
Targeting Ribosome Biogenesis in Cancer: Lessons Learned and Way Forward



Abstract
:Simple Summary
Abstract
1. Introduction
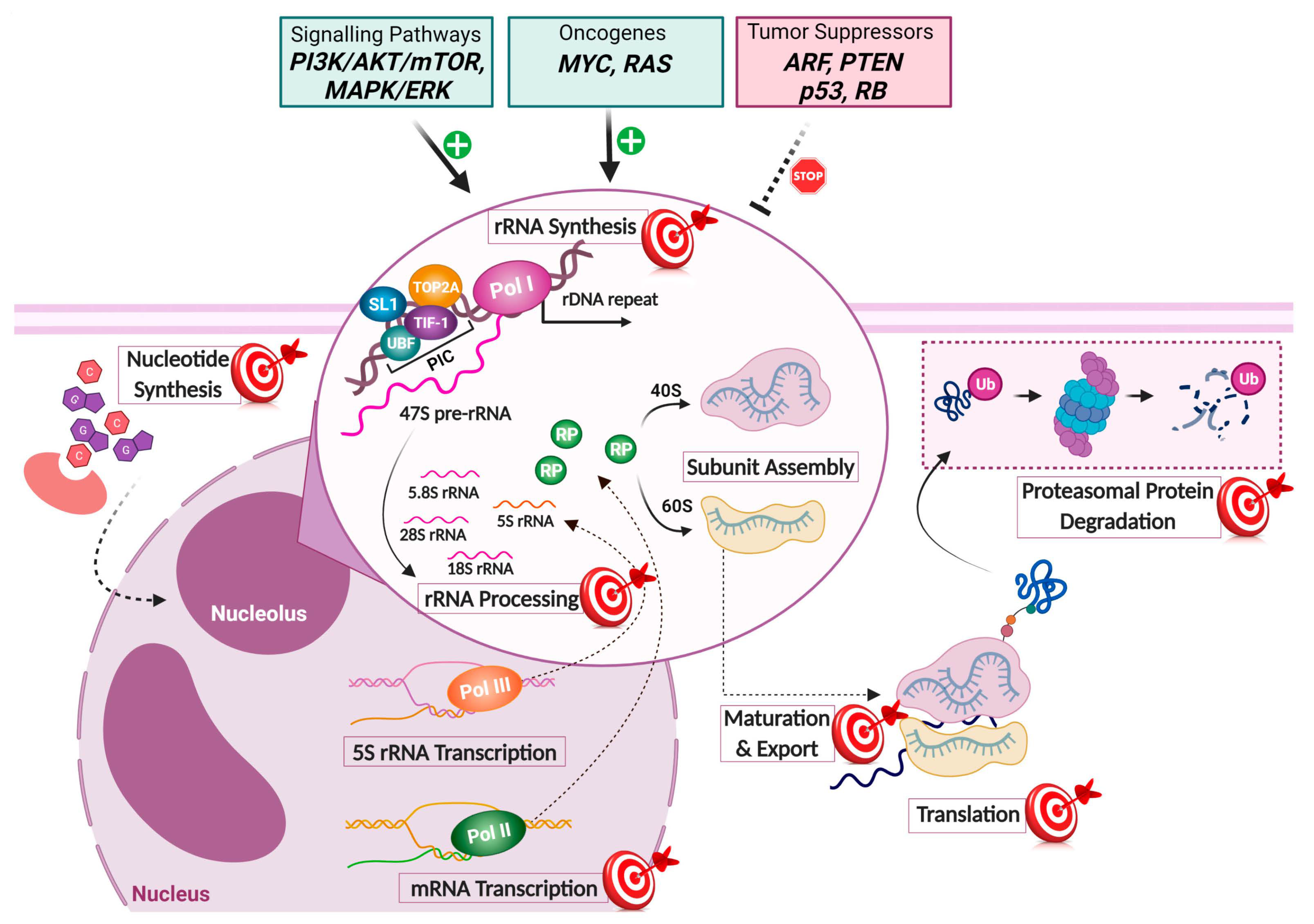
2. Ribosome Biogenesis as a Target in Cancer Cells
2.1. Ribosome Biogenesis Is Often Increased in Cancer Cells
2.2. Chemotherapy Often Targets Ribosome Biogenesis
2.3. Ribosome Biogenesis Dysfunction Often Leads to p53 Activation
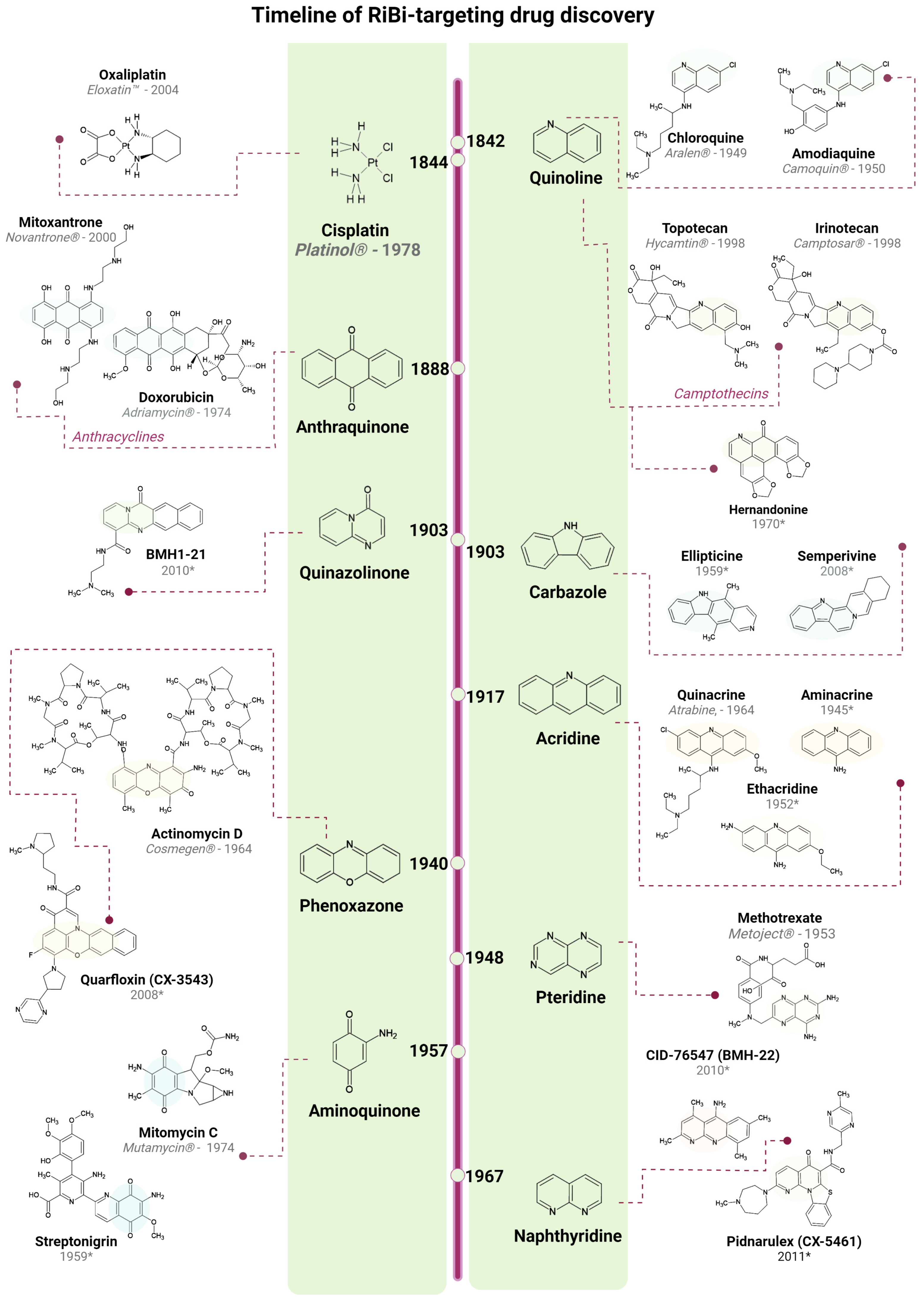
3. Clinically Approved Drugs and Their Effect on RiBi
3.1. DNA Intercalators
3.2. DNA Alkylating Agents

{kind=link}
{kind=link}
{kind=link}
| Compound | Mechanism | RiBi Target | Reference |
|---|---|---|---|
| Actinomycin D | DNA Intercalator | rRNA synthesis | [67,68] |
| Mitoxantrone | DNA Damage, TOP2 inhibitor. | rRNA synthesis | [34,89] |
| Doxorubicin | DNA Intercalator, TOP2 inhibitor | rRNA synthesis | [34,90,104] |
| Oxaliplatin | DNA Cross-linker | rRNA synthesis, processing | [94,99,105] |
| Cisplatin | DNA Cross-linker | rRNA synthesis | [93,95,96] |
| Carboplatin | DNA Cross-linker | rRNA synthesis | [94] |
| Mitomycin C | DNA Alkylator, TOP2 inhibitor | rRNA synthesis | [106,107,108] |
| 5-Fluorouracil | Antimetabolite | rRNA processing | [34,109,110] |
| Methotrexate | Antimetabolite | rRNA synthesis | [111,112] |
| Camptothecin | TOP1 Inhibitor | rRNA synthesis | [113,114] |
| Etoposide | TOP2 Inhibitor | rRNA processing | [115,116] |
| Aminoacridine | DNA Intercalator | rRNA synthesis | [81] |
| Ethacridine | DNA Intercalator | rRNA synthesis, processing | [81] |
| Amodiaquine | Several + Autophagy Inhibitor | rRNA synthesis | [86] |
| Rapamycin | mTOR Inhibitor | rRNA synthesis | [31,117] |
| Mycophenolic acid | IMPDH2 Inhibitor | rRNA synthesis | [118] |
3.3. Antimetabolites
3.4. Plant-Derived Alkaloids
3.5. Non-Intercalating Antibiotics
3.6. Other Compound Classes That May Affect Ribosome Biogenesis
4. Drug Discovery: Identification and Development of molecules That Inhibit Ribosome Biogenesis
4.1. Quarfloxin, CX-3543
4.2. Pidnarulex, CX-5461
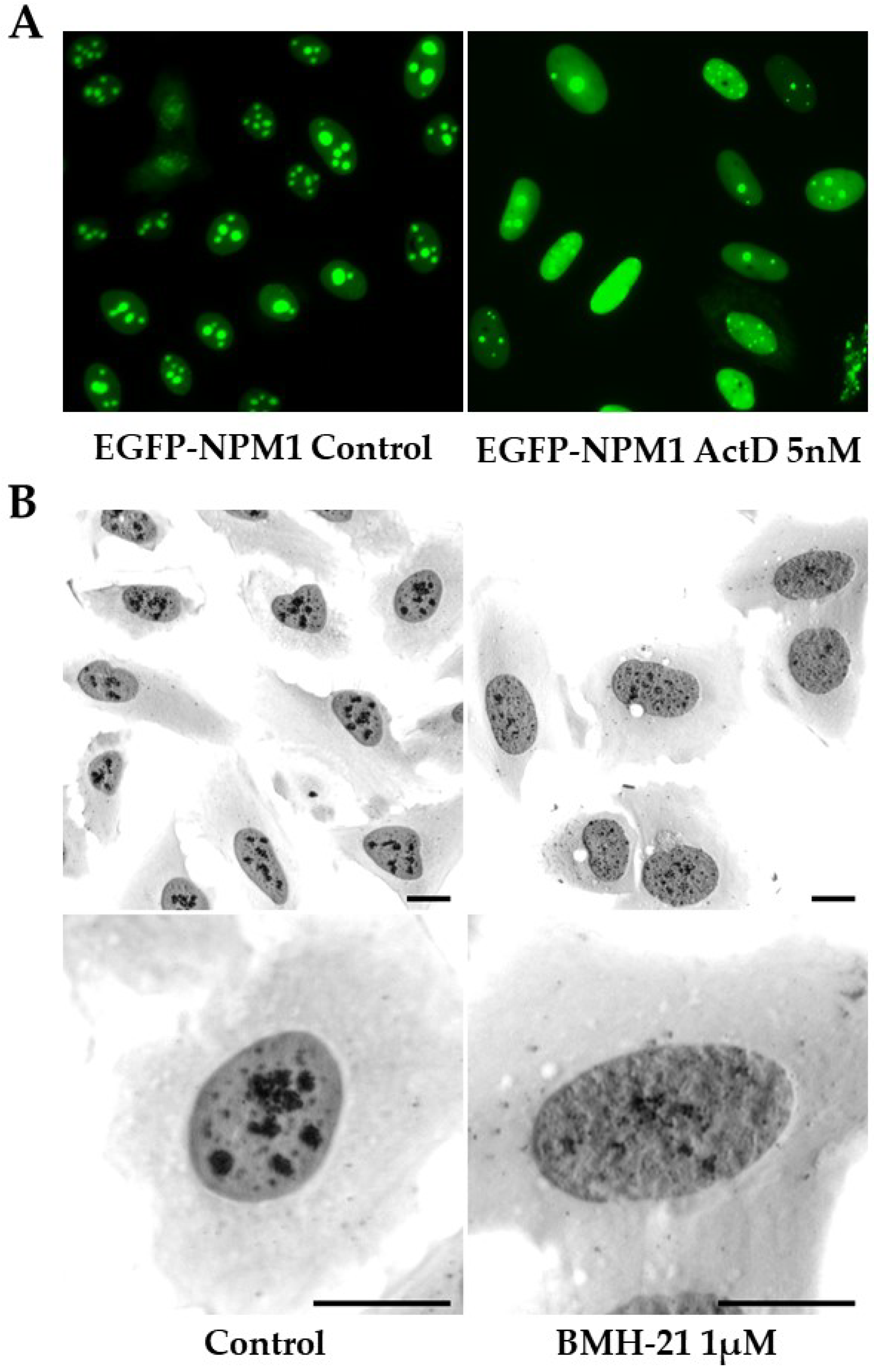
4.3. BMH-21 and CID-765471
4.4. Alkaloids and Lactones with RiBi Inhibiting Activity
4.5. Metarrestin
4.6. Additional RiBi Targeting Compounds
4.7. Targeting Other Cellular Processes Impacting on RiBi
4.8. Inhibitors of RiBi in Yeast
4.9. Nanoparticles and RNA Binding Compounds
5. Preclinical and Clinical Applications on Certain Cancer Types
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ActD | Actinomycin D |
| AAA-ATPases | ATPases Associated with diverse cellular Activities |
| CDK | Cyclin Dependent Kinase |
| CRC | Colorectal Cancer |
| DDR | DNA Damage Response |
| DHODH | Dihydroorotate Dehydrogenase |
| EMT | Epithelial to Mesenchymal Transition |
| hERG | human Ether-a-go-go Related gene |
| HMG | High-Mobility Group |
| IC50 | Half Maximal Inhibitory Concentration |
| IMPDH | Inosine-5’-monophosphate dehydrogenase |
| IRBC | Impaired Ribosome Biogenesis Checkpoint |
| MAPK | Mitogen-activated protein kinase |
| MDM2 | Mouse Double Minute 2 |
| mTOR | Mechanistic Target of Rapamycin |
| PIC | Pre-Initiation Complex |
| Pol I | RNA Polymerase I |
| Pol II | RNA Polymerase II |
| PTEN | Phosphatase and Tensin Homolog |
| RB | Retinoblastoma protein |
| rDNA | Ribosomal DNA |
| RiBi | Ribosome Biogenesis |
| RP | Ribosomal Protein |
| rRNA | Ribosomal RNA |
| RTK | Receptor Tyrosine Kinase |
| SL1 | Selectivity Factor 1 |
| TIF-I | Transcription Initiation Factor I |
| TOP1/TOP2 | Topoisomerase 1 and 2 |
| UBF | upstream binding factor |
| 5-FU | 5-fluorouracil |
| 5S RNP | 5S ribonucleoprotein |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Goodfellow, S.J.; Zomerdijk, J.C.B.M. Basic mechanisms in RNA polymerase I transcription of the ribosomal RNA genes. Subcell. Biochem. 2013, 61, 211–236. [Google Scholar] [CrossRef] [Green Version]
- Bywater, M.J.; Pearson, R.B.; McArthur, G.A.; Hannan, R.D. Dysregulation of the basal RNA polymerase transcription apparatus in cancer. Nat. Rev. Cancer 2013, 13, 299–314. [Google Scholar] [CrossRef]
- Drygin, D.; Rice, W.G.; Grummt, I. The RNA Polymerase I Transcription Machinery: An Emerging Target for the Treatment of Cancer. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 131–156. [Google Scholar] [CrossRef]
- Panov, K.I.; Hannan, K.; Hannan, R.D.; Hein, N. The Ribosomal Gene Loci—The Power behind the Throne. Genes 2021, 12, 763. [Google Scholar] [CrossRef]
- Thomson, E.; Ferreira-Cerca, S.; Hurt, E. Eukaryotic ribosome biogenesis at a glance. J. Cell Sci. 2013, 126, 4815–4821. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, R.; Schneekloth, J.S.; Panov, K.I.; Hannan, K.M.; Hannan, R.D. Targeting the RNA Polymerase I Transcription for Cancer Therapy Comes of Age. Cells 2020, 9, 266. [Google Scholar] [CrossRef] [Green Version]
- Drygin, D.; O’Brien, S.E.; Hannan, R.D.; McArthur, G.A.; Von Hoff, D.D. Targeting the nucleolus for cancer-specific activation of p53. Drug Discov. Today 2014, 19, 259–265. [Google Scholar] [CrossRef]
- Quin, J.E.; Devlin, J.R.; Cameron, D.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D. Targeting the nucleolus for cancer intervention. Biochim. Biophys. Acta 2014, 1842, 802–816. [Google Scholar] [CrossRef] [Green Version]
- Derenzini, E.; Rossi, A.; Treré, D. Treating hematological malignancies with drugs inhibiting ribosome biogenesis: When and why. J. Hematol. Oncol. 2018, 11, 75. [Google Scholar] [CrossRef]
- Bursać, S.; Prodan, Y.; Pullen, N.; Bartek, J.; Volarević, S. Dysregulated Ribosome Biogenesis Reveals Therapeutic Liabilities in Cancer. Trends Cancer 2021, 7, 57–76. [Google Scholar] [CrossRef] [PubMed]
- Carotenuto, P.; Pecoraro, A.; Palma, G.; Russo, G.; Russo, A. Therapeutic Approaches Targeting Nucleolus in Cancer. Cells 2019, 8, 1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robichaud, N.; Sonenberg, N.; Ruggero, D.; Schneider, R.J. Translational Control in Cancer. Cold Spring Harb. Perspect. Biol. 2019, 11, a032896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albihn, A.; Johnsen, J.I.; Henriksson, M.A. MYC in oncogenesis and as a target for cancer therapies. Adv. Cancer Res. 2010, 107, 163–224. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, D. The role of Myc-induced protein synthesis in cancer. Cancer Res. 2009, 69, 8839–8843. [Google Scholar] [CrossRef] [Green Version]
