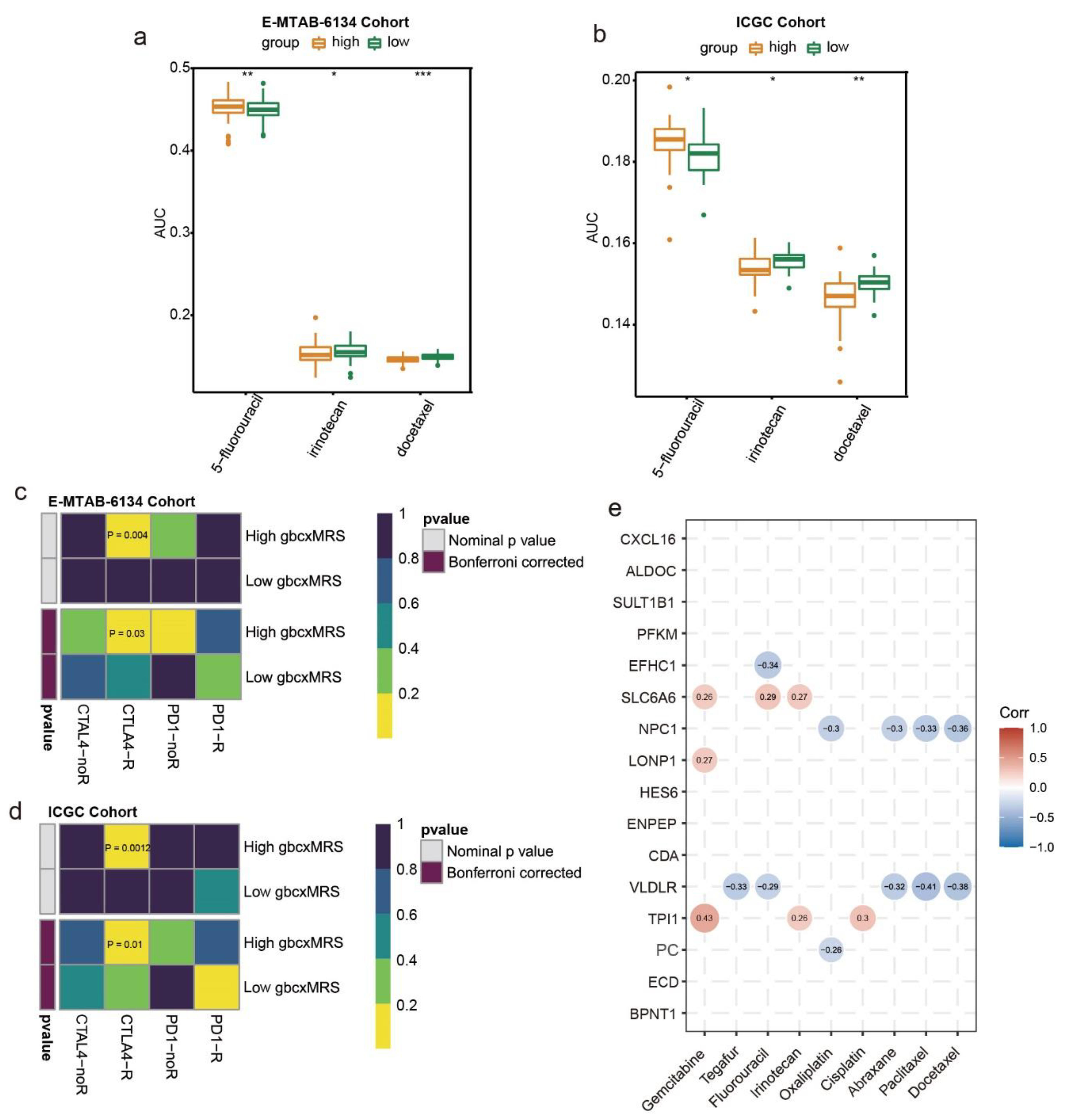
3.7. GbcxMRS Predicted Drug Sensitivity in PDAC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Khorolsky, C.; Benipal, B. Incidence of Pancreatic Adenocarcinoma in the United States from 2001 to 2015: A United States Cancer Statistics Analysis of 50 States. Cureus 2018, 10, e3796. [Google Scholar] [CrossRef] [Green Version]
- Bosetti, C.; Lucenteforte, E.; Silverman, D.T.; Petersen, G.; Bracci, P.M.; Ji, B.T.; Negri, E.; Li, D.; Risch, H.A.; Olson, S.H.; et al. Cigarette smoking and pancreatic cancer: An analysis from the International Pancreatic Cancer Case-Control Consortium (Panc4). Ann. Oncol. 2012, 23, 1880–1888. [Google Scholar] [CrossRef]
- Klein, A.P.; Brune, K.A.; Petersen, G.M.; Goggins, M.; Tersmette, A.C.; Offerhaus, G.J.; Griffin, C.; Cameron, J.L.; Yeo, C.J.; Kern, S.; et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004, 64, 2634–2638. [Google Scholar] [CrossRef] [Green Version]
- Bosetti, C.; Bertuccio, P.; Negri, E.; La Vecchia, C.; Zeegers, M.P.; Boffetta, P. Pancreatic cancer: Overview of descriptive epidemiology. Mol. Carcinog. 2012, 51, 3–13. [Google Scholar] [CrossRef]
- Oberstein, P.E.; Olive, K.P. Pancreatic cancer: Why is it so hard to treat? Therap. Adv. Gastroenterol. 2013, 6, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Lambe, M.; Eloranta, S.; Wigertz, A.; Blomqvist, P. Pancreatic cancer; reporting and long-term survival in Sweden. Acta Oncol. 2011, 50, 1220–1227. [Google Scholar] [CrossRef]
- Hardie, R.A.; van Dam, E.; Cowley, M.; Han, T.L.; Balaban, S.; Pajic, M.; Pinese, M.; Iconomou, M.; Shearer, R.F.; McKenna, J.; et al. Mitochondrial mutations and metabolic adaptation in pancreatic cancer. Cancer Metab. 2017, 5, 2. [Google Scholar] [CrossRef]
- Bilimoria, K.Y.; Bentrem, D.J.; Ko, C.Y.; Ritchey, J.; Stewart, A.K.; Winchester, D.P.; Talamonti, M.S. Validation of the 6th edition AJCC Pancreatic Cancer Staging System: Report from the National Cancer Database. Cancer 2007, 110, 738–744. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Lohr, J.M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.L.; Heinemann, V. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardi, N.; Ketkar, M.; McKinnon, R.A.; Pandol, S.J.; Dutt, S.; Barreto, S.G. Down-regulation of metabolic pathways could offset the poor prognosis conferred by co-existent diabetes mellitus in pancreatic (head) adenocarcinoma. ANZ J. Surg. 2021, 91, 2466–2474. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.P.; Sharma, A.; Nagpal, S.; Patlolla, S.H.; Sharma, A.; Kandlakunta, H.; Anani, V.; Angom, R.S.; Kamboj, A.K.; Ahmed, N.; et al. Phases of Metabolic and Soft Tissue Changes in Months Preceding a Diagnosis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2019, 156, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Baek, G.; Tse, Y.F.; Hu, Z.; Cox, D.; Buboltz, N.; McCue, P.; Yeo, C.J.; White, M.A.; DeBerardinis, R.J.; Knudsen, E.S.; et al. MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep. 2014, 9, 2233–2249. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 392. [Google Scholar] [CrossRef]
