1. Introduction
Hepatocellular carcinoma (HCC) is a dominant health problem. Detection of HCC at an early stage is difficult and most tumors are detected at an advanced stage due to their rapid progression and high metastasis, resulting in a poor prognosis for affected individuals. The main options for the clinical treatment of HCC include surgery, radiation therapy, transarterial chemoembolization, and molecular targeted therapy. First-line drugs include sorafenib (approved in 2007) and lenvatinib (approved in 2018), as well as the most recently approved combination therapy of atezolizumab and bevacizumab (approved in 2020). In addition, a number of second-line or tertiary drugs, which are targeted small molecular drugs, such as cabozantinib (2019), regorafenib (2017), the monoclonal antibody ramucirumab (2019), and the immune checkpoint inhibitors nivolumab (2017) and pembrolizumab (2018), have been approved by the U.S. Food and Drug Administration (FDA) as a follow-up treatment for patients pretreated with sorafenib [
1]. However, there are limitations in the treatment of HCC. Most patients develop resistance to sorafenib within 6 months, resulting in a poor response rate. In the case of lenvatinib, follow-up treatment is impossible due to the absence of an approved second-line treatment. As immune checkpoint inhibitors have few side effects, their demand has surged worldwide over the past 5 years. However, even these have now been associated with a novel spectrum of side-effects, such as autoimmune-related adverse events [
2]. Despite all these possible treatments, the median progression-free survival (PFS) in patients with HCC is less than 1 year. Existing treatments do not completely treat liver cancer, but only prolong survival. Although advances in medical technology have increased the promising 5-year survival rate of patients with HCC, long-term survival rates after surgical resection remain low due to high recurrence and metastasis rates [
3,
4]. Thus, there is an urgent need to develop new evidence-based therapeutics for the treatment of HCC.
In general, the pathogenesis of HCC includes the induction of tumor growth, metastasis, and angiogenesis through the binding of growth factors to cell surface receptors, which activate the proliferative signaling system. The development of therapeutics targeting such signal transduction pathways has been actively studied over the last years. For instance, a multiple kinase inhibitor (TKI), sorafenib (Nexavar
®), was reported to prolong the survival period of patients with advanced HCC [
5]. In addition, various targeted therapeutics have been studied, raising expectations for the treatment of refractory HCC. The advent of TKIs has become a promising targeted therapeutic strategy [
6,
7,
8]. Briefly, TKIs enter cells and interact with several receptors and other intracellular signaling molecules to block the phosphorylation of tyrosine residues and hence the activation of various downstream signaling pathways, such as Ras/Raf/MEK/MAPK and PI3K/AKT/mTOR [
9].
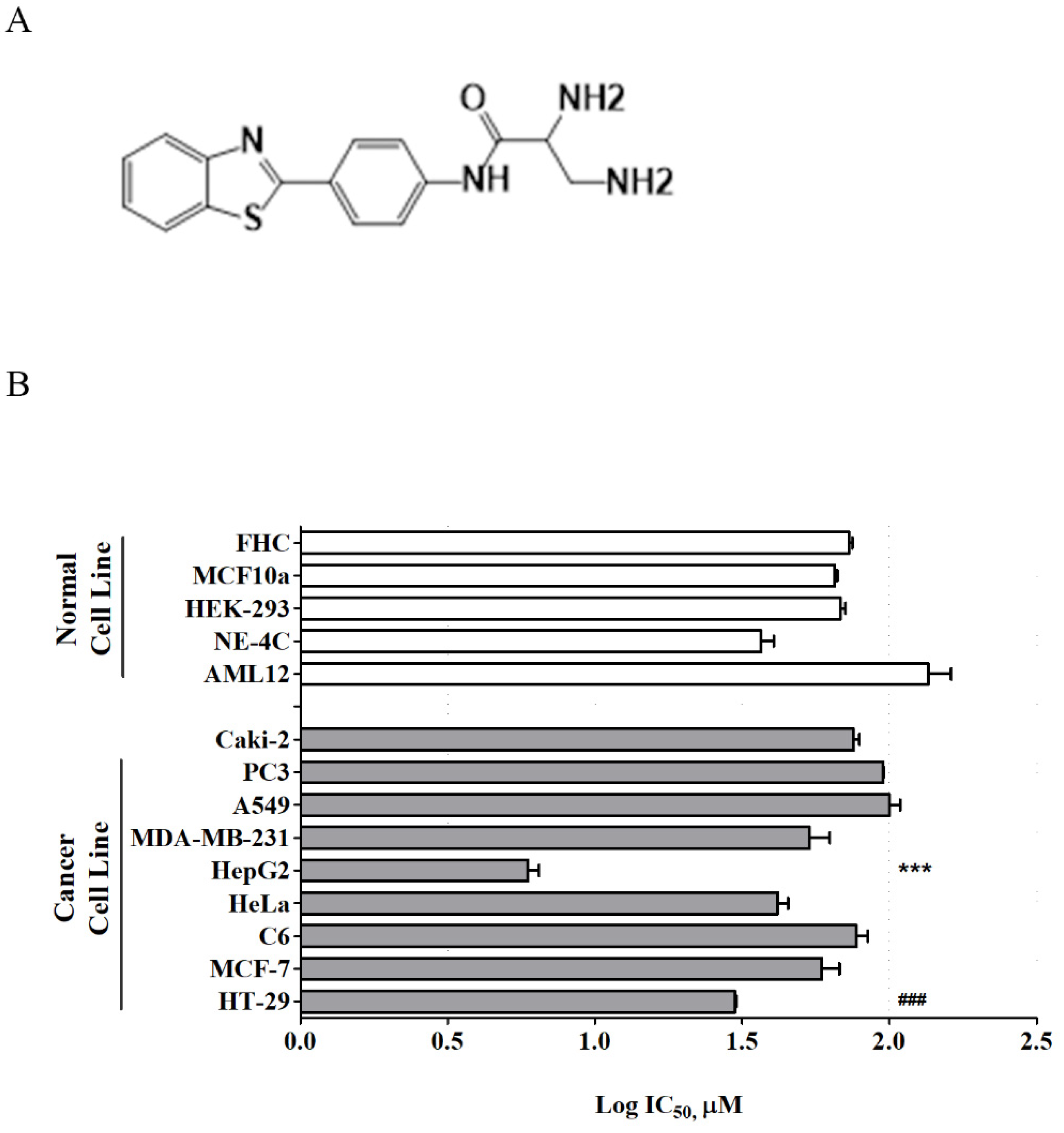
Recently, a new series of benzothiazole aniline derivatives have been synthesized in our laboratory, and among them, MBP-11901 (
Figure 1A) showed excellent inhibitory activity against HCC cell growth in vitro [
10]. The present study investigated the efficacy of this novel orally administered TKI, MBP-11901, against human HCC in vitro and in vivo with the aim of ultimately characterizing its mechanism of action and its therapeutic potential in treating HCC.
2. Materials and Methods
2.1. Chemicals
MBP-11901 (
Figure 1) was prepared at 99.64% purity as previously reported [
10] with further modifications for pilot-scale production. HPLC analysis was performed using a Prominence model (Shimadzu Corporation, Kyoto, Japan) equipped with a photodiode array detector (SPD-M20A) to confirm the purity of MBP-11901 (
Figure S1). Test sample solution was prepared in water at a concentration of 200 ppm, and a SunFire C18 column (100 Å, 5 µm, 4.6 mm × 250 mm) was used for analysis. Mobile phases consisted of 0.1% formic acid-buffered water (A) and acetonitrile (B), with the gradient mode at a flow rate of 1 mL/min. Injection volume was 10 µL and the wavelength was fixed at 310 nm for UV detection. Data were acquired based on the following gradient system: T
min/B%—T
0/10, T
1.5/10, T
4.5/60, T
10.5/80, T
15/80, T
28.5/10, T
30/10.