- Boon, K.; Caron, H.N.; van Asperen, R.; Valentijn, L.; Hermus, M.C.; van Sluis, P.; Roobeek, I.; Weis, I.; Voûte, P.A.; Schwab, M.; et al. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 2001, 20, 1383–1393. [Google Scholar] [CrossRef]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef]
- Ruggero, D.; Pandolfi, P.P. Does the ribosome translate cancer? Nat. Rev. Cancer 2003, 3, 179–192. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Derenzini, M.; Trerè, D.; Pession, A.; Montanaro, L.; Sirri, V.; Ochs, R.L. Nucleolar function and size in cancer cells. Am. J. Pathol. 1998, 152, 1291–1297. [Google Scholar]
- Montanaro, L.; Treré, D.; Derenzini, M. Nucleolus, ribosomes, and cancer. Am. J. Pathol. 2008, 173, 301–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeks, S.E.; Metge, B.J.; Samant, R.S. The nucleolus: A central response hub for the stressors that drive cancer progression. Cell. Mol. Life Sci. 2019, 76, 4511–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pederson, T. The nucleolus. Cold Spring Harb. Perspect. Biol. 2011, 3, a000638. [Google Scholar] [CrossRef] [PubMed]
- Lindström, M.S.; Jurada, D.; Bursac, S.; Orsolic, I.; Bartek, J.; Volarevic, S. Nucleolus as an emerging hub in maintenance of genome stability and cancer pathogenesis. Oncogene 2018, 37, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.-M.; van Koningsbruggen, S.; Navascués, J.; Lamond, A.I. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007, 8, 574–585. [Google Scholar] [CrossRef]
- Correll, C.C.; Bartek, J.; Dundr, M. The Nucleolus: A Multiphase Condensate Balancing Ribosome Synthesis and Translational Capacity in Health, Aging and Ribosomopathies. Cells 2019, 8, 869. [Google Scholar] [CrossRef] [Green Version]
- Lafontaine, D.L.J.; Riback, J.A.; Bascetin, R.; Brangwynne, C.P. The nucleolus as a multiphase liquid condensate. Nat. Rev. Mol. Cell Biol. 2021, 22, 165–182. [Google Scholar] [CrossRef]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef]
- Arabi, A.; Wu, S.; Ridderstråle, K.; Bierhoff, H.; Shiue, C.; Fatyol, K.; Fahlén, S.; Hydbring, P.; Söderberg, O.; Grummt, I.; et al. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat. Cell Biol. 2005, 7, 303–310. [Google Scholar] [CrossRef]
- Mayer, C.; Grummt, I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 2006, 25, 6384–6391. [Google Scholar] [CrossRef] [Green Version]
- Iadevaia, V.; Huo, Y.; Zhang, Z.; Foster, L.J.; Proud, C.G. Roles of the mammalian target of rapamycin, mTOR, in controlling ribosome biogenesis and protein synthesis. Biochem. Soc. Trans. 2012, 40, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Bursac, S.; Brdovcak, M.C.; Donati, G.; Volarevic, S. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim. Biophys. Acta 2014, 1842, 817–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckerman, R.; Prives, C. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a000935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, K.; Mühl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Hölzel, M.; et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustelo, X.R.; Dosil, M. Ribosome biogenesis and cancer: Basic and translational challenges. Curr. Opin. Genet. Dev. 2018, 48, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Holmberg Olausson, K.; Nistér, M.; Lindström, M.S. p53 -Dependent and -Independent Nucleolar Stress Responses. Cells 2012, 1, 774–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golomb, L.; Volarevic, S.; Oren, M. p53 and ribosome biogenesis stress: The essentials. FEBS Lett. 2014, 588, 2571–2579. [Google Scholar] [CrossRef]
- Nicolas, E.; Parisot, P.; Pinto-Monteiro, C.; de Walque, R.; De Vleeschouwer, C.; Lafontaine, D.L.J. Involvement of human ribosomal proteins in nucleolar structure and p53-dependent nucleolar stress. Nat. Commun. 2016, 7, 11390. [Google Scholar] [CrossRef]
- Turi, Z.; Senkyrikova, M.; Mistrik, M.; Bartek, J.; Moudry, P. Perturbation of RNA Polymerase I transcription machinery by ablation of HEATR1 triggers the RPL5/RPL11-MDM2-p53 ribosome biogenesis stress checkpoint pathway in human cells. Cell Cycle 2018, 17, 92–101. [Google Scholar] [CrossRef]
- Fumagalli, S.; Di Cara, A.; Neb-Gulati, A.; Natt, F.; Schwemberger, S.; Hall, J.; Babcock, G.F.; Bernardi, R.; Pandolfi, P.P.; Thomas, G. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat. Cell Biol. 2009, 11, 501–508. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, S.; Thomas, G. The role of p53 in ribosomopathies. Semin. Hematol. 2011, 48, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Moudry, P.; Chroma, K.; Bursac, S.; Volarevic, S.; Bartek, J. RNA-interference screen for p53 regulators unveils a role of WDR75 in ribosome biogenesis. Cell Death Differ. 2022, 29, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Tafforeau, L.; Zorbas, C.; Langhendries, J.-L.; Mullineux, S.-T.; Stamatopoulou, V.; Mullier, R.; Wacheul, L.; Lafontaine, D.L.J. The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of Pre-rRNA processing factors. Mol. Cell 2013, 51, 539–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volarevic, S.; Stewart, M.J.; Ledermann, B.; Zilberman, F.; Terracciano, L.; Montini, E.; Grompe, M.; Kozma, S.C.; Thomas, G. Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science 2000, 288, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Pestov, D.G.; Strezoska, Z.; Lau, L.F. Evidence of p53-dependent cross-talk between ribosome biogenesis and the cell cycle: Effects of nucleolar protein Bop1 on G(1)/S transition. Mol. Cell. Biol. 2001, 21, 4246–4255. [Google Scholar] [CrossRef] [Green Version]
- Marechal, V.; Elenbaas, B.; Piette, J.; Nicolas, J.C.; Levine, A.J. The ribosomal L5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol. Cell. Biol. 1994, 14, 7414–7420. [Google Scholar] [CrossRef]
- Sloan, K.E.; Bohnsack, M.T.; Watkins, N.J. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep. 2013, 5, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wolf, G.W.; Bhat, K.; Jin, A.; Allio, T.; Burkhart, W.A.; Xiong, Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell. Biol. 2003, 23, 8902–8912. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.-S.; Lu, H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J. Biol. Chem. 2004, 279, 44475–44482. [Google Scholar] [CrossRef] [Green Version]