- Reske, S.N.; Grillenberger, K.G.; Glatting, G.; Port, M.; Hildebrandt, M.; Gansauge, F.; Beger, H.G. Overexpression of glucose transporter 1 and increased FDG uptake in pancreatic carcinoma. J. Nucl. Med. 1997, 38, 1344–1348. [Google Scholar] [PubMed]
- Scafoglio, C.; Hirayama, B.A.; Kepe, V.; Liu, J.; Ghezzi, C.; Satyamurthy, N.; Moatamed, N.A.; Huang, J.; Koepsell, H.; Barrio, J.R.; et al. Functional expression of sodium-glucose transporters in cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4111–E4119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Raj, P.; Yao, W.; Ying, H. Glucose Metabolism in Pancreatic Cancer. Cancers 2019, 11, 1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, H.; Bernstein, C.; Payne, C.M.; Dvorakova, K.; Garewal, H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat. Res.-Rev. Mutat. 2005, 589, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.Y.; Chen, Y.C. Role of bile acids in carcinogenesis of pancreatic cancer: An old topic with new perspective. World J. Gastroenterol. 2016, 22, 7463–7477. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Grunebach, F.; Schmidt, S.M.; Heine, A.; Hantschel, M.; Stevanovic, S.; Rammensee, H.G.; Brossart, P. Matrilysin (MMP-7) is a novel broadly expressed tumor antigen recognized by antigen-specific T cells. Clin. Cancer Res. 2008, 14, 5503–5511. [Google Scholar] [CrossRef] [Green Version]
- Harder, J.; Waiz, O.; Otto, F.; Geissler, M.; Olschewski, M.; Weinhold, B.; Blum, H.E.; Schmitt-Graeff, A.; Opitz, O.G. EGFR and HER2 expression in advanced biliary tract cancer. World J. Gastroenterol. 2009, 15, 4511–4517. [Google Scholar] [CrossRef]
- Pignochino, Y.; Sarotto, I.; Peraldo-Neia, C.; Penachioni, J.Y.; Cavalloni, G.; Migliardi, G.; Casorzo, L.; Chiorino, G.; Risio, M.; Bardelli, A.; et al. Targeting EGFR/HER2 pathways enhances the antiproliferative effect of gemcitabine in biliary tract and gallbladder carcinomas. BMC Cancer 2010, 10, 631. [Google Scholar] [CrossRef] [Green Version]
- Komoto, M.; Nakata, B.; Amano, R.; Yamada, N.; Yashiro, M.; Ohira, M.; Wakasa, K.; Hirakawa, K. HER2 overexpression correlates with survival after curative resection of pancreatic cancer. Cancer Sci. 2009, 100, 1243–1247. [Google Scholar] [CrossRef]
- Troiani, T.; Martinelli, E.; Capasso, A.; Morgillo, F.; Orditura, M.; De Vita, F.; Ciardiello, F. Targeting EGFR in pancreatic cancer treatment. Curr. Drug Targets 2012, 13, 802–810. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Lok, V.; Ngai, C.H.; Zhang, L.; Yuan, J.; Lao, X.Q.; Ng, K.; Chong, C.; Zheng, Z.J.; Wong, M.C.S. Worldwide Burden of, Risk Factors for, and Trends in Pancreatic Cancer. Gastroenterology 2021, 160, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gu, D.; Lee, S.S.; Song, B.; Bandyopadhyay, S.; Chen, S.; Konieczny, S.F.; Ratliff, T.L.; Liu, X.; Xie, J.; et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene 2016, 35, 6378–6388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigelsen, S. Evidence-based complementary treatment of pancreatic cancer: A review of adjunct therapies including paricalcitol, hydroxychloroquine, intravenous vitamin C, statins, metformin, curcumin, and aspirin. Cancer Manag. Res. 2018, 10, 2003–2018. [Google Scholar] [CrossRef] [Green Version]