2.2. Reagents
Eagle’s minimum essential medium (EMEM) was obtained from ATCC (Cat. 30-2003, Manassas, VA, USA). RPMI-1640 medium was obtained from Welgene (Cat. LM011-01, Daegu, Republic of Korea). Fetal bovine serum (FBS) was obtained from Hyclone (Cat. SH30919.03, Waltham, MA, USA). Antibiotic–antimycotic solution was obtained from Hyclone (Cat. SV30079.01). Antibodies against β-actin (Cat. sc-47778) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies against phospho-FLT3 (Cat. #3461), Ras (Cat. #3965), phospho-c-Raf (Cat. #9421), phospho-MEK1/2 (Cat. #9121), phospho-ERK1/2 (Cat. #9101), phospho-Akt (Cat. #9271), phospho-mTOR (Cat. #5536), phospho-VEGFR2 (Cat. #3770), caspase-3 (Cat. #9662), cleaved caspase-3 (Cat. #9661), and PARP (Cat. #9542) were purchased from Cell Signaling Technology (Beverly, MA, USA). Phospho-PDGFRβ (Cat. LF-PA0035) was obtained from BioVendor Laboratory Medicine Inc. (Brno, Czech Republic).
2.3. Cell Lines and Culture
All cancer cell lines and corresponding normal cell lines were purchased from the American Type Culture Collection (ATCC). Hepatocellular carcinoma cell lines, HepG2 (HBV(−), p53 wt, ATCC® HB-8065™) and Hep3B (HBV(+), p53 null, ATCC® HB-8064™) were obtained from ATCC. Huh-7 (HBV(−), p53 mut., KCLB No. 60104) and PLC/PRF.5 (HBV(+), p53 mut., KCLB No. 28024) cells were obtained from the Korean Cell Line Bank (Seoul National University, Seoul, Korea).
Human colorectal adenocarcinoma cells (HT-29, ATCC® HTB-38™), adenocarcinoma human alveolar basal epithelial cells (A549, ATCC® CCL-185), and prostate cancer cells (PC-3, ATCC® CRL-1435) were cultured in RPMI1640 medium. Human breast adenocarcinoma cells (MCF-7, ATCC® HTB-22™ and MDA-MB-231, ATCC® HTB-26™), rattus brain glioma cells (C6, ATCC® CRL-2303™), and human renal carcinoma cells (Caki-2, ATCC® HTB-47™) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Cat. LM001-05, WelGENE, Daegu, Korea). Mouse brain neural cells (NE-4C, ATCC® CRL-2925™) and human cervix adenocarcinoma cells (HeLa, ATCC® CCL-2™) were cultured in EMEM (ATCC® 30-2003™), while human embryonic kidney cells (HEK-293, ATCC® CRL-1573™) were cultured in minimum essential medium (MEM, Cat. LM007-09, WelGENE). All media contained 10% (v/v) FBS and antibiotic–antimycotic solution (1 mL per 100 mL of cell culture medium). Mouse liver normal cells (AML12, ATCC® CRL-2254™) were cultured in Dulbecco’s modified Eagle medium/nutrient mixture F-12 (DMEM/F-12, Cat. LM002-04, WelGENE) containing 10% FBS (Hyclone), 1× ITS (10 μg/mL insulin, 5.5 μg/mL transferrin, and 6.7 ng/mL selenium; Cat. 41400-045, Gibco, Carlsbad, CA, USA), 40 ng/mL dexamethasone (Cat. D4902, Sigma-Aldrich, St. Louis, MO, USA), and 1% antibiotic–antimycotic solution (Hyclone). Human breast epithelial cells (MCF 10A, ATCC® CRL-10317™) were cultured in DMEM/F-12 (WelGENE) supplemented with 5% horse serum (Cat. 16050-122, Gibco), 20 ng/mL epidermal growth factor (EGF; Peprotech Inc., Rocky Hill, NJ, USA), 500 μg/mL hydrocortisone (Sigma-Aldrich), 100 ng/mL cholera toxin (Sigma-Aldrich), 10 μg/mL insulin (Cat. I9278, Sigma-Aldrich), and 1% antibiotic–antimycotic solution (Hyclone). Human colon normal epithelial cells (FHC, ATCC® CRL-1831™) were cultured in DMEM/F-12 (WelGENE) supplemented with 10% FBS (Hyclone), 10 ng/mL cholera toxin (Sigma-Aldrich), 20 ng/mL epidermal growth factor (EGF; Peprotech Inc., Rocky Hill, NJ, USA), 100 μg/mL hydrocortisone (Sigma-Aldrich), 1× ITS (10 μg/mL insulin, 5.5 μg/mL transferrin, and 6.7 ng/mL selenium; Gibco) and 1% antibiotic–antimycotic solution (Hyclone). HepG2 and Hep3B were cultured in EMEM medium, whereas Huh-7 and PLC.PRF.5 cells were cultured in RPMI-1640 medium. All media contained 10% (v/v) FBS and antibiotic–antimycotic solution (1 mL per 100 mL of cell culture medium). Cells were maintained at 37 °C in a humidified incubator under an atmosphere of 5% CO2.
2.4. Cell Proliferation Assay
Cell proliferation was detected using the Cell Counting Kit-8 (CCK-8, Cat. CK04-13; Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instruction. To evaluate cytotoxicity, cells were seeded in a 96-well plate (FHC, 1 × 104 cells/well; Caki-2, PLC/PFR.5, and MCF 10A, 1 × 104 cells/well; AML12, HepG2, Hep3B, and Huh-7 1.2 × 104 cells/well; HeLa, HEK-293, A549, and PC-3, 1.5 × 104 cells/well; HT-29, MCF-7, C6, and NE-4C, 2 × 104 cells/well). After attachment and stabilization of cells for 24 h, the medium was switched with fresh containing various concentrations (0, 1, 5, 10, 25, 50, and 100 μM) of MBP-11901, and cells were incubated for 22 h. Next, CCK-8 solution was added to each well, and cells were incubated for another 2 h. Absorbance was then measured at 450 nm using a microplate reader (SpectraMax i3, Molecular Devices, San Jose, CA, USA). The IC50 and log IC50 values were calculated using GraphPad Prism (version 5.02; GraphPad PRISM software Inc. San Diego, CA, USA). All experiments were independently performed 3 times. The graph of log IC50 values represents average values.
2.5. Western Blot Analysis
HepG2 cells were seeded in a 60 mm
2 culture dish at a density of 1.5 × 10
6 cells/dish and incubated for 24 h. Cells were treated with MBP-11901 for 24 h. Cells were harvested and protein extraction and Western blotting methods were performed as previously described [
11]. Membranes were incubated using the following diluted primary antibodies; PARP (1:1000 dilution), caspase-3 (1:1000 dilution), cleaved caspase-3 (1:1000 dilution), pFLT3 (1:500 dilution), Ras (1:1000 dilution), pc-Raf (1:1000 dilution), pMEK1/2 (1:1000 dilution), pERK1/2 (1:1000 dilution), pAKT (1:1000 dilution), pmTOR (1:1000 dilution), pVEGFR2 (1:1000 dilution), pPDGFR β (1:2000 dilution), and β-actin (1:1000 dilution) in Tris-HCl-based buffer containing 0.2% Tween-20 (T1027, Biosesang, Seongnam, Korea) (TBS-T; pH 7.5). Membranes were then incubated with horseradish-conjugated secondary antibodies (1:5000, Cell Signaling Technologies, Beverly, MA, USA) for 1 h at 25 °C. After washing with TBS-T, immunoreactive bands were visualized using the chemiluminescence western imaging system (Supernova-Q1800TM, Centronics, Daejeon, Korea). Band intensities were measured using ImageJ (Version 1.50i; US National Institutes of Health, Bethesda, MD, USA) and were normalized to β-actin.
2.6. Transwell Migration and Invasion Assay
For the migration assay, HepG2 cells were seeded into the upper Transwell chamber at a density of 2 × 104 cells and incubated for 24 h. For the invasion assay, 100 μL of Matrigel: serum-free media (1:4) mixture was placed on the filter membrane in the upper chamber and incubated (37 °C, 4 h) to seed cells. Cells were exposed to MBP-11901 and sorafenib for 2 h. Then, the medium in the upper chamber was changed to serum-free medium, whereas the medium in the lower chamber was changed to a medium containing 10% FBS. The medium was replaced once every 2–3 d. Following 6 d incubation, the upper surfaces of the filters were scraped 5 times with cotton swabs to remove nonmigrated cells, whereas cells crossing the membrane into the lower chamber were stained with 1% (w/v) crystal violet for 30 min. Crystal violet was removed and cells were washed with tap water. For the migration assay, after drying at 25 °C, 10% acetic acid solution was added to cells, and left to react in a shaker for 30 min. Absorbance was measured at 590 nm using a microplate reader (SpectraMax i3, Molecular Devices). For the invasion assay, the number of migrated cells was counted.