- Donati, G.; Peddigari, S.; Mercer, C.A.; Thomas, G. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 2013, 4, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Macias, E.; Jin, A.; Deisenroth, C.; Bhat, K.; Mao, H.; Lindström, M.S.; Zhang, Y. An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 Interaction. Cancer Cell 2010, 18, 231–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindström, M.S.; Deisenroth, C.; Zhang, Y. Putting a finger on growth surveillance: Insight into MDM2 zinc finger-ribosomal protein interactions. Cell Cycle 2007, 6, 434–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubbi, C.P.; Milner, J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003, 22, 6068–6077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, K.; Eick, D. Functional ribosome biogenesis is a prerequisite for p53 destabilization: Impact of chemotherapy on nucleolar functions and RNA metabolism. Biol. Chem. 2013, 394, 1133–1143. [Google Scholar] [CrossRef]
- Ladds, M.J.G.W.; Laín, S. Small molecule activators of the p53 response. J. Mol. Cell Biol. 2019, 11, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Karni-Schmidt, O.; Lokshin, M.; Prives, C. The Roles of MDM2 and MDMX in Cancer. Annu. Rev. Pathol. 2016, 11, 617–644. [Google Scholar] [CrossRef]
- Oršolić, I.; Bursać, S.; Jurada, D.; Drmić Hofman, I.; Dembić, Z.; Bartek, J.; Mihalek, I.; Volarević, S. Cancer-associated mutations in the ribosomal protein L5 gene dysregulate the HDM2/p53-mediated ribosome biogenesis checkpoint. Oncogene 2020, 39, 3443–3457. [Google Scholar] [CrossRef]
- Fancello, L.; Kampen, K.R.; Hofman, I.J.F.; Verbeeck, J.; De Keersmaecker, K. The ribosomal protein gene RPL5 is a haploinsufficient tumor suppressor in multiple cancer types. Oncotarget 2017, 8, 14462–14478. [Google Scholar] [CrossRef] [Green Version]
- James, A.; Wang, Y.; Raje, H.; Rosby, R.; DiMario, P. Nucleolar stress with and without p53. Nucleus 2014, 5, 402–426. [Google Scholar] [CrossRef] [Green Version]
- Lessard, F.; Igelmann, S.; Trahan, C.; Huot, G.; Saint-Germain, E.; Mignacca, L.; Del Toro, N.; Lopes-Paciencia, S.; Le Calvé, B.; Montero, M.; et al. Senescence-associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nat. Cell Biol. 2018, 20, 789–799. [Google Scholar] [CrossRef]
- Lessard, F.; Brakier-Gingras, L.; Ferbeyre, G. Ribosomal Proteins Control Tumor Suppressor Pathways in Response to Nucleolar Stress. Bioessays 2019, 41, e1800183. [Google Scholar] [CrossRef] [PubMed]
- Donati, G.; Brighenti, E.; Vici, M.; Mazzini, G.; Treré, D.; Montanaro, L.; Derenzini, M. Selective inhibition of rRNA transcription downregulates E2F-1: A new p53-independent mechanism linking cell growth to cell proliferation. J. Cell Sci. 2011, 124, 3017–3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.P. The cellular sites of synthesis of ribosomal and 4S RNA. Proc. Natl. Acad. Sci. USA 1962, 48, 2179–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoefl, G.I. The effect of actinomycin d on the fine structure of the nucleolus. J. Ultrastruct. Res. 1964, 10, 224–243. [Google Scholar] [CrossRef]
- Reynolds, R.C.; Montgomery, P.O.; Hughes, B. Nucleolar “caps” produced by actinomycind. Cancer Res. 1964, 24, 1269–1277. [Google Scholar]
- Perry, R.P.; Kelley, D.E. Inhibition of RNA synthesis by actinomycin D: Characteristic dose-response of different RNA species. J. Cell. Physiol. 1970, 76, 127–139. [Google Scholar] [CrossRef]
- Trask, D.K.; Muller, M.T. Stabilization of type I topoisomerase-DNA covalent complexes by actinomycin D. Proc. Natl. Acad. Sci. USA 1988, 85, 1417–1421. [Google Scholar] [CrossRef] [Green Version]
- Hudson, J.S.; Brooks, S.C.; Graves, D.E. Interactions of actinomycin D with human telomeric G-quadruplex DNA. Biochemistry 2009, 48, 4440–4447. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.-J.; Park, H.-J. Novel molecular mechanism for actinomycin D activity as an oncogenic promoter G-quadruplex binder. Biochemistry 2009, 48, 7392–7398. [Google Scholar] [CrossRef]
- Niknezhad, Z.; Hassani, L.; Norouzi, D. Investigating actinomycin D binding to G-quadruplex, i-motif and double-stranded DNA in 27-nt segment of c-MYC gene promoter. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 58, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Mischo, H.E.; Hemmerich, P.; Grosse, F.; Zhang, S. Actinomycin D induces histone gamma-H2AX foci and complex formation of gamma-H2AX with Ku70 and nuclear DNA helicase II. J. Biol. Chem. 2005, 280, 9586–9594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arima, Y.; Nitta, M.; Kuninaka, S.; Zhang, D.; Fujiwara, T.; Taya, Y.; Nakao, M.; Saya, H. Transcriptional blockade induces p53-dependent apoptosis associated with translocation of p53 to mitochondria. J. Biol. Chem. 2005, 280, 19166–19176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [CrossRef] [Green Version]
- Lohrum, M.A.E.; Ludwig, R.L.; Kubbutat, M.H.G.; Hanlon, M.; Vousden, K.H. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 2003, 3, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.T.; Ellison, S.; Pandele, A.; Wood, S.; Nathan, E.; Forte, G.; Parker, H.; Zindy, E.; Elvin, M.; Dickson, A.; et al. Actinomycin D downregulates Sox2 and improves survival in preclinical models of recurrent glioblastoma. Neuro-Oncol. 2020, 22, 1289–1301. [Google Scholar] [CrossRef]
- Das, T.; Nair, R.R.; Green, R.; Padhee, S.; Howell, M.; Banerjee, J.; Mohapatra, S.S.; Mohapatra, S. Actinomycin D Down-regulates SOX2 Expression and Induces Death in Breast Cancer Stem Cells. Anticancer Res. 2017, 37, 1655–1663. [Google Scholar] [CrossRef]
- Ferguson, D.M.; Jacobson, B.A.; Jay-Dixon, J.; Patel, M.R.; Kratzke, R.A.; Raza, A. Targeting Topoisomerase II Activity in NSCLC with 9-Aminoacridine Derivatives. Anticancer Res. 2015, 7, 5211–5217. [Google Scholar]
- Ehsanian, R.; Van Waes, C.; Feller, S.M. Beyond DNA binding—A review of the potential mechanisms mediating quinacrine’s therapeutic activities in parasitic infections, inflammation, and cancers. Cell Commun. Signal. 2011, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Ho, W.C.; Dicker, D.T.; MacKinnon, C.; Winkler, J.D.; Marmorstein, R.; El-Deiry, W.S. Acridine derivatives activate p53 and induce tumor cell death through Bax. Cancer Biol. Ther. 2005, 4, 893–898. [Google Scholar] [CrossRef] [Green Version]