- Shaw, V.; Srivastava, S.; Srivastava, S.K. Repurposing antipsychotics of the diphenylbutylpiperidine class for cancer therapy. Semin. Cancer Biol. 2021, 68, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.Z.; Chang, J.I.; Li, E.; Xiang, A.H.; Wu, B.U. Influence of Statins and Cholesterol on Mortality Among Patients With Pancreatic Cancer. J. Natl. Cancer Inst. 2017, 109, 275. [Google Scholar] [CrossRef] [PubMed]
- Dale, K.M.; Coleman, C.I.; Henyan, N.N.; Kluger, J.; White, C.M. Statins and cancer risk: A meta-analysis. JAMA 2006, 295, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Gabitova-Cornell, L.; Surumbayeva, A.; Peri, S.; Franco-Barraza, J.; Restifo, D.; Weitz, N.; Ogier, C.; Goldman, A.R.; Hartman, T.R.; Francescone, R.; et al. Cholesterol Pathway Inhibition Induces TGF-beta Signaling to Promote Basal Differentiation in Pancreatic Cancer. Cancer Cell 2020, 38, 567–583.e11. [Google Scholar] [CrossRef]
- Adams, C.R.; Htwe, H.H.; Marsh, T.; Wang, A.L.; Montoya, M.L.; Subbaraj, L.; Tward, A.D.; Bardeesy, N.; Perera, R.M. Transcriptional control of subtype switching ensures adaptation and growth of pancreatic cancer. eLife 2019, 8, 45313. [Google Scholar] [CrossRef]
- Shi, X.H.; Li, X.; Zhang, H.; He, R.Z.; Zhao, Y.; Zhou, M.; Pan, S.T.; Zhao, C.L.; Feng, Y.C.; Wang, M.; et al. A Five-microRNA Signature for Survival Prognosis in Pancreatic Adenocarcinoma based on TCGA Data. Sci. Rep. 2018, 8, 7638. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Xu, Q.; Zhang, H.; Yin, X.; Zhu, C.; Zhao, K.; Zhu, J. Five key lncRNAs considered as prognostic targets for predicting pancreatic ductal adenocarcinoma. J. Cell Biochem. 2018, 119, 4559–4569. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Wan, H.; Hao, X.; Lan, T.; Li, W.; Xu, L.; Yuan, K.; Wu, H. Importance of gene expression signatures in pancreatic cancer prognosis and the establishment of a prediction model. Cancer Manag. Res. 2019, 11, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Z.; Lei, Y.; Xu, J.; Shi, S.; Hua, J.; Zhang, B.; Meng, Q.; Liu, J.; Zhang, Y.; Wei, M.; et al. The value of a metabolic reprogramming-related gene signature for pancreatic adenocarcinoma prognosis prediction. Aging 2020, 12, 24228–24241. [Google Scholar] [CrossRef] [PubMed]
- Anslan, S.; Bahram, M.; Hiiesalu, I.; Tedersoo, L. PipeCraft: Flexible open-source toolkit for bioinformatics analysis of custom high-throughput amplicon sequencing data. Mol. Ecol. Resour. 2017, 17, e234–e240. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.K.; Ramaker, R.C.; Gertz, J.; Davis, N.S.; Johnston, B.E.; Oliver, P.G.; Sexton, K.C.; Greeno, E.W.; Christein, J.D.; Heslin, M.J.; et al. RNA sequencing of pancreatic adenocarcinoma tumors yields novel expression patterns associated with long-term survival and reveals a role for ANGPTL4. Mol. Oncol. 2016, 10, 1169–1182. [Google Scholar] [CrossRef] [Green Version]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Yang, S.; He, P.; Wang, J.; Schetter, A.; Tang, W.; Funamizu, N.; Yanaga, K.; Uwagawa, T.; Satoskar, A.R.; Gaedcke, J.; et al. A Novel MIF Signaling Pathway Drives the Malignant Character of Pancreatic Cancer by Targeting NR3C2. Cancer Res. 2016, 76, 3838–3850. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; He, P.; Tan, H.; Budhu, A.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Yfantis, H.G.; Lee, D.H.; Maitra, A.; et al. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin. Cancer Res. 2013, 19, 4983–4993. [Google Scholar] [CrossRef] [Green Version]
- Reinhold, W.C.; Varma, S.; Sunshine, M.; Elloumi, F.; Ofori-Atta, K.; Lee, S.; Trepel, J.B.; Meltzer, P.S.; Doroshow, J.H.; Pommier, Y. RNA Sequencing of the NCI-60: Integration into CellMiner and CellMiner CDB. Cancer Res. 2019, 79, 3514–3524. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [Green Version]
- Reinhold, W.C.; Sunshine, M.; Varma, S.; Doroshow, J.H.; Pommier, Y. Using CellMiner 1.6 for Systems Pharmacology and Genomic Analysis of the NCI-60. Clin. Cancer Res. 2015, 21, 3841–3852. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Huang, X.; Li, Y.; Chen, J.; Lv, Y.; Dai, S. Prognosis and personalized treatment prediction in TP53-mutant hepatocellular carcinoma: An in silico strategy towards precision oncology. Brief Bioinform. 2021, 22, 164. [Google Scholar] [CrossRef] [PubMed]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef]
- Gu, W.; Kim, M.; Wang, L.; Yang, Z.; Nakajima, T.; Tsushima, Y. Multi-omics Analysis of Ferroptosis Regulation Patterns and Characterization of Tumor Microenvironment in Patients with Oral Squamous Cell Carcinoma. Int. J. Biol. Sci. 2021, 17, 3476–3492. [Google Scholar] [CrossRef]
- Hoshida, Y.; Brunet, J.P.; Tamayo, P.; Golub, T.R.; Mesirov, J.P. Subclass mapping: Identifying common subtypes in independent disease data sets. PLoS ONE 2007, 2, e1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e3. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Erkan, M.; Kurtoglu, M.; Kleeff, J. The role of hypoxia in pancreatic cancer: A potential therapeutic target? Expert Rev. Gastroenterol. Hepatol. 2016, 10, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Yang, G.; Zhou, W.; Qiu, J.; Chen, G.; Luo, W.; Zhao, F.; You, L.; Zheng, L.; Zhang, T.; et al. Targeting hypoxic tumor microenvironment in pancreatic cancer. J. Hematol. Oncol. 2021, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Abou Khouzam, R.; Rao, S.P.; Venkatesh, G.H.; Zeinelabdin, N.A.; Buart, S.; Meylan, M.; Nimmakayalu, M.; Terry, S.; Chouaib, S. An Eight-Gene Hypoxia Signature Predicts Survival in Pancreatic Cancer and Is Associated With an Immunosuppressed Tumor Microenvironment. Front. Immunol. 2021, 12, 680435. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Peng, C.; Zhu, C.; Nie, S.; Qian, X.; Shi, Z.; Shi, M.; Liang, Y.; Ding, X.; Zhang, S.; et al. Hypoxia promotes the metastasis of pancreatic cancer through regulating NOX4/KDM5A-mediated histone methylation modification changes in a HIF1A-independent manner. Clin. Epigenet. 2021, 13, 18. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Rosario, S.R.; Long, M.D.; Affronti, H.C.; Rowsam, A.M.; Eng, K.H.; Smiraglia, D.J. Pan-cancer analysis of transcriptional metabolic dysregulation using The Cancer Genome Atlas. Nat. Commun. 