2.7. Flow Cytometry Analysis of Cell Death
Approximately 1.5 × 106 HepG2 cells were suspended in 100 mL PBS, and then 200 mL 95% ethanol was added while vortexing. Cells were incubated at 4 °C for 1 h, washed with PBS, and resuspended in 250 mL 1.12% sodium citrate buffer (pH 8.4, C8532, Sigma-Aldrich) supplemented with 12.5 μg RNase. Incubation was continued at 37 °C for 30 min. Cellular DNA was then stained by applying 250 mL propidium iodide (50 μg/mL) for 30 min at 25 °C. Stained cells were analyzed on a FACScan flow cytometer for relative DNA content based on increased red fluorescence.
2.8. LDH Cytotoxicity Assay
The LDH assay was performed using the D-Plus™ LDH cell cytotoxicity assay kit (Cat. LDH-1000; Dongin Biotech, Seoul, Korea). Briefly, HepG2 cells were seeded at 1.2 × 104 cells per well and AML12 cells at 2.0 × 104 cells per well in 96-well plates. After 24 h of incubation, MBP-11901 at indicated concentrations was added to each well in a final volume of 100 μL. After 24 h, floating cells in the supernatants were removed by centrifugation at 600× g for 5 min. Control cells were lysed by adding 10 µL lysis buffer before centrifugation. Each supernatant (10 µL) was transferred to a new well in a 96-well plate. Finally, 100 µL LDH reaction mixture (1:50 ratio of WST substrate to LDH assay buffer) was added and samples were incubated at 25 °C for 30 min. Absorbance at 450 nm was measured using a microplate reader. Three independent experiments were performed in duplicate at different time points.
2.9. Colony Formation Assay
HepG2 cells were seeded in a 6-well plate at a density of 2 × 104 cells/well and incubated for 24 h. Cells were treated with MBP-11901 and sorafenib for 2 h and media was changed with fresh one. Cells were incubated for 8 d and the medium was replaced once every 2–3 d. The medium was carefully removed, cells were washed with PBS, and fixed with 100% methanol for 30 min. After removing methanol, cells were stained with 0.5% (w/v) crystal violet for 30 min, and washed with tap water. The plate was dried at 25 °C and images were captured under a microscope (Nikon Eclipse Ts2, Tokyo, Japan). The stained area was automatically measured using ImageJ. This experiment was independently performed 3 times. The area of the colony was calculated as a percentage of the total area of the well.
2.10. JC-1 Mitochondrial Membrane Potential Assay
Alterations in MMP were investigated utilizing the cationic dye tetraethylbenzimidazolylcarbocyanine iodide (JC-1) according to the manufacturer’s instructions (Cat. ab113850, Abcam, Cambridge, MA, USA). HepG2 cells were seeded in a 35 mm2 dish at a density of 7.5 × 105 cells/dish and incubated for 24 h. Cells were treated with MBP-11901 for 2 h and media was changed with fresh one. Briefly, after 2 d incubation, cells were washed with 1× dilution buffer, and incubated with 1 μM JC-1 in 1× dilution buffer for 30 min at 37 °C. Cells were then washed 3 times with 1× dilution buffer. Fluorescence was measured under a fluorescence microscope (ECLIPSE Ti, Nikon, Melville, NY, USA) using an excitation filter of 530 nm ± 15 nm and an emission filter of 590 nm ± 17.5 nm. The fluorescence intensity of immunostaining was quantified using ImageJ.
2.11. Immunofluorescence Staining
Sectioned tumor tissues (5 μm) were blocked with TBS supplemented with 5% normal goat serum (NGS) and 5% BSA for 2 h, and incubated overnight with primary antibodies (anti-caspase-3; diluted 1:100) in blocking solution (5% NGS and BSA) at 4 °C. Sections were then washed 3 times in TBS, and incubated for 1 h in the presence of Alexa Fluor-conjugated IgG-labeled secondary antibodies (Invitrogen, Carlsbad, CA, USA). Sections were washed and mounted with Vectashield mounting medium containing DAPI (Vectashield H-1500; Vector Laboratories, Inc., Burlingame, CA, USA). Images were captured using a fluorescence microscope (ECLIPSE Ti, Nikon) and fluorescence intensity of immunostaining was quantified using ImageJ.
2.12. Immunohistochemistry Staining
Sectioned tumor tissues (5 μm) were deparaffinized in xylene (Junsei Chemical, Tokyo, Japan) and rehydrated through an ethanol series. Endogenous peroxidase activity was inactivated by incubation in 0.3% H2O2 (Sigma-Aldrich) in methanol for 10 min. Sections were then rinsed in 0.1 M TBS (pH 7.4) and boiled in citrate buffer (pH 6.0) containing 0.03% Tween-20 for 4 min. Finally, sections were incubated with blocking solution (5% NGS and BSA in TBS) at 25 °C for 1 h, and subjected to indirect immunohistochemistry using an antibody against anti-Ki-67(diluted 1:100) for 1 h. For the negative control, the primary antibody was omitted and slides were incubated with a blocking solution. Sections were then incubated with HRP-conjugated anti-rabbit IgG for 1 h at 25 °C, stained with VECTOR1 NovaRED (Vector Laboratories, Inc.), and counterstained with hematoxylin (BBC Biochemical, Mount Vernon, WA, USA). Sections were dehydrated, cleared, and mounted with Permount (Fisher, Fair Lawn, NJ, USA). Images were captured under a fluorescence microscope (ECLIPSE Ti, Nikon) and the fluorescence intensity of immunostaining was quantified using ImageJ.
2.13. Animals
Male Balb/c-nude mouse weighing 20–22 g (Nara Biotech, Seoul, Republic of Korea) were used for all experiments. Animals were housed under controlled environmental conditions in an ambient temperature of 23 ± 1 °C and relative humidity of 50 ± 10% under a 12 h light/dark cycle and were given ad libitum access to food and water. All animal experiments were approved by and performed in accordance with the guidelines issued by the Institutional Animal Care and Use Committee (IACUC) of Kyungpook National University (No. KNU-2018-0086; Daegu, Korea).
2.14. In Vivo Tumor Growth Assay Using Xenograft Mouse Model
For the generation of a tumor xenograft, HepG2 cells (5 × 106) were suspended in 100 μL serum free EMEM:Matrigel (1:1) and then subcutaneously injected into the upper-left flank region of nude mice. When the tumor reached a mean size of about 100–150 mm3, mice were randomized into each experimental group according to tumor size. Mice were orally administered Nexavar® (82, or 164 mg/kg, daily or once every 2 d, respectively), MBP-11901 (40, 60, or 82 mg/kg, once every 2, 3, or 4 d, respectively), or saline as control (once every 2 d). Tumor size was measured with a caliper rule twice a week and body weight was measured simultaneously. The tumor size was calculated as follows: tumor size (mm3) = (L × W2)/2, where L is the longest and W is the shortest radius of the tumor in millimeters. At the end of the experiment, mice were euthanized, blood samples were collected via the abdominal vena cava, and tumors were removed and weighed.
Tumor, liver, and kidney tissues were harvested for fixation in 4% paraformaldehyde (PFA, BP031, Bio-solution, Suwon-si, Gyeonggi-do, Korea) for histologic examination and immunohistochemical staining or immediately frozen at −80 °C in a deep-freezer and stored until protein extraction.
Drug efficacy was expressed as the percentage of tumor growth inhibition (% TGI), calculated using the equation: 100 − (T/C × 100), where T is the mean relative tumor volume (RTV) of the treated tumor and C is the mean RTV in the vehicle control group at the time of sacrifice. RTV = Vx/V1, where Vx is the volume in mm3 at a given time and V1 at the start of treatment. Mean TGI (%) and standard deviation were calculated for each group.