- Morgado-Palacin, L.; Llanos, S.; Urbano-Cuadrado, M.; Blanco-Aparicio, C.; Megias, D.; Pastor, J.; Serrano, M. Non-genotoxic activation of p53 through the RPL11-dependent ribosomal stress pathway. Carcinogenesis 2014, 35, 2822–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anikin, L.; Pestov, D.G. 9-Aminoacridine Inhibits Ribosome Biogenesis by Targeting Both Transcription and Processing of Ribosomal RNA. Int. J. Mol. Sci. 2022, 23, 1260. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Kundu, C.N. Anti-Cancer Stem Cells Potentiality of an Anti-Malarial Agent Quinacrine: An Old Wine in a New Bottle. Anticancer Agents Med. Chem. 2021, 21, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, A.; Österroos, A.; Hassan, S.; Gullbo, J.; Rickardson, L.; Jarvius, M.; Nygren, P.; Fryknäs, M.; Höglund, M.; Larsson, R. Drug screen in patient cells suggests quinacrine to be repositioned for treatment of acute myeloid leukemia. Blood Cancer J. 2015, 5, e307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oien, D.B.; Ray, U.; Pathoulas, C.L.; Jin, L.; Thirusangu, P.; Jung, D.; Kumka, J.E.; Xiao, Y.; Sarkar Bhattacharya, S.; Montoya, D.; et al. Quinacrine Induces Nucleolar Stress in Treatment-Refractory Ovarian Cancer Cell Lines. Cancers 2021, 13, 4645. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.A.; Zisi, A.; Kanellis, D.C.; Carreras-Puigvert, J.; Henriksson, M.; Hühn, D.; Watanabe, K.; Helleday, T.; Lindström, M.S.; Bartek, J. The antimalarial drug amodiaquine stabilizes p53 through ribosome biogenesis stress, independently of its autophagy-inhibitory activity. Cell Death Differ. 2020, 27, 773–789. [Google Scholar] [CrossRef] [Green Version]
- Qiao, S.; Tao, S.; Rojo de la Vega, M.; Park, S.L.; Vonderfecht, A.A.; Jacobs, S.L.; Zhang, D.D.; Wondrak, G.T. The antimalarial amodiaquine causes autophagic-lysosomal and proliferative blockade sensitizing human melanoma cells to starvation- and chemotherapy-induced cell death. Autophagy 2013, 9, 2087–2102. [Google Scholar] [CrossRef] [Green Version]
- Sohn, T.A.; Bansal, R.; Su, G.H.; Murphy, K.M.; Kern, S.E. High-throughput measurement of the Tp53 response to anticancer drugs and random compounds using a stably integrated Tp53-responsive luciferase reporter. Carcinogenesis 2002, 23, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Damiani, R.M.; Moura, D.J.; Viau, C.M.; Caceres, R.A.; Henriques, J.A.P.; Saffi, J. Pathways of cardiac toxicity: Comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch. Toxicol. 2016, 90, 2063–2076. [Google Scholar] [CrossRef]
- Zhu, H.; Sarkar, S.; Scott, L.; Danelisen, I.; Trush, M.A.; Jia, Z.; Li, Y.R. Doxorubicin Redox Biology: Redox Cycling, Topoisomerase Inhibition, and Oxidative Stress. React. Oxyg. Species (Apex) 2016, 1, 189–198. [Google Scholar] [CrossRef]
- Pang, B.; Qiao, X.; Janssen, L.; Velds, A.; Groothuis, T.; Kerkhoven, R.; Nieuwland, M.; Ovaa, H.; Rottenberg, S.; van Tellingen, O.; et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat. Commun. 2013, 4, 1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, S.; Panova, T.; Miller, G.; Volkov, A.; Porter, A.C.G.; Russell, J.; Panov, K.I.; Zomerdijk, J.C.B.M. Topoisomerase IIα promotes activation of RNA polymerase I transcription by facilitating pre-initiation complex formation. Nat. Commun. 2013, 4, 1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, P.; Carmo-Fonseca, M. Cisplatin inhibits synthesis of ribosomal RNA in vivo. Nucleic Acids Res. 1998, 26, 2831–2836. [Google Scholar] [CrossRef] [Green Version]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef]
- Zhai, X.; Beckmann, H.; Jantzen, H.M.; Essigmann, J.M. Cisplatin-DNA adducts inhibit ribosomal RNA synthesis by hijacking the transcription factor human upstream binding factor. Biochemistry 1998, 37, 16307–16315. [Google Scholar] [CrossRef] [PubMed]
- Treiber, D.K.; Zhai, X.; Jantzen, H.M.; Essigmann, J.M. Cisplatin-DNA adducts are molecular decoys for the ribosomal RNA transcription factor hUBF (human upstream binding factor). Proc. Natl. Acad. Sci. USA 1994, 91, 5672–5676. [Google Scholar] [CrossRef] [Green Version]
- Hamdane, N.; Herdman, C.; Mars, J.-C.; Stefanovsky, V.; Tremblay, M.G.; Moss, T. Depletion of the cisplatin targeted HMGB-box factor UBF selectively induces p53-independent apoptotic death in transformed cells. Oncotarget 2015, 6, 27519–27536. [Google Scholar] [CrossRef] [Green Version]
- Sutton, E.C.; DeRose, V.J. Early nucleolar responses differentiate mechanisms of cell death induced by oxaliplatin and cisplatin. J. Biol. Chem. 2021, 296, 100633. [Google Scholar] [CrossRef]
- Sutton, E.C.; McDevitt, C.E.; Prochnau, J.Y.; Yglesias, M.V.; Mroz, A.M.; Yang, M.C.; Cunningham, R.M.; Hendon, C.H.; DeRose, V.J. Nucleolar Stress Induction by Oxaliplatin and Derivatives. J. Am. Chem. Soc. 2019, 141, 18411–18415. [Google Scholar] [CrossRef] [PubMed]
- Tomasz, M. Mitomycin C: Small, fast and deadly (but very selective). Chem. Biol. 1995, 2, 575–579. [Google Scholar] [CrossRef] [Green Version]
- Raymond, E.; Faivre, S.; Woynarowski, J.M.; Chaney, S.G. Oxaliplatin: Mechanism of action and antineoplastic activity. Semin. Oncol. 1998, 25, 4–12. [Google Scholar] [PubMed]
- Awad, D.; Prattes, M.; Kofler, L.; Rössler, I.; Loibl, M.; Pertl, M.; Zisser, G.; Wolinski, H.; Pertschy, B.; Bergler, H. Inhibiting eukaryotic ribosome biogenesis. BMC Biol. 2019, 17, 46. [Google Scholar] [CrossRef] [PubMed]
- Bolzán, A.D.; Bianchi, M.S. Genotoxicity of streptonigrin: A review. Mutat. Res. 2001, 488, 25–37. [Google Scholar] [CrossRef]
- Chan, P.K.; Aldrich, M.B.; Yung, B.Y. Nucleolar protein B23 translocation after doxorubicin treatment in murine tumor cells. Cancer Res. 1987, 47, 3798–3801. [Google Scholar]
- McKeage, M.J.; Hsu, T.; Screnci, D.; Haddad, G.; Baguley, B.C. Nucleolar damage correlates with neurotoxicity induced by different platinum drugs. Br J Cancer. 2001, 85, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- Snodgrass, R.G.; Collier, A.C.; Coon, A.E.; Pritsos, C.A. Mitomycin C inhibits ribosomal RNA: A novel cytotoxic mechanism for bioreductive drugs. J. Biol. Chem. 2010, 285, 19068–19075. [Google Scholar] [CrossRef] [Green Version]
- Lapis, K.; Bernhard, W. The effect of mitomycin-c on the nucleolar fine structure of kb cells in cell culture. Cancer Res. 1965, 25, 628–645. [Google Scholar]
- Chan, P.K.; Aldrich, M.B.; Chakrabarty, S. Assessment of tumor cell sensitivity to mitomycin C by “B23 translocation” assay. Cancer Lett. 1988, 40, 143–149. [Google Scholar] [CrossRef]