2018, 9, 5330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandimalla, R.; Tomihara, H.; Banwait, J.K.; Yamamura, K.; Singh, G.; Baba, H.; Goel, A. A 15-Gene Immune, Stromal, and Proliferation Gene Signature that Significantly Associates with Poor Survival in Patients with Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2020, 26, 3641–3648. [Google Scholar] [CrossRef] [Green Version]
- Dey, P.; Rachagani, S.; Chakraborty, S.; Singh, P.K.; Zhao, X.; Gurumurthy, C.B.; Anderson, J.M.; Lele, S.; Hollingsworth, M.A.; Band, V.; et al. Overexpression of ecdysoneless in pancreatic cancer and its role in oncogenesis by regulating glycolysis. Clin. Cancer Res. 2012, 18, 6188–6198. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Han, J.; Long, Y.S.; Epstein, P.N.; Liu, Y.Q. The role of pyruvate carboxylase in insulin secretion and proliferation in rat pancreatic beta cells. Diabetologia 2008, 51, 2022–2030. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, M.J.; Efendić, S.; Ostenson, C.G. Normalization by insulin treatment of low mitochondrial glycerol phosphate dehydrogenase and pyruvate carboxylase in pancreatic islets of the GK rat. Diabetes 1996, 45, 886–890. [Google Scholar] [CrossRef]
- Lau, A.N.; Li, Z.; Danai, L.V.; Westermark, A.M.; Darnell, A.M.; Ferreira, R.; Gocheva, V.; Sivanand, S.; Lien, E.C.; Sapp, K.M.; et al. Dissecting cell-type-specific metabolism in pancreatic ductal adenocarcinoma. eLife 2020, 9, 56782. [Google Scholar] [CrossRef] [PubMed]
- Tanner, L.B.; Goglia, A.G.; Wei, M.H.; Sehgal, T.; Parsons, L.R.; Park, J.O.; White, E.; Toettcher, J.E.; Rabinowitz, J.D. Four Key Steps Control Glycolytic Flux in Mammalian Cells. Cell Syst. 2018, 7, 49–62.e8. [Google Scholar] [CrossRef] [PubMed]
- Cortese, N.; Capretti, G.; Barbagallo, M.; Rigamonti, A.; Takis, P.G.; Castino, G.F.; Vignali, D.; Maggi, G.; Gavazzi, F.; Ridolfi, C.; et al. Metabolome of Pancreatic Juice Delineates Distinct Clinical Profiles of Pancreatic Cancer and Reveals a Link between Glucose Metabolism and PD-1+ Cells. Cancer Immunol. Res. 2020, 8, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.M.; Tien, S.C.; Hsieh, P.K.; Jeng, Y.M.; Chang, M.C.; Chang, Y.T.; Chen, Y.J.; Chen, Y.J.; Lee, E.Y.P.; Lee, W.H. High Glucose Triggers Nucleotide Imbalance through O-GlcNAcylation of Key Enzymes and Induces KRAS Mutation in Pancreatic Cells. Cell Metab. 2019, 29, 1334–1349.e10. [Google Scholar] [CrossRef]
- Obata, A.; Kimura, T.; Obata, Y.; Shimoda, M.; Kinoshita, T.; Kohara, K.; Okauchi, S.; Hirukawa, H.; Kamei, S.; Nakanishi, S.; et al. Vascular endothelial PDPK1 plays a pivotal role in the maintenance of pancreatic beta cell mass and function in adult male mice. Diabetologia 2019, 62, 1225–1236. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.L.; Yu, G.; Dong, H.; Chen, K.; Xie, J.; Yu, H.; Ji, Y.; Yang, G.S.; Li, A.J.; Cong, W.M.; et al. Proteomics analysis identified TPI1 as a novel biomarker for predicting recurrence of intrahepatic cholangiocarcinoma. J. Gastroenterol. 2020, 55, 1171–1182. [Google Scholar] [CrossRef]