2.15. Measurement of Glutamic-Oxaloacetic Transaminase (GOT) and Glutamic-Pyruvic Transaminase (GPT)
The levels of GOT and GPT were measured using enzymatic assay kits (Asan Pharm, Seoul, Korea). The collected blood was allowed to clot and the serum, which was separated by centrifugation at 4000 rpm for 15 min after 2 h at 25 °C, was used for analysis.
2.16. In Vivo Tumor Growth Assay Using an Orthotopic Mouse Model
The orthotopic HCC mouse model was established by placing a small piece of tumor-derived HepG2-Luc cells. Mice were randomly distributed into 3 groups upon bioluminescence signal reaching 107. The first group served as the saline control. The second and third groups were orally administered MBP-11901 (82 mg/kg) and Nexavar® (164 mg/kg) once every other day for 4 weeks. Tumor progression/regression was recorded once weekly by quantifying the bioluminescence signals. Experimental animals were sacrificed by CO2 inhalation.
2.17. Histological Analysis
Tumors were fixed in 4% PFA for 72 h, and treated with an alcohol concentration gradient (50, 70, 95, and 100%), xylene (Junsei Chemical), and paraffin for 30 min, respectively. Sectioned tumor tissues (5 μm) were treated with xylene and an alcohol concentration gradient (100, 95, 70, and 50%) for 1 h and 10 min, respectively, in an oven at 65 °C. Hematoxylin and eosin (H&E) staining (BBC Biochemical, Mount Vernon, WA, USA) was performed according to the manufacturer’s instructions and stained tissues were observed under a microscope (ECLIPSE Ti, Nikon). For Sirius Red staining, tissues were immersed in Picro Sirius Red Stain (ab150681; Abcam) for 30 min, washed for 1 min in 0.1 N HCl, followed by dehydration and mounting of slides.
2.18. In Silico Molecular Docking Analysis
Crystal structures of FLT3, VEGFR2, MEK5, and KIT were obtained from the RCSB Protein Data Bank website (
https://www.rcsb.org) (accessed on 21 July 2021) under the PDB IDs of 4RT7, 3VHE, 3SLS, and 4U0I, respectively. Due to the lack of an experimentally revealed structure, the three-dimensional structure of PDGFRβ was obtained from the AlphaFold Protein structure database (
https://alphafold.ebi.ac.uk/entry/P09619) (accessed on 21 July 2021). These structures were optimized for in silico simulation using the Prime protein modeling application [
12]. Protein–ligand docking simulation was performed using the Glide docking application [
13]. Glide searches all possible binding poses of a given ligand and calculates the binding energy of the ligand using the GlideScore scoring function. For docking simulation, the flexible ligand sampling method was applied, using the SP (Standard Precision) mode of Glide.
2.19. KINOMEscan Profiling of MBP-11901
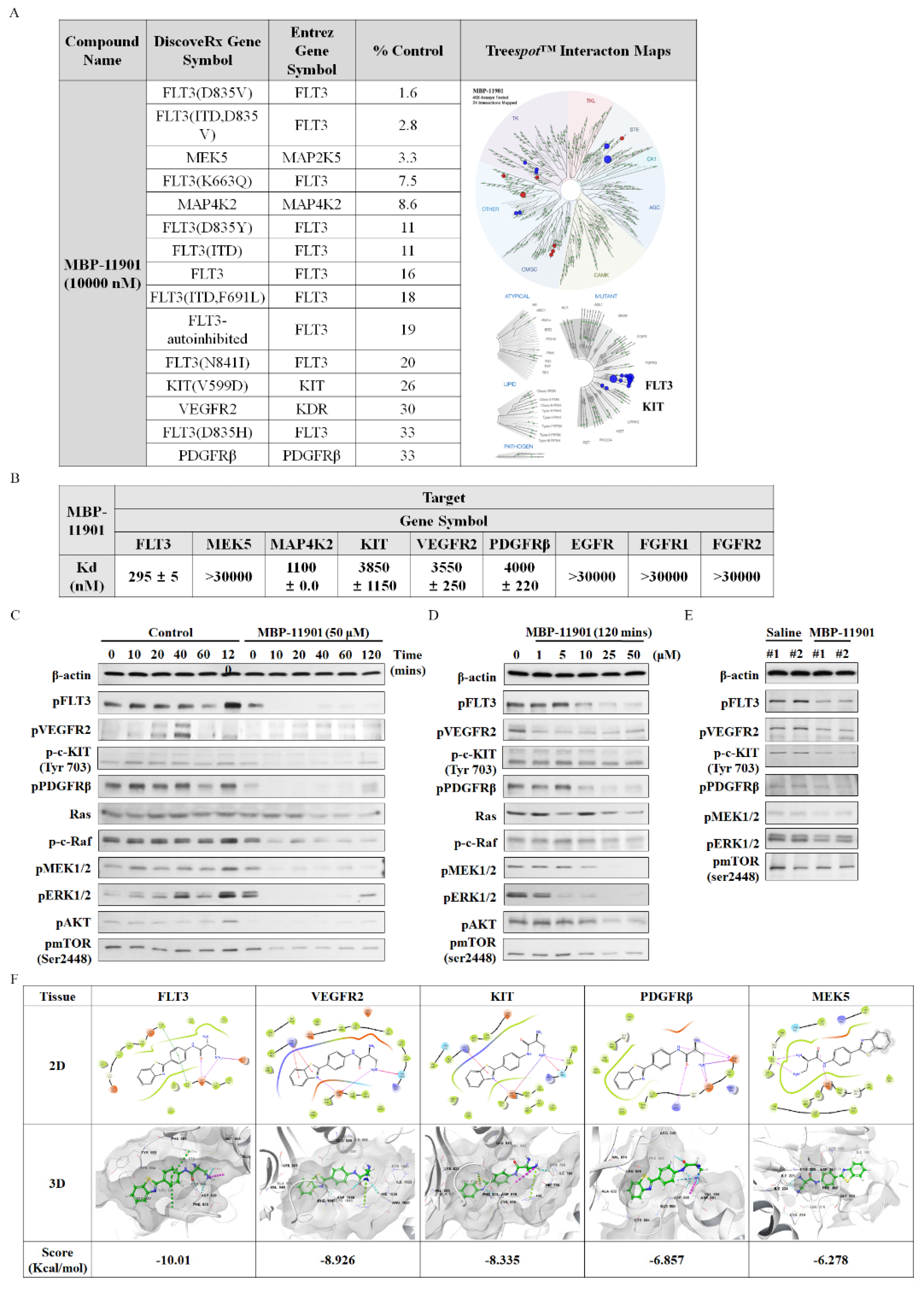
The profiling of MBP-11901 against a panel of 468 kinases was performed by DiscoverX using KINOMEscan technology, an active-site-dependent competition-binding assay (Eurofins DiscoverX Corporation, San Diego, CA, USA). Binding constants (Kd) were calculated by DiscoverX using KdELECT technology, a widely used assay for comparing selectivity across the kinome. The DiscoverX platform reports the thermodynamic Kd to facilitate direct comparison of inhibitor affinity across the kinome, independent of ATP concentration.
2.20. Statistical Analysis
Data were evaluated using one-way or two-way ANOVA followed by Tukey’s or Dunnett’s test. Analyses were performed using GraphPad Prism (version 5.02; GraphPad Prism Software Inc.). Data are expressed as the mean ± SD (standard deviation) or standard error of the mean (SEM), and p < 0.05 was considered statistically significant.
4. Discussion
Advanced or unresectable HCC is a malignant tumor with poor prognosis and few available treatment options [
15]. Multikinase inhibitors such as sorafenib, lenvatinib, cabozantinib, and regorafenib have been suggested as first- and second-line therapy; however, the response to these drugs is low and limited. Recently, the number of options for the treatment of HCC has increased, with an added combination therapy with atezolizumab and bevacizumab [
16]. Moreover, rather than reaching a full cure, these treatments merely prolong the overall survival period of patients by several months [
16,
17]. Surprisingly, immunotherapy of hepatocellular carcinoma has been reported to lead to the development of hyperprogressive cancer [
18]. Therefore, it is necessary to carefully select patients with HCC for immunotherapy. In addition, there is still an unmet need for innovative therapeutic agents for the cure of HCC.