- Sun, X.-X.; Dai, M.-S.; Lu, H. 5-fluorouracil activation of p53 involves an MDM2-ribosomal protein interaction. J. Biol. Chem. 2007, 282, 8052–8059. [Google Scholar] [CrossRef] [Green Version]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef]
- Chan, P.K. Characterization and cellular localization of nucleophosmin/B23 in HeLa cells treated with selected cytotoxic agents (studies of B23-translocation mechanism). Exp. Cell Res. 1992, 203, 174–181. [Google Scholar] [CrossRef]
- Kaminskas, E. Effects of methotrexate on ribonucleotide pools in growing and in growth-arrested tumor cells and antagonism by RNA synthesis inhibitors. J. Biol. Chem. 1982, 257, 4279–4284. [Google Scholar] [CrossRef]
- Wu, R.S.; Kumar, A.; Warner, J.R. Ribosome formation is blocked by camptothecin, a reversible inhibitor of RNA synthesis. Proc. Natl. Acad. Sci. USA 1971, 68, 3009–3014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisenberger, D.; Scheer, U.; Benavente, R. The DNA topoisomerase I inhibitor camptothecin blocks postmitotic reformation of nucleoli in mammalian cells. Eur. J. Cell Biol. 1993, 61, 189–192. [Google Scholar] [PubMed]
- Pietrzak, M.; Smith, S.C.; Geralds, J.T.; Hagg, T.; Gomes, C.; Hetman, M. Nucleolar disruption and apoptosis are distinct neuronal responses to etoposide-induced DNA damage. J. Neurochem. 2011, 117, 1033–1046. [Google Scholar] [CrossRef] [Green Version]
- Montecucco, A.; Biamonti, G. Cellular response to etoposide treatment. Cancer Lett. 2007, 252, 9–18. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.-X.; Dai, M.-S.; Lu, H. Mycophenolic acid activation of p53 requires ribosomal proteins L5 and L11. J. Biol. Chem. 2008, 283, 12387–12392. [Google Scholar] [CrossRef] [Green Version]
- Almqvist, H.; Axelsson, H.; Jafari, R.; Dan, C.; Mateus, A.; Haraldsson, M.; Larsson, A.; Martinez Molina, D.; Artursson, P.; Lundbäck, T.; et al. CETSA screening identifies known and novel thymidylate synthase inhibitors and slow intracellular activation of 5-fluorouracil. Nat. Commun. 2016, 7, 11040. [Google Scholar] [CrossRef] [Green Version]
- Azwar, S.; Seow, H.F.; Abdullah, M.; Faisal Jabar, M.; Mohtarrudin, N. Recent Updates on Mechanisms of Resistance to 5-Fluorouracil and Reversal Strategies in Colon Cancer Treatment. Biology 2021, 10, 854. [Google Scholar] [CrossRef]
- Liang, Y.Y.; Bacanu, S.; Sreekumar, L.; Ramos, A.D.; Dai, L.; Michaelis, M.; Cinatl, J.; Seki, T.; Cao, Y.; Coffill, C.R.; et al. CETSA interaction proteomics define specific RNA-modification pathways as key components of fluorouracil-based cancer drug cytotoxicity. Cell Chem. Biol. 2022, 29, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Bash-Imam, Z.; Thérizols, G.; Vincent, A.; Lafôrets, F.; Polay Espinoza, M.; Pion, N.; Macari, F.; Pannequin, J.; David, A.; Saurin, J.-C.; et al. Translational reprogramming of colorectal cancer cells induced by 5-fluorouracil through a miRNA-dependent mechanism. Oncotarget 2017, 8, 46219–46233. [Google Scholar] [CrossRef] [PubMed]
- Andrews, W.J.; Ray, S.; Panova, T.; Engel, C.; Panov, K.I. DNA Intercalators Inhibit Eukaryotic Ribosomal RNA Synthesis by Impairing the Initiation of Transcription. Genes 2021, 12, 1412. [Google Scholar] [CrossRef] [PubMed]
- Buckwalter, C.A.; Lin, A.H.; Tanizawa, A.; Pommier, Y.G.; Cheng, Y.C.; Kaufmann, S.H. RNA synthesis inhibitors alter the subnuclear distribution of DNA topoisomerase I. Cancer Res. 1996, 56, 1674–1681. [Google Scholar]
- Zhang, H.; Wang, J.C.; Liu, L.F. Involvement of DNA topoisomerase I in transcription of human ribosomal RNA genes. Proc. Natl. Acad. Sci. USA 1988, 85, 1060–1064. [Google Scholar] [CrossRef] [Green Version]
- Colis, L.; Peltonen, K.; Sirajuddin, P.; Liu, H.; Sanders, S.; Ernst, G.; Barrow, J.C.; Laiho, M. DNA intercalator BMH-21 inhibits RNA polymerase I independent of DNA damage response. Oncotarget 2014, 5, 4361–4369. [Google Scholar] [CrossRef] [Green Version]
- Peltonen, K.; Colis, L.; Liu, H.; Jäämaa, S.; Zhang, Z.; Af Hällström, T.; Moore, H.M.; Sirajuddin, P.; Laiho, M. Small molecule BMH-compounds that inhibit RNA polymerase I and cause nucleolar stress. Mol. Cancer Ther. 2014, 13, 2537–2546. [Google Scholar] [CrossRef] [Green Version]
- Peltonen, K.; Colis, L.; Liu, H.; Jäämaa, S.; Moore, H.M.; Enbäck, J.; Laakkonen, P.; Vaahtokari, A.; Jones, R.J.; af Hällström, T.M.; et al. Identification of novel p53 pathway activating small-molecule compounds reveals unexpected similarities with known therapeutic agents. PLoS ONE 2010, 5, e12996. [Google Scholar] [CrossRef]
- Kofuji, S.; Hirayama, A.; Eberhardt, A.O.; Kawaguchi, R.; Sugiura, Y.; Sampetrean, O.; Ikeda, Y.; Warren, M.; Sakamoto, N.; Kitahara, S.; et al. IMP dehydrogenase-2 drives aberrant nucleolar activity and promotes tumorigenesis in glioblastoma. Nat. Cell Biol. 2019, 21, 1003–1014. [Google Scholar] [CrossRef]
- Badertscher, L.; Wild, T.; Montellese, C.; Alexander, L.T.; Bammert, L.; Sarazova, M.; Stebler, M.; Csucs, G.; Mayer, T.U.; Zamboni, N.; et al. Genome-wide RNAi Screening Identifies Protein Modules Required for 40S Subunit Synthesis in Human Cells. Cell Rep. 2015, 13, 2879–2891. [Google Scholar] [CrossRef] [Green Version]
- Dörner, K.; Badertscher, L.; Horváth, B.; Hollandi, R.; Molnár, C.; Fuhrer, T.; Meier, R.; Sárazová, M.; van den Heuvel, J.; Zamboni, N.; et al. Genome-wide RNAi screen identifies novel players in human 60S subunit biogenesis including key enzymes of polyamine metabolism. Nucleic Acids Res. 2022, 50, 2872–2888. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, V.; Kinor, N.; Shav-Tal, Y.; Biggiogera, M.; Brüning, A. The stress-inducible transcription factor ATF4 accumulates at specific rRNA-processing nucleolar regions after proteasome inhibition. Eur. J. Cell Biol. 2016, 95, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Latonen, L.; Moore, H.M.; Bai, B.; Jäämaa, S.; Laiho, M. Proteasome inhibitors induce nucleolar aggregation of proteasome target proteins and polyadenylated RNA by altering ubiquitin availability. Oncogene 2011, 30, 790–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latonen, L. Phase-to-Phase With Nucleoli–Stress Responses, Protein Aggregation and Novel Roles of RNA. Front. Cell. Neurosci. 2019, 13, 151. [Google Scholar] [CrossRef]