- Qiao, G.; Li, J.; Wang, J.; Wang, Z.; Bian, W. miR-381 functions as a tumor suppressor by targeting ETS1 in pancreatic cancer. Int. J. Mol. Med. 2019, 44, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.; Feng, L.; Yu, S.; Yu, C.; Chi, N. MicroRNA-769-5p Inhibits Pancreatic Ductal Adenocarcinoma Progression by Directly Targeting and Downregulating ETS Proto-Oncogene 1. Oncol. Targets Ther. 2019, 12, 11737–11750. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Duxbury, M.; Benoit, E.; Clancy, T.E.; Zinner, M.J.; Ashley, S.W.; Whang, E.E. Prostaglandin E2 enhances pancreatic cancer invasiveness through an Ets-1-dependent induction of matrix metalloproteinase-2. Cancer Res. 2004, 64, 7439–7446. [Google Scholar] [CrossRef] [Green Version]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Bjånes, T.K.; Jordheim, L.P.; Schjøtt, J.; Kamceva, T.; Cros-Perrial, E.; Langer, A.; Ruiz de Garibay, G.; Kotopoulis, S.; McCormack, E.; Riedel, B. Intracellular Cytidine Deaminase Regulates Gemcitabine Metabolism in Pancreatic Cancer Cell Lines. Drug Metab. Dispos. 2020, 48, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawai, Y.; Kodama, Y.; Shimizu, T.; Ota, Y.; Maruno, T.; Eso, Y.; Kurita, A.; Shiokawa, M.; Tsuji, Y.; Uza, N.; et al. Activation-Induced Cytidine Deaminase Contributes to Pancreatic Tumorigenesis by Inducing Tumor-Related Gene Mutations. Cancer Res. 2015, 75, 3292–3301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A. Tracking down the hedgehog’s lair in the pancreas. Gastroenterology 2010, 138, 823–825. [Google Scholar] [CrossRef] [Green Version]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Sousa, B.; Pereira, J.; Paredes, J. The Crosstalk Between Cell Adhesion and Cancer Metabolism. Int. J. Mol. Sci. 2019, 20, 1933. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, W.J.; Mullen, P.J.; Schmid, E.W.; Flores, A.; Momcilovic, M.; Sharpley, M.S.; Jelinek, D.; Whiteley, A.E.; Maxwell, M.B.; Wilde, B.R.; et al. Extracellular Matrix Remodeling Regulates Glucose Metabolism through TXNIP Destabilization. Cell 2018, 175, 117–132.e1. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Li, J.; Yu, Z.; Rao, H.; Wang, S.; Lan, H. HMGB1 promotes HLF-1 proliferation and ECM production through activating HIF1-α-regulated aerobic glycolysis. Pulm. Pharmacol. Ther. 2017, 45, 136–141. [Google Scholar] [CrossRef]
- Nokin, M.J.; Bellier, J.; Durieux, F.; Peulen, O.; Rademaker, G.; Gabriel, M.; Monseur, C.; Charloteaux, B.; Verbeke, L.; van Laere, S.; et al. Methylglyoxal, a glycolysis metabolite, triggers metastasis through MEK/ERK/SMAD1 pathway activation in breast cancer. Breast Cancer Res. 2019, 21, 11. [Google Scholar] [CrossRef]
- Huang, C.; Hays, F.A.; Tomasek, J.J.; Benyajati, S.; Zhang, X.A. Tetraspanin CD82 interaction with cholesterol promotes extracellular vesicle-mediated release of ezrin to inhibit tumour cell movement. J. Extracell. Vesicles 2020, 9, 1692417. [Google Scholar] [CrossRef]