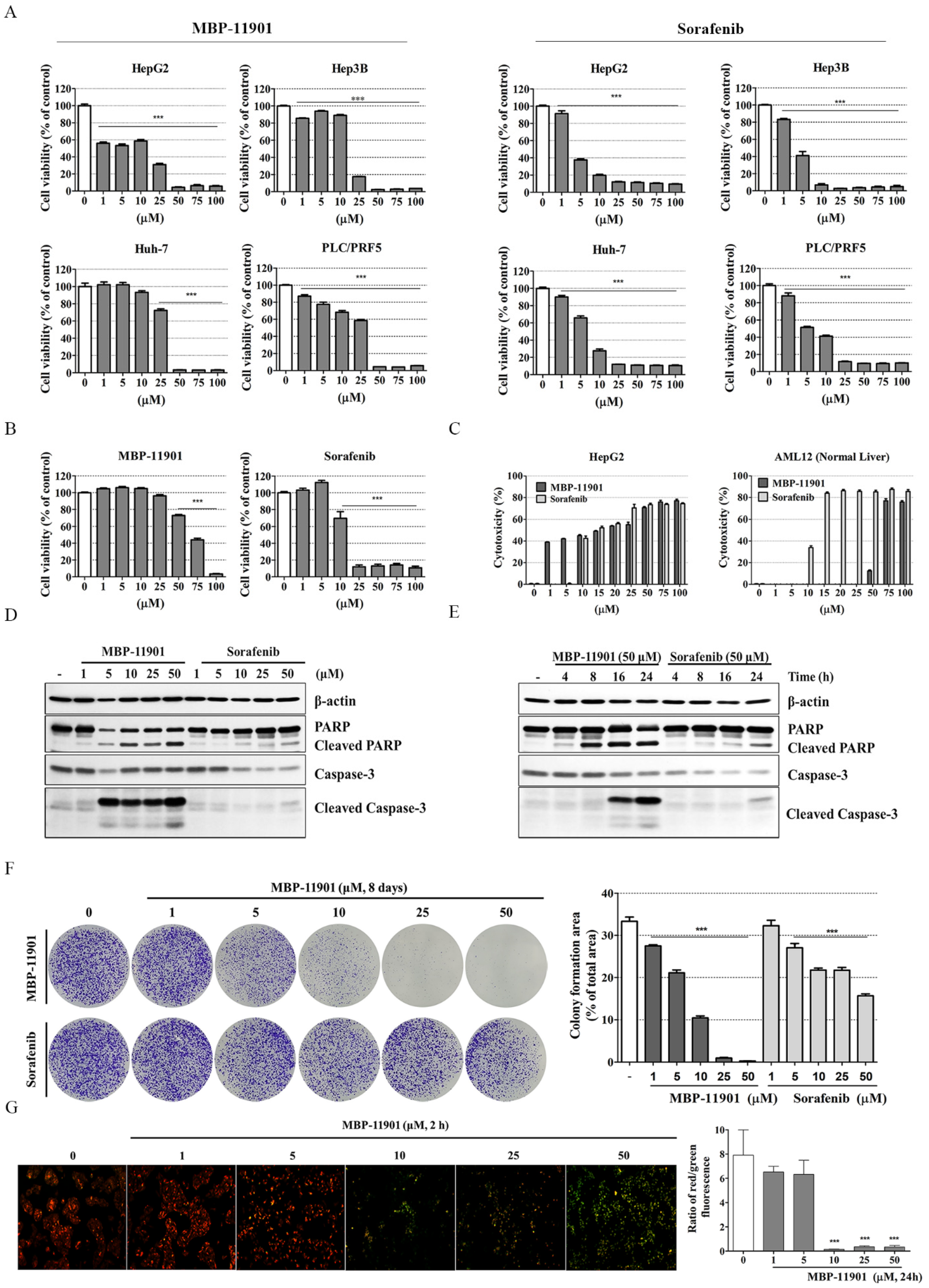
Here, we presented the excellent therapeutic effect of MBP-11901 on HCC using an animal model. We confirmed the HCC-specific efficacy of MBP-11901 through in vitro screening using various human cancer cell lines (
Figure 1B), and verified its effectiveness in various HCC cell lines (
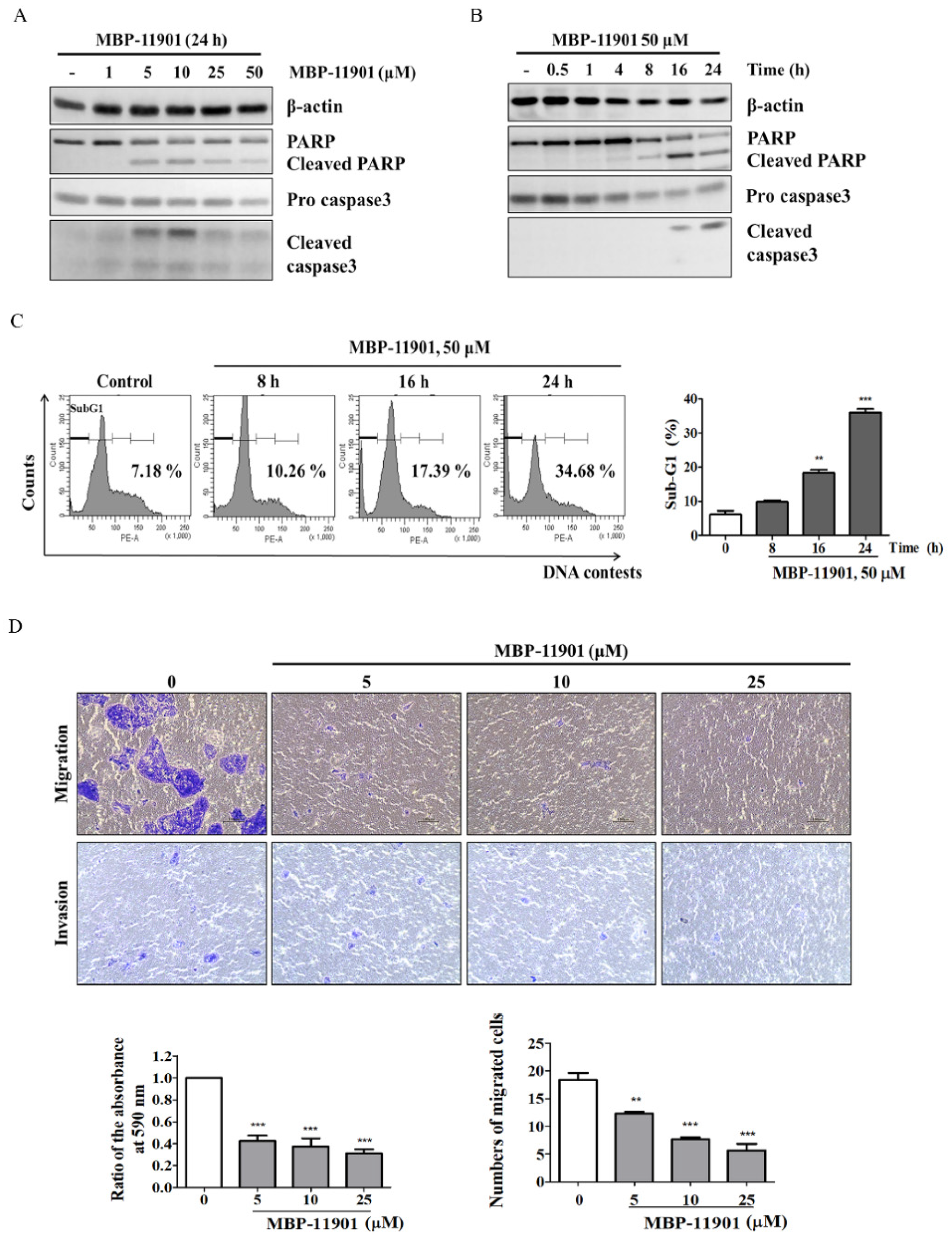
Table 1). Through various experimental techniques, we verified that MBP-11901 not only induced the apoptosis of HCC cells, but also inhibited their proliferation, metastasis, and invasion (
Figure 2 and
Figure 3).