- Zhou, D.C.; Zittoun, R.; Marie, J.P. Homoharringtonine: An effective new natural product in cancer chemotherapy. Bull. Cancer 1995, 82, 987–995. [Google Scholar]
- Zhu, H.-H.; Jiang, H.; Jiang, Q.; Jia, J.-S.; Qin, Y.-Z.; Huang, X.-J. Homoharringtonine, aclarubicin and cytarabine (HAA) regimen as the first course of induction therapy is highly effective for acute myeloid leukemia with t (8;21). Leuk. Res. 2016, 44, 40–44. [Google Scholar] [CrossRef]
- Scull, C.E.; Zhang, Y.; Tower, N.; Rasmussen, L.; Padmalayam, I.; Hunter, R.; Zhai, L.; Bostwick, R.; Schneider, D.A. Discovery of novel inhibitors of ribosome biogenesis by innovative high throughput screening strategies. Biochem. J. 2019, 476, 2209–2219. [Google Scholar] [CrossRef]
- Yung, B.Y.; Busch, H.; Chan, P.K. Translocation of nucleolar phosphoprotein B23 (37 kDa/pI 5.1) induced by selective inhibitors of ribosome synthesis. Biochim. Biophys. Acta 1985, 826, 167–173. [Google Scholar] [CrossRef]
- Yung, B.Y.; Bor, A.M.; Chan, P.K. Short exposure to actinomycin D induces “reversible” translocation of protein B23 as well as “reversible” inhibition of cell growth and RNA synthesis in HeLa cells. Cancer Res. 1990, 50, 5987–5991. [Google Scholar]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef] [Green Version]
- Ide, S.; Imai, R.; Ochi, H.; Maeshima, K. Transcriptional suppression of ribosomal DNA with phase separation. Sci. Adv. 2020, 6, eabb5953. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef] [Green Version]
- Haddach, M.; Schwaebe, M.K.; Michaux, J.; Nagasawa, J.; O’Brien, S.E.; Whitten, J.P.; Pierre, F.; Kerdoncuff, P.; Darjania, L.; Stansfield, R.; et al. Discovery of CX-5461, the First Direct and Selective Inhibitor of RNA Polymerase I, for Cancer Therapeutics. ACS Med. Chem. Lett. 2012, 3, 602–606. [Google Scholar] [CrossRef] [Green Version]
- Quin, J.; Chan, K.T.; Devlin, J.R.; Cameron, D.P.; Diesch, J.; Cullinane, C.; Ahern, J.; Khot, A.; Hein, N.; George, A.J.; et al. Inhibition of RNA polymerase I transcription initiation by CX-5461 activates non-canonical ATM/ATR signaling. Oncotarget 2016, 7, 49800–49818. [Google Scholar] [CrossRef] [Green Version]
- El Hassouni, B.; Mantini, G.; Immordino, B.; Peters, G.J.; Giovannetti, E. CX-5461 Inhibits Pancreatic Ductal Adenocarcinoma Cell Growth, Migration and Induces DNA Damage. Molecules 2019, 24, 4445. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.S.; Zeki, J.; Ornell, K.; Coburn, J.; Shimada, H.; Ikegaki, N.; Chiu, B. Down-regulation of MYCN protein by CX-5461 leads to neuroblastoma tumor growth suppression. J. Pediatr. Surg. 2019, 54, 1192–1197. [Google Scholar] [CrossRef]
- Negi, S.S.; Brown, P. rRNA synthesis inhibitor, CX-5461, activates ATM/ATR pathway in acute lymphoblastic leukemia, arrests cells in G2 phase and induces apoptosis. Oncotarget 2015, 6, 18094–18104. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.; Wang, H.; Baladandayuthapani, V.; Lin, H.; He, J.; Jones, R.J.; Kuiatse, I.; Gu, D.; Wang, Z.; Ma, W.; et al. RNA Polymerase I Inhibition with CX-5461 as a Novel Therapeutic Strategy to Target MYC in Multiple Myeloma. Br. J. Haematol. 2017, 177, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Khot, A.; Brajanovski, N.; Cameron, D.P.; Hein, N.; Maclachlan, K.H.; Sanij, E.; Lim, J.; Soong, J.; Link, E.; Blombery, P.; et al. First-in-Human RNA Polymerase I Transcription Inhibitor CX-5461 in Patients with Advanced Hematologic Cancers: Results of a Phase I Dose-Escalation Study. Cancer Discov. 2019, 9, 1036–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achiron, A.; Zilkha-Falb, R.; Mashiach, R.; Gurevich, M. RAM-589.555 a new Polymerase-1 inhibitor as innovative targeted-treatment for multiple sclerosis. J. Neuroimmunol. 2017, 302, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Bruno, P.M.; Lu, M.; Dennis, K.A.; Inam, H.; Moore, C.J.; Sheehe, J.; Elledge, S.J.; Hemann, M.T.; Pritchard, J.R. The primary mechanism of cytotoxicity of the chemotherapeutic agent CX-5461 is topoisomerase II poisoning. Proc. Natl. Acad. Sci. USA 2020, 117, 4053–4060. [Google Scholar] [CrossRef]
- Bossaert, M.; Pipier, A.; Riou, J.-F.; Noirot, C.; Nguyên, L.-T.; Serre, R.-F.; Bouchez, O.; Defrancq, E.; Calsou, P.; Britton, S.; et al. Transcription-associated topoisomerase 2α (TOP2A) activity is a major effector of cytotoxicity induced by G-quadruplex ligands. eLife 2021, 10, e65184. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Wright, W.C.; Chapple, R.H.; Zubair, A.; Sandhu, M.; Batchelder, J.E.; Huddle, B.C.; Low, J.; Blankenship, K.B.; Wang, Y.; et al. The chemotherapeutic CX-5461 primarily targets TOP2B and exhibits selective activity in high-risk neuroblastoma. Nat. Commun. 2021, 12, 6468. [Google Scholar] [CrossRef]
- Wei, T.; Najmi, S.M.; Liu, H.; Peltonen, K.; Kucerova, A.; Schneider, D.A.; Laiho, M. Small-Molecule Targeting of RNA Polymerase I Activates a Conserved Transcription Elongation Checkpoint. Cell Rep. 2018, 23, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Peltonen, K.; Colis, L.; Liu, H.; Trivedi, R.; Moubarek, M.S.; Moore, H.M.; Bai, B.; Rudek, M.A.; Bieberich, C.J.; Laiho, M. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell 2014, 25, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, R.Q.; Huffines, A.K.; Laiho, M.; Schneider, D.A. The small-molecule BMH-21 directly inhibits transcription elongation and DNA occupancy of RNA polymerase I in vivo and in vitro. J. Biol. Chem. 2022, 298, 101450. [Google Scholar] [CrossRef]
- Musso, L.; Mazzini, S.; Rossini, A.; Castagnoli, L.; Scaglioni, L.; Artali, R.; Di Nicola, M.; Zunino, F.; Dallavalle, S. c-MYC G-quadruplex binding by the RNA polymerase I inhibitor BMH-21 and analogues revealed by a combined NMR and biochemical Approach. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 615–629. [Google Scholar] [CrossRef]
- Masud, T.; Soong, C.; Xu, H.; Biele, J.; Bjornson, S.; McKinney, S.; Aparicio, S. Ubiquitin-mediated DNA damage response is synthetic lethal with G-quadruplex stabilizer CX-5461. Sci. Rep. 2021, 11, 9812. [Google Scholar] [CrossRef]