- Caldieri, G.; Giacchetti, G.; Beznoussenko, G.; Attanasio, F.; Ayala, I.; Buccione, R. Invadopodia biogenesis is regulated by caveolin-mediated modulation of membrane cholesterol levels. J. Cell Mol. Med. 2009, 13, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, W.; Li, B.; Zhang, D. Activation of G-protein-coupled bile acid receptor Gpbar1 (TGR5) inhibits degradation of type II collagen and aggrecan in human chondrocytes. Eur. J. Pharmacol. 2019, 856, 172387. [Google Scholar] [CrossRef]
- Dreyer, S.B.; Jamieson, N.B.; Upstill-Goddard, R.; Bailey, P.J.; McKay, C.J.; Biankin, A.V.; Chang, D.K. Defining the molecular pathology of pancreatic body and tail adenocarcinoma. Br. J. Surg. 2018, 105, e183–e191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Kane, G.M.; Grünwald, B.T.; Jang, G.H.; Masoomian, M.; Picardo, S.; Grant, R.C.; Denroche, R.E.; Zhang, A.; Wang, Y.; Lam, B.; et al. GATA6 Expression Distinguishes Classical and Basal-like Subtypes in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4901–4910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.S.; Gupta, V.K.; Garrido, V.T.; Hadad, R.; Durden, B.C.; Kesh, K.; Giri, B.; Ferrantella, A.; Dudeja, V.; Saluja, A.; et al. Targeting tumor-intrinsic hexosamine biosynthesis sensitizes pancreatic cancer to anti-PD1 therapy. J. Clin. Investig. 2020, 130, 451–465. [Google Scholar] [CrossRef] [Green Version]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8+ T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156.e5. [Google Scholar] [CrossRef]
- Fritschi, L.; Benke, G.; Risch, H.A.; Schulte, A.; Webb, P.M.; Whiteman, D.C.; Fawcett, J.; Neale, R.E. Occupational exposure to N-nitrosamines and pesticides and risk of pancreatic cancer. Occup. Environ. Med. 2015, 72, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Bengsch, F.; Knoblock, D.M.; Liu, A.; McAllister, F.; Beatty, G.L. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol. Immunother. 2017, 66, 1609–1617. [Google Scholar] [CrossRef]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Zappasodi, R.; Serganova, I.; Cohen, I.J.; Maeda, M.; Shindo, M.; Senbabaoglu, Y.; Watson, M.J.; Leftin, A.; Maniyar, R.; Verma, S.; et al. CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature 2021, 591, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liang, X.; Zhang, L.; Wang, D. Metabolic characterization and metabolism-score of tumor to predict the prognosis in prostate cancer. Sci. Rep. 2021, 11, 22486. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, 8399. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Features | gbcxMRS High | gbcxMRS Low | p Value |
|---|---|---|---|
| n = 130 | n = 127 | ||
| OS.time | 18.5 (14.2) | 36.7 (27.7) | <0.001 |
| OS: | <0.001 | ||
| Alive | 23 (17.7%) | 72 (56.7%) | |
| Dead | 107 (82.3%) | 55 (43.3%) | |
| Gender: | 0.242 | ||
| Female | 48 (36.9%) | 57 (44.9%) | |
| Male | 82 (63.1%) | 70 (55.1%) | |
| Grade: | <0.001 | ||
| G1 | 38 (29.2%) | 65 (51.2%) | |
| G2 | 61 (46.9%) | 53 (41.7%) | |
| G3 | 31 (23.8%) | 9 (7.09%) | |
| T stage: | 0.196 | ||
| T1 | 3 (2.31%) | 8 (6.30%) | |
| T2 | 21 (16.2%) | 15 (11.8%) | |
| T3 | 106 (81.5%) | 104 (81.9%) | |
| N stage: | 0.065 | ||
| N0 | 25 (19.2%) | 38 (29.9%) | |
| N1 | 105 (80.8%) | 89 (70.1%) | |
| Resection margin: | 0.177 | ||