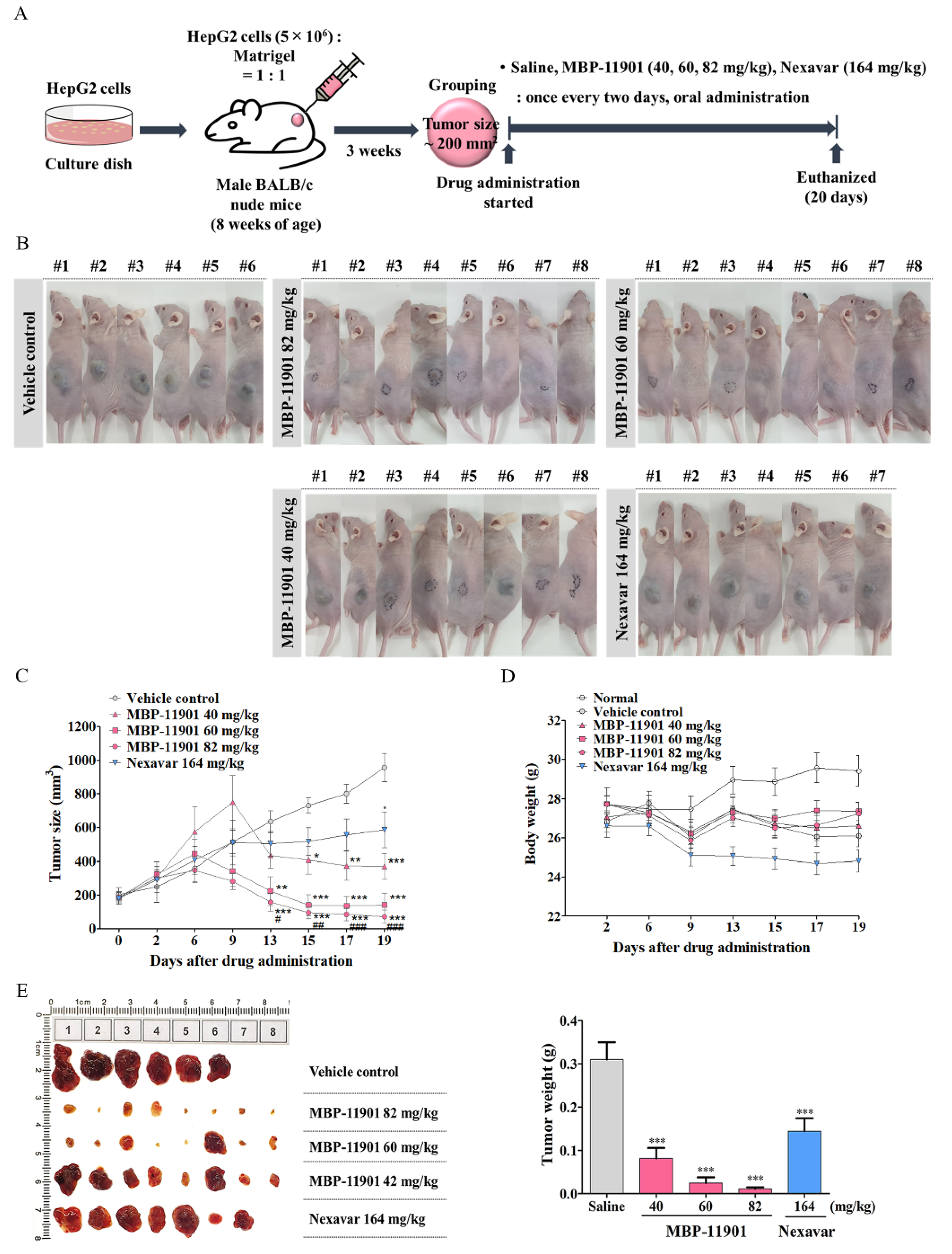
A noteworthy aspect of our study was the complete therapeutic effect of MBP-11901 in subcutaneous and orthotopic transplantation models. At the start of drug administration, the average tumor volume in each group was 200 mm
3, as shown in
Figure 4. was After oral administration once every 2 d, the average tumor volume in the Saline group reached approximately 1000 mm
3 at the end of a total of 9 administrations, approximately 600 mm
3 in the Nexavar
® group, whereas it reached approximately 350, 150, and 70 mm
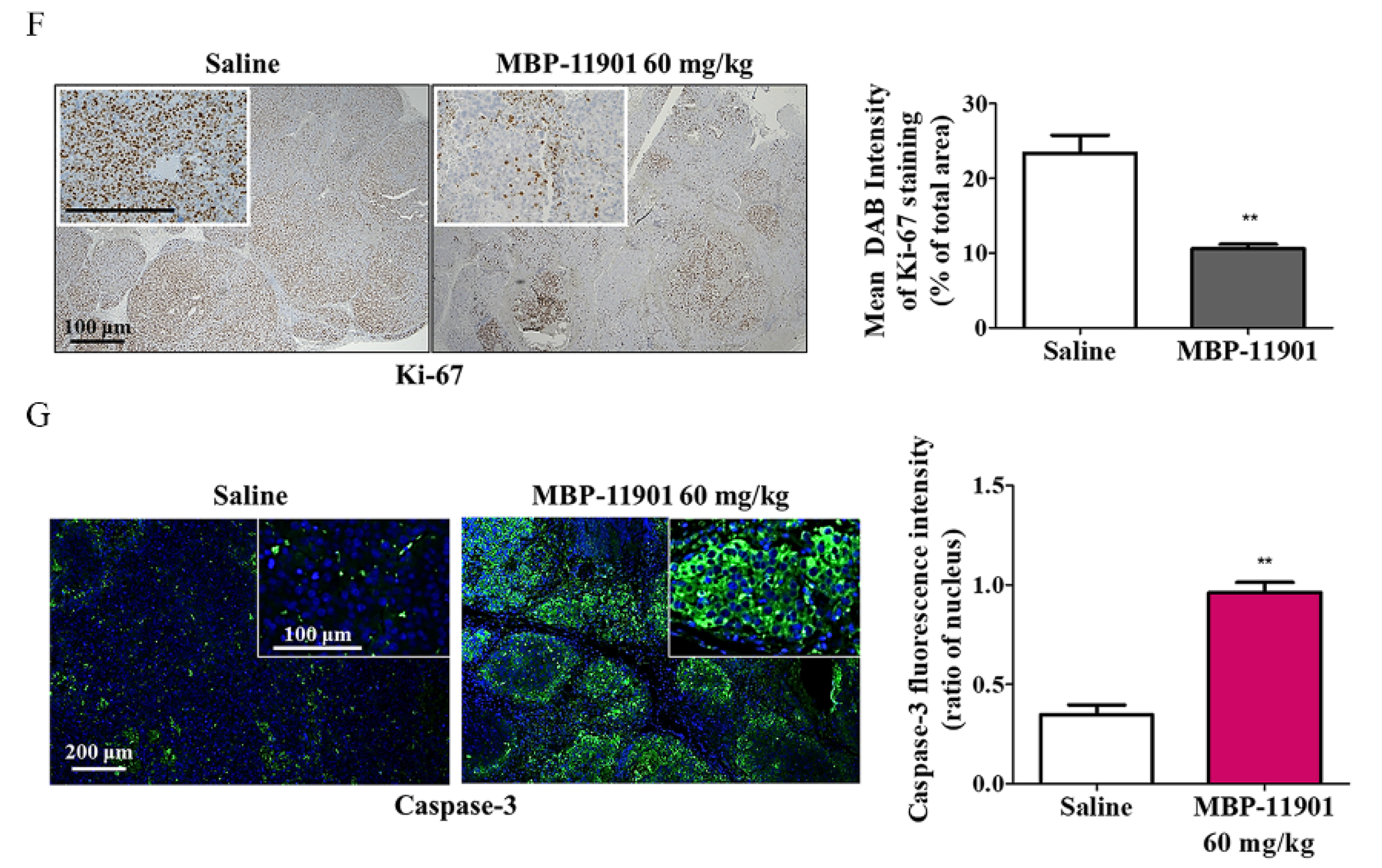
3, in the 40, 60, and 82 mg/kg MBP-11901 groups, respectively. In particular, in the 82 mg/kg MBP-11901 group, the tumor completely disappeared in 4 out of 8 animals; the same effect was observed in 3 out of 8 animals in the 60 mg/kg group. Histological analysis of MBP-11901-treated tumor masses revealed a decrease in the levels of proliferation proteins and an increase in those of apoptosis proteins.