- Dorado, T.E.; de Leon, P.; Begum, A.; Liu, H.; Chen, D.; Rajeshkumar, N.V.; Rey-Rordiguez, R.; Horeau-Aveilla, C.; Alcouffe, C.; Laiho, M.; et al. Discovery and evaluation of novel angular fused pyridoquinazolinecarboxamides as RNA polymerase I inhibitors. ACS Med. Chem. Lett. 2022, 13, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Andrews, W.J.; Panova, T.; Normand, C.; Gadal, O.; Tikhonova, I.G.; Panov, K.I. Old drug, new target: Ellipticines selectively inhibit RNA polymerase I transcription. J. Biol. Chem. 2013, 288, 4567–4582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-T.; Chen, J.-J.; Wang, H.-T. Targeting RNA Polymerase I with Hernandonine Inhibits Ribosomal RNA Synthesis and Tumor Cell Growth. Mol. Cancer Res. 2019, 17, 2294–2305. [Google Scholar] [CrossRef] [PubMed]
- Caggiano, C.; Guida, E.; Todaro, F.; Bielli, P.; Mori, M.; Ghirga, F.; Quaglio, D.; Botta, B.; Moretti, F.; Grimaldi, P.; et al. Sempervirine inhibits RNA polymerase I transcription independently from p53 in tumor cells. Cell Death Discov. 2020, 6, 111. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z.; Li, F.; Chen, J.; Gong, X.; Cao, B.; Wang, W. Triptolide interrupts rRNA synthesis and induces the RPL23-MDM2-p53 pathway to repress lung cancer cells. Oncol. Rep. 2020, 43, 1863–1874. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, J.; He, L.; Yu, Q. Triptolide (TPL) inhibits global transcription by inducing proteasome-dependent degradation of RNA polymerase II (Pol II). PLoS ONE 2011, 6, e23993. [Google Scholar] [CrossRef] [Green Version]
- Titov, D.V.; Gilman, B.; He, Q.-L.; Bhat, S.; Low, W.-K.; Dang, Y.; Smeaton, M.; Demain, A.L.; Miller, P.S.; Kugel, J.F.; et al. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat. Chem. Biol. 2011, 7, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Manzo, S.G.; Zhou, Z.-L.; Wang, Y.-Q.; Marinello, J.; He, J.-X.; Li, Y.-C.; Ding, J.; Capranico, G.; Miao, Z.-H. Natural product triptolide mediates cancer cell death by triggering CDK7-dependent degradation of RNA polymerase II. Cancer Res. 2012, 72, 5363–5373. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Xie, R.; Su, J.; Ye, B.; Wei, S.; Liang, Z.; Bai, R.; Chen, Z.; Li, Z.; Gao, X. Inhibition of RNA polymerase III transcription by Triptolide attenuates colorectal tumorigenesis. J. Exp. Clin. Cancer Res. 2019, 38, 217. [Google Scholar] [CrossRef]
- Frankowski, K.J.; Wang, C.; Patnaik, S.; Schoenen, F.J.; Southall, N.; Li, D.; Teper, Y.; Sun, W.; Kandela, I.; Hu, D.; et al. Metarrestin, a perinucleolar compartment inhibitor, effectively suppresses metastasis. Sci. Transl. Med. 2018, 10, eaap8307. [Google Scholar] [CrossRef] [Green Version]
- Slusarczyk, A.; Kamath, R.; Wang, C.; Anchel, D.; Pollock, C.; Lewandowska, M.A.; Fitzpatrick, T.; Bazett-Jones, D.P.; Huang, S. Structure and function of the perinucleolar compartment in cancer cells. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 599–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, V.; Carson, B.B.; Feenstra, J.M.; Dass, R.A.; Sekyrova, P.; Hoshino, A.; Petersen, J.; Guo, Y.; Parks, M.M.; Kurylo, C.M.; et al. Ribosome biogenesis during cell cycle arrest fuels EMT in development and disease. Nat. Commun. 2019, 10, 2110. [Google Scholar] [CrossRef] [PubMed]
- Elhamamsy, A.R.; Metge, B.J.; Alsheikh, H.A.; Shevde, L.A.; Samant, R.S. Ribosome biogenesis: A central player in cancer metastasis and therapeutic resistance. Cancer Res. 2022, in press. [Google Scholar] [CrossRef]
- Tan, X.; Awuah, S.G. A cell-based screening system for RNA polymerase I inhibitors. MedChemComm 2019, 10, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Rothblum, K.; Hu, Q.; Penrod, Y.; Rothblum, L.I. Selective inhibition of rDNA transcription by a small-molecule peptide that targets the interface between RNA polymerase I and Rrn3. Mol. Cancer Res. 2014, 12, 1586–1596. [Google Scholar] [CrossRef] [Green Version]
- Caudron-Herger, M.; Pankert, T.; Seiler, J.; Németh, A.; Voit, R.; Grummt, I.; Rippe, K. Alu element-containing RNAs maintain nucleolar structure and function. EMBO J. 2015, 34, 2758–2774. [Google Scholar] [CrossRef] [Green Version]
- Abraham, K.J.; Khosraviani, N.; Chan, J.N.Y.; Gorthi, A.; Samman, A.; Zhao, D.Y.; Wang, M.; Bokros, M.; Vidya, E.; Ostrowski, L.A.; et al. Nucleolar RNA polymerase II drives ribosome biogenesis. Nature 2020, 585, 298–302. [Google Scholar] [CrossRef]
- Haaf, T.; Ward, D.C. Inhibition of RNA polymerase II transcription causes chromatin decondensation, loss of nucleolar structure, and dispersion of chromosomal domains. Exp. Cell Res. 1996, 224, 163–173. [Google Scholar] [CrossRef]
- David-Pfeuty, T.; Nouvian-Dooghe, Y.; Sirri, V.; Roussel, P.; Hernandez-Verdun, D. Common and reversible regulation of wild-type p53 function and of ribosomal biogenesis by protein kinases in human cells. Oncogene 2001, 20, 5951–5963. [Google Scholar] [CrossRef] [Green Version]
- Sirri, V.; Hernandez-Verdun, D.; Roussel, P. Cyclin-dependent kinases govern formation and maintenance of the nucleolus. J. Cell Biol. 2002, 156, 969–981. [Google Scholar] [CrossRef] [Green Version]
- Burger, K.; Mühl, B.; Rohrmoser, M.; Coordes, B.; Heidemann, M.; Kellner, M.; Gruber-Eber, A.; Heissmeyer, V.; Strässer, K.; Eick, D. Cyclin-dependent kinase 9 links RNA polymerase II transcription to processing of ribosomal RNA. J. Biol. Chem. 2013, 288, 21173–21183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanellis, D.C.; Espinoza, J.A.; Zisi, A.; Sakkas, E.; Bartkova, J.; Katsori, A.-M.; Boström, J.; Dyrskjøt, L.; Broholm, H.; Altun, M.; et al. The exon-junction complex helicase eIF4A3 controls cell fate via coordinated regulation of ribosome biogenesis and translational output. Sci. Adv. 2021, 7, eabf7561. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Riaño-Canalias, F.; Almacellas, E.; Mauvezin, C.; Samino, S.; Feu, S.; Menoyo, S.; Domostegui, A.; Garcia-Cajide, M.; Salazar, R.; et al. Nucleotide depletion reveals the impaired ribosome biogenesis checkpoint as a barrier against DNA damage. EMBO J. 2020, 39, e103838. [Google Scholar] [CrossRef] [PubMed]
- Grummt, I.; Grummt, F. Control of nucleolar RNA synthesis by the intracellular pool sizes of ATP and GTP. Cell 1976, 7, 447–453. [Google Scholar] [CrossRef]
- Lafita-Navarro, M.C.; Conacci-Sorrell, M. Nucleolar stress: From development to cancer. Semin. Cell Dev. Biol. 2022, in press. [CrossRef]
- Lafita-Navarro, M.C.; Venkateswaran, N.; Kilgore, J.A.; Kanji, S.; Han, J.; Barnes, S.; Williams, N.S.; Buszczak, M.; Burma, S.; Conacci-Sorrell, M. Inhibition of the de novo pyrimidine biosynthesis pathway limits ribosomal RNA transcription causing nucleolar stress in glioblastoma cells. PLoS Genet. 2020, 16, e1009117. [Google Scholar] [CrossRef]