| resection margin R0 | 101 (77.7%) | 108 (85.0%) | |
| resection margin R1 | 29 (22.3%) | 19 (15.0%) | |
| KRAS mutation: | 0.093 | ||
| mutation in KRAS | 110 (84.6%) | 117 (92.1%) | |
| no mutation in KRAS | 20 (15.4%) | 10 (7.87%) | |
| TP53 mutation: | 0.14 | ||
| mutation in TP53 | 96 (73.8%) | 82 (64.6%) | |
| no mutation in TP53 | 34 (26.2%) | 45 (35.4%) | |
| CDKN2A mutation: | 0.261 | ||
| mutation in CDKN2A | 24 (18.5%) | 16 (12.6%) | |
| no mutation in CDKN2A | 106 (81.5%) | 111 (87.4%) |
| Clinical Features | gbcxMRS Low n = 17 | gbcxMRS High n = 17 | p Value |
|---|---|---|---|
| Gender | 0.084 | ||
| Female | 11 (32.4%) | 5 (14.7%) | |
| Male | 6 (17.6%) | 12 (35.3%) | |
| Grade | 1.000 | ||
| High--middle | 13 (38.2%) | 12 (35.3%) | |
| Low | 4 (11.8%) | 5 (14.7%) | |
| Tissue invasion | 0.265 | ||
| NO | 3 (9.4%) | 7 (21.9%) | |
| YES | 12 (37.5%) | 10 (31.2%) | |
| Lymph node metastasis | 0.084 | ||
| NO | 12 (35.3%) | 6 (17.6%) | |
| YES | 5 (14.7%) | 11 (32.4%) | |
| Tumor thrombus | 0.688 | ||
| NO | 12 (35.3%) | 14 (41.2%) | |
| YES | 5 (14.7%) | 3 (8.8%) | |
| Neural invasion | 1.000 | ||
| NO | 2 (5.9%) | 1 (2.9%) | |
| YES | 15 (44.1%) | 16 (47.1%) | |
| Recurrence | 0.017 | ||
| NO | 8 (23.5%) | 1 (2.9%) | |
| YES | 9 (26.5%) | 16 (47.1%) | |
| Age (mean ± SD) | 59.82 ± 9.82 | 61.88 ± 7.51 | 0.497 |
| Tumor size (mean ± SD) | 3.63 ± 1.94 | 4.06 ± 1.49 | 0.475 |
| CA19-9, median | 51.2 (17.34, 153.2) | 448.3 (38.75, 727.1) | 0.042 |
| CA125, median | 21.06 (15.02, 30.89) | 18.69 (11.65, 23.87) | 0.357 |
| CA50, median | 13.92 (5.41, 83.47) | 154.74 (14.65, 327.13) | 0.063 |
| CA242, median | 7.91 (4.11, 22.47) | 52.89 (13.83, 150) | 0.025 |
| CEA, median | 2.11 (1.71, 2.82) | 4.52 (2.67, 7.43) | 0.046 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, W.; Mo, S.; Wang, Y.; Kawabata-Iwakawa, R.; Zhang, W.; Yang, Z.; Sun, C.; Tsushima, Y.; Xu, H.; Nakajima, T. Robust Validation and Comprehensive Analysis of a Novel Signature Derived from Crucial Metabolic Pathways of Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 1825. https://doi.org/10.3390/cancers14071825
Gu W, Mo S, Wang Y, Kawabata-Iwakawa R, Zhang W, Yang Z, Sun C, Tsushima Y, Xu H, Nakajima T. Robust Validation and Comprehensive Analysis of a Novel Signature Derived from Crucial Metabolic Pathways of Pancreatic Ductal Adenocarcinoma. Cancers. 2022; 14(7):1825. https://doi.org/10.3390/cancers14071825
Chicago/Turabian StyleGu, Wenchao, Shaocong Mo, Yulin Wang, Reika Kawabata-Iwakawa, Wei Zhang, Zongcheng Yang, Chenyu Sun, Yoshito Tsushima, Huaxiang Xu, and Takahito Nakajima. 2022. "Robust Validation and Comprehensive Analysis of a Novel Signature Derived from Crucial Metabolic Pathways of Pancreatic Ductal Adenocarcinoma" Cancers 14, no. 7: 1825. https://doi.org/10.3390/cancers14071825
APA StyleGu, W., Mo, S., Wang, Y., Kawabata-Iwakawa, R., Zhang, W., Yang, Z., Sun, C., Tsushima, Y., Xu, H., & Nakajima, T. (2022). Robust Validation and Comprehensive Analysis of a Novel Signature Derived from Crucial Metabolic Pathways of Pancreatic Ductal Adenocarcinoma. Cancers, 14(7), 1825. https://doi.org/10.3390/cancers14071825