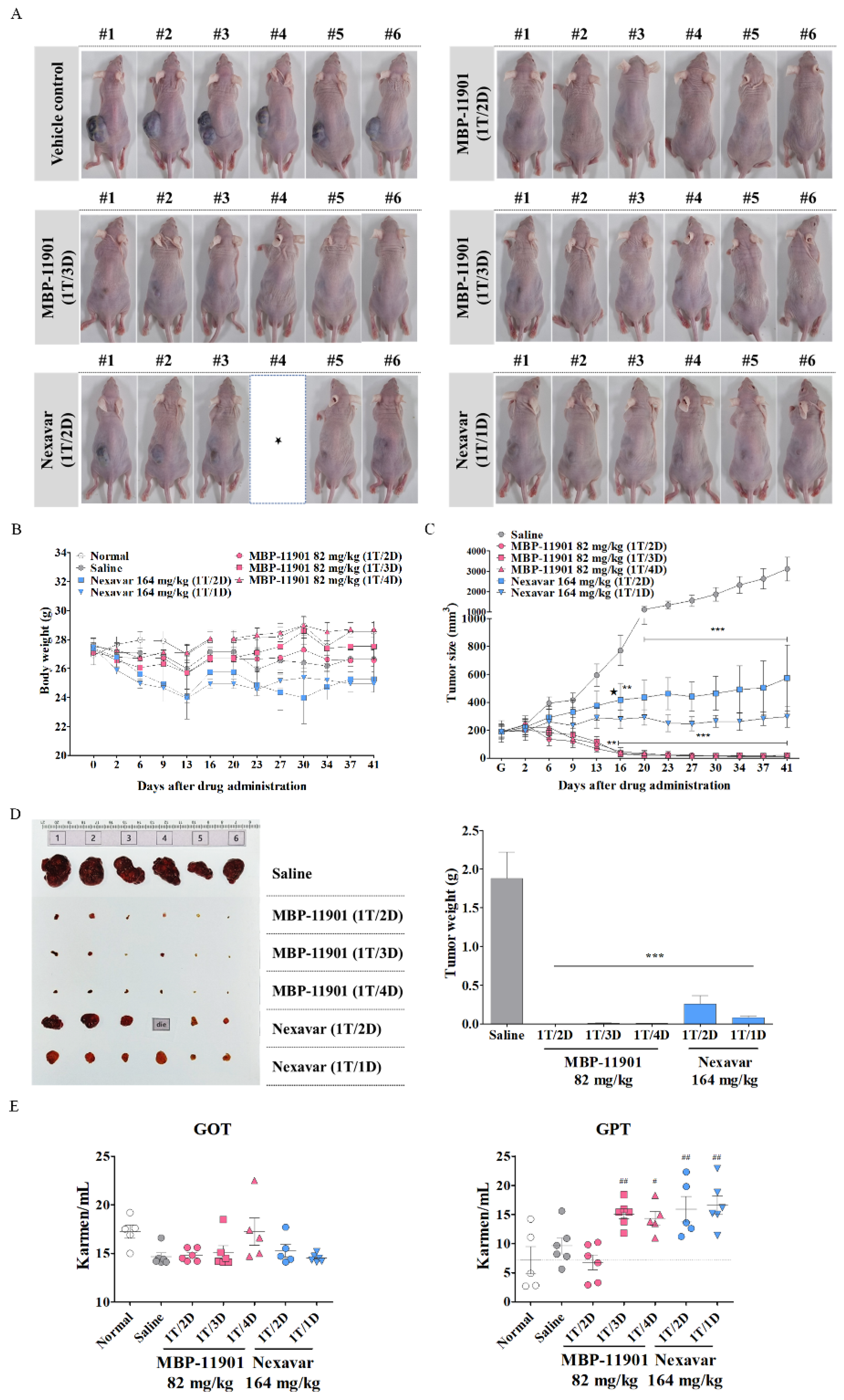
Another noteworthy result shown in
Figure 5 was that there was no difference in efficacy even if the dosing interval was changed. In this experiment, administration was initiated at an average tumor volume of approximately 190 mm
3. Following termination of treatment after 42 d, tumors had disappeared in 3–4 out of a total of 6 animals in all groups to which MBP-11901 was applied. In the case of Nexavar
®, the tumor suppression rate was approximately 2 times greater in the daily administration group than in the group treated once every 2 d. However, this effect was clearly lower than that observed in the group administered with MBP-11901 once every 4 d. When the interval between the administration of MBP-11901 was once every 3 d and once every 4 d, a statistically significant increase was found in the levels of GPT compared with normal values (
Figure 5E); however, the weight loss rate was maintained at a similar level to normal (
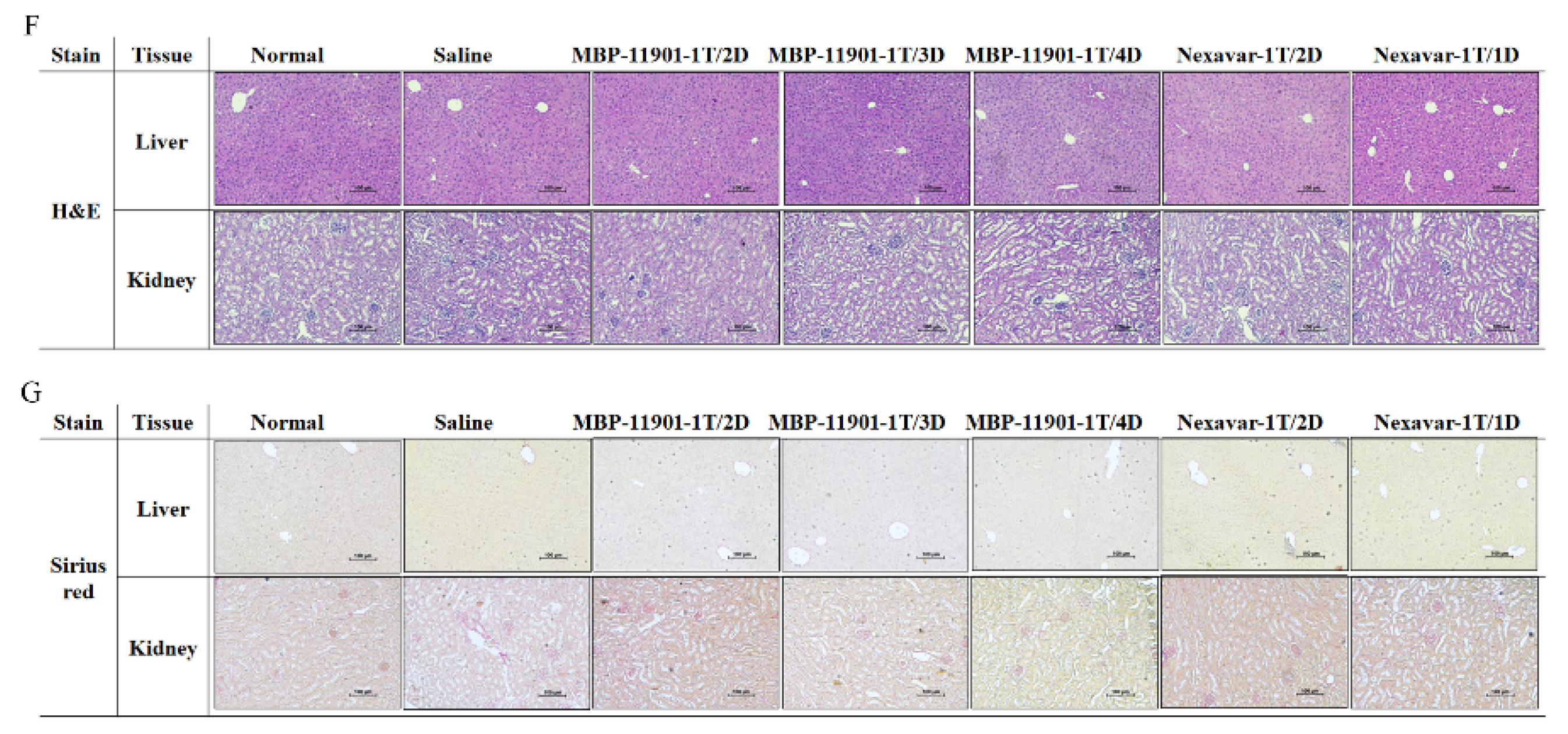
Table 4). Moreover, histological analysis of the liver and kidney showed no apparent toxicity (
Figure 5F,G).
We further verified the effectiveness of MBP-11901 by constructing a mouse model in which Hep3B and Huh-7 were subcutaneously transplanted in addition to the human liver cancer cell line HepG2. In the case of Hep3B, the administration was initiated at a tumor volume of about 300 mm
3, once every 2 d, and the experiment was terminated after a total of 12 administrations (
Figure S5). The average tumor volume was measured to be approximately 1400 mm
3 in the Saline group, whereas the Nexavar
® group had a tumor volume of about 900 mm
3. In contrast, tumors had completely disappeared in 1 out of 6 animals in the MBP-11901 group; 1 had only a trace of a tumor measuring approximately 30 mm
3, whereas the average tumor volume in the 6 animals was approximately 100 mm
3. Compared with HepG2 or Hep3B, administration of MBP-11901 to the Huh-7 subcutaneously implanted model did not result in tumor disappearance, although the tumor weight was significantly reduced (
Figure S6). These results showed that MBP-11901 did not show specificity for human HCC cell lines, but rather was effective against all evaluated HCC cell lines. Unlike Nexavar
®, there were no side-effects relating to drug resistance or rapid tumor growth.
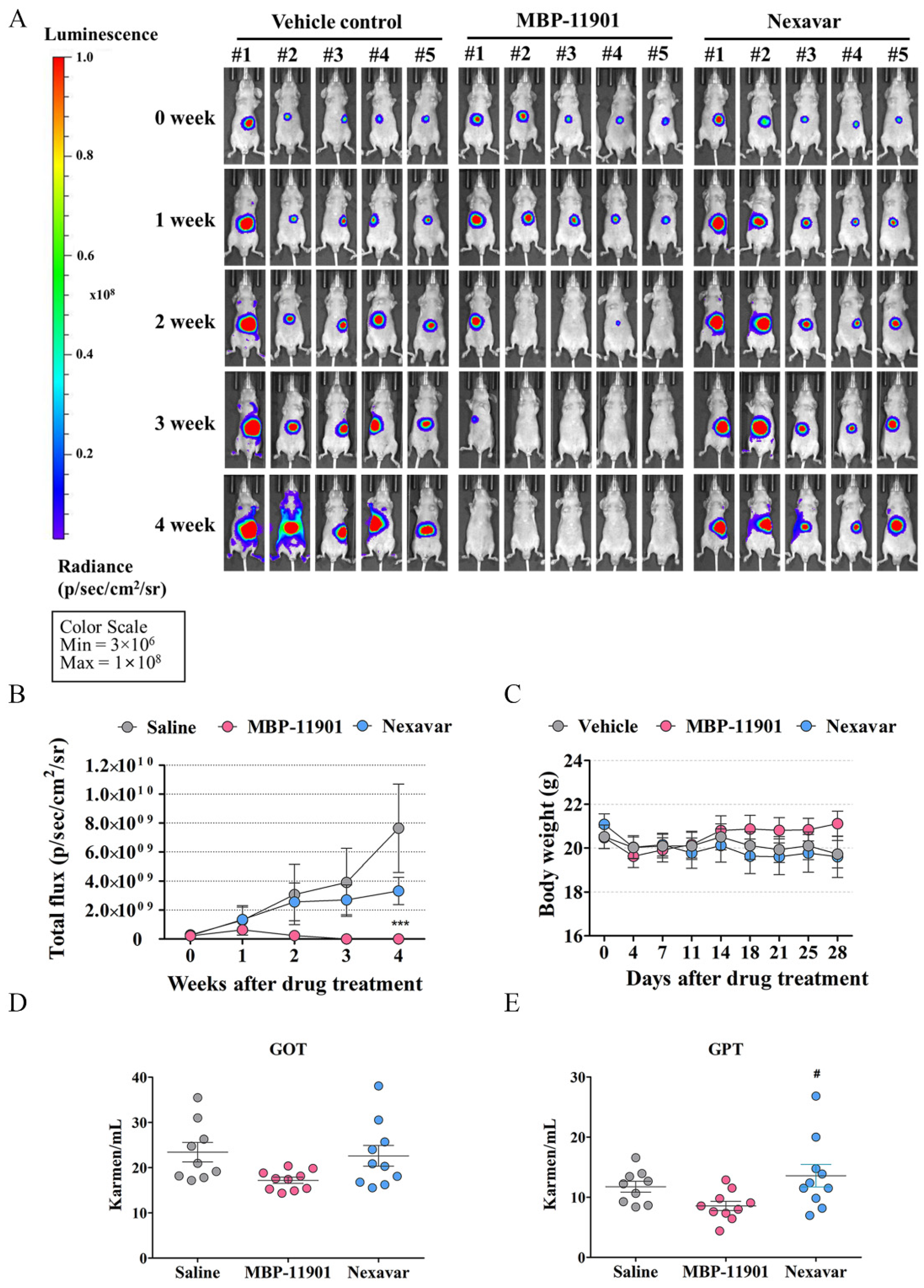
In addition to the HCC subcutaneous transplantation model, administration of MBP-11901 led to similar results of tumor elimination in the orthotopic mouse model as well (
Figure 6). In order to determine the presence of a tumor, we obtained liver tissues at the end of the experiment and performed histological analysis of the orthotopic graft sites (
Figure S9). When saline was administered, the boundary between the mouse liver and transplanted liver was clearly observed, as well as a portion positive for the antibody against Ki-67, a proliferation marker. In contrast, when MBP-11901 was administered, the boundary between the mouse liver and transplanted tissue was indistinguishable, and almost no cells were observed to be stained with Ki-67 and hematoxylin. Sirius Red staining confirmed that the traces still remaining were collagen fibers similar to scars. In the case of the HepG2 subcutaneous implant, when the skin in the area where the tumor was present was incised and observed with the naked eye at autopsy, no tissue mass was detected and only traces remained similar to the wound (
Figure S10). These findings suggested the complete disappearance of HCC in animal models due to the application of MBP-11901. We plan to discontinue administration of MBP-11901 to animals in which the tumor has completely disappeared, and further investigate whether the tumor recurs through continuous follow-up.
MBP-11901, a multi-RTK inhibitor, was shown to inhibit tumor growth and kill HCC by targeting VEGFR2, c-KIT, PDGFRβ, and FLT3. Kinase profiling confirmed that there was almost no binding affinity for each of these Ras/Raf/MEK/ERK subsignals, which was further confirmed by in silico binding assays (
Figure S12). Remarkably, among the various RTKs, the binding affinity to FLT3 was the strongest. Mutations in the activation loop of the tyrosine kinase domain (TKD1) of FLT3 are present in up to 30% of patients with acute myeloid leukemia (AML), and FLT3 has been utilized as a potential target for kinase inhibitor therapy [
19,
20]. However, FLT3 has not received much attention in HCC. A recent study showed that the reactivity of sorafenib was significantly increased in HCC with strong FLT3 expression [
21]. Thus, we should focus on FLT3 as a biomarker to increase the reactivity to sorafenib, a target anticancer drug for HCC.
MBP-11901 is similar to sorafenib or cabozantinib in targeting FLT3, VEGFR2, PDGFRβ, and c-KIT. Nevertheless, it is thought that there might be additional reasons for MBP-11901 not having significant toxicity, driving tumor reduction in all mice, and leading to the complete disappearance of tumors. MBP-11901 exhibited significantly high bioavailability and long half-life pharmacokinetics in mice (data not shown). Presumably, for this reason, it has the advantage of exhibiting sufficient efficacy even at low concentrations and with no associated toxicity. We are currently conducting additional research for conducting phase 1 clinical trials.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}