- Hubackova, S.; Davidova, E.; Boukalova, S.; Kovarova, J.; Bajzikova, M.; Coelho, A.; Terp, M.G.; Ditzel, H.J.; Rohlena, J.; Neuzil, J. Replication and ribosomal stress induced by targeting pyrimidine synthesis and cellular checkpoints suppress p53-deficient tumors. Cell Death Dis. 2020, 11, 110. [Google Scholar] [CrossRef]
- Ladds, M.J.G.W.; van Leeuwen, I.M.M.; Drummond, C.J.; Chu, S.; Healy, A.R.; Popova, G.; Pastor Fernández, A.; Mollick, T.; Darekar, S.; Sedimbi, S.K.; et al. A DHODH inhibitor increases p53 synthesis and enhances tumor cell killing by p53 degradation blockage. Nat. Commun. 2018, 9, 1107. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Copeland, C.; Le, A. Glutamine Metabolism in Cancer. Adv. Exp. Med. Biol. 2021, 1311, 17–38. [Google Scholar] [CrossRef]
- Skrott, Z.; Mistrik, M.; Andersen, K.K.; Friis, S.; Majera, D.; Gursky, J.; Ozdian, T.; Bartkova, J.; Turi, Z.; Moudry, P.; et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature 2017, 552, 194–199. [Google Scholar] [CrossRef]
- Loibl, M.; Klein, I.; Prattes, M.; Schmidt, C.; Kappel, L.; Zisser, G.; Gungl, A.; Krieger, E.; Pertschy, B.; Bergler, H. The drug diazaborine blocks ribosome biogenesis by inhibiting the AAA-ATPase Drg1. J. Biol. Chem. 2014, 289, 3913–3922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashima, S.A.; Chen, Z.; Aoi, Y.; Patgiri, A.; Kobayashi, Y.; Nurse, P.; Kapoor, T.M. Potent, Reversible, and Specific Chemical Inhibitors of Eukaryotic Ribosome Biogenesis. Cell 2016, 167, 512–524.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrino, S.; Meyer, M.; Zorbas, C.; Bouchta, S.A.; Saraf, K.; Pelly, S.C.; Yusupova, G.; Evidente, A.; Mathieu, V.; Kornienko, A.; et al. The Amaryllidaceae Alkaloid Haemanthamine Binds the Eukaryotic Ribosome to Repress Cancer Cell Growth. Structure 2018, 26, 416–425.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brighenti, E.; Treré, D.; Derenzini, M. Targeted cancer therapy with ribosome biogenesis inhibitors: A real possibility? Oncotarget 2015, 6, 38617–38627. [Google Scholar] [CrossRef] [Green Version]
- Gilles, A.; Frechin, L.; Natchiar, K.; Biondani, G.; von Loeffelholz, O.; Holvec, S.; Malaval, J.-L.; Winum, J.-Y.; Klaholz, B.P.; Peyron, J.-F. Targeting the Human 80S Ribosome in Cancer: From Structure to Function and Drug Design for Innovative Adjuvant Therapeutic Strategies. Cells 2020, 9, 629. [Google Scholar] [CrossRef] [Green Version]
- Catez, F.; Dalla Venezia, N.; Marcel, V.; Zorbas, C.; Lafontaine, D.L.J.; Diaz, J.-J. Ribosome biogenesis: An emerging druggable pathway for cancer therapeutics. Biochem. Pharmacol. 2019, 159, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Kodiha, M.; Mahboubi, H.; Maysinger, D.; Stochaj, U. Gold Nanoparticles Impinge on Nucleoli and the Stress Response in MCF7 Breast Cancer Cells. Nanobiomedicine 2016, 3, 3. [Google Scholar] [CrossRef]
- Chen, M.; von Mikecz, A. Formation of nucleoplasmic protein aggregates impairs nuclear function in response to SiO2 nanoparticles. Exp. Cell Res. 2005, 305, 51–62. [Google Scholar] [CrossRef]
- Falese, J.P.; Donlic, A.; Hargrove, A.E. Targeting RNA with small molecules: From fundamental principles towards the clinic. Chem. Soc. Rev. 2021, 50, 2224–2243. [Google Scholar] [CrossRef]
- Warner, K.D.; Hajdin, C.E.; Weeks, K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef]
- Wirth, R.; White, J.D.; Moghaddam, A.D.; Ginzburg, A.L.; Zakharov, L.N.; Haley, M.M.; DeRose, V.J. Azide vs Alkyne Functionalization in Pt(II) Complexes for Post-treatment Click Modification: Solid-State Structure, Fluorescent Labeling, and Cellular Fate. J. Am. Chem. Soc. 2015, 137, 15169–15175. [Google Scholar] [CrossRef] [PubMed]
- Law, A.S.-Y.; Lee, L.C.-C.; Lo, K.K.-W.; Yam, V.W.-W. Aggregation and Supramolecular Self-Assembly of Low-Energy Red Luminescent Alkynylplatinum(II) Complexes for RNA Detection, Nucleolus Imaging, and RNA Synthesis Inhibitor Screening. J. Am. Chem. Soc. 2021, 143, 5396–5405. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, V.; Schneider, D.A.; Ortiz-Gonzalez, M.; Soriano-Lerma, A.; Linde-Rodriguez, A.; Perez-Carrasco, V.; Gutierrez-Fernandez, J.; Cuadros, M.; González, C.; Soriano, M.; et al. Targeting ribosomal G-quadruplexes with naphthalene-diimides as RNA polymerase I inhibitors for colorectal cancer treatment. Cell Chem. Biol. 2021, 28, 1590–1601.e4. [Google Scholar] [CrossRef] [PubMed]
- Pickard, A.J.; Bierbach, U. The Cell’s Nucleolus: An Emerging Target for Chemotherapeutic Intervention. ChemMedChem 2013, 8, 1441–1449. [Google Scholar] [CrossRef] [Green Version]
- Pich, A.; Chiusa, L.; Margaria, E. Prognostic relevance of AgNORs in tumor pathology. Micron 2000, 31, 133–141. [Google Scholar] [CrossRef]
- Guner, G.; Sirajuddin, P.; Zheng, Q.; Bai, B.; Brodie, A.; Liu, H.; Af Hällström, T.; Kulac, I.; Laiho, M.; De Marzo, A.M. Novel Assay to Detect RNA Polymerase I Activity In Vivo. Mol. Cancer Res. 2017, 15, 577–584. [Google Scholar] [CrossRef] [Green Version]
- Son, J.; Hannan, K.M.; Poortinga, G.; Hein, N.; Cameron, D.P.; Ganley, A.R.D.; Sheppard, K.E.; Pearson, R.B.; Hannan, R.D.; Sanij, E. rDNA Chromatin Activity Status as a Biomarker of Sensitivity to the RNA Polymerase I Transcription Inhibitor CX-5461. Front. Cell Dev. Biol. 2020, 8, 568. [Google Scholar] [CrossRef]
- Scala, F.; Brighenti, E.; Govoni, M.; Imbrogno, E.; Fornari, F.; Treré, D.; Montanaro, L.; Derenzini, M. Direct relationship between the level of p53 stabilization induced by rRNA synthesis-inhibiting drugs and the cell ribosome biogenesis rate. Oncogene 2016, 35, 977–989. [Google Scholar] [CrossRef]
- Derenzini, M.; Donati, G.; Mazzini, G.; Montanaro, L.; Vici, M.; Ceccarelli, C.; Santini, D.; Taffurelli, M.; Treré, D. Loss of retinoblastoma tumor suppressor protein makes human breast cancer cells more sensitive to antimetabolite exposure. Clin. Cancer Res. 2008, 14, 2199–2209. [Google Scholar] [CrossRef] [Green Version]
- Montanaro, L.; Mazzini, G.; Barbieri, S.; Vici, M.; Nardi-Pantoli, A.; Govoni, M.; Donati, G.; Treré, D.; Derenzini, M. Different effects of ribosome biogenesis inhibition on cell proliferation in retinoblastoma protein- and p53-deficient and proficient human osteosarcoma cell lines. Cell Prolif. 2007, 40, 532–549. [Google Scholar] [CrossRef]
- Treré, D.; Brighenti, E.; Donati, G.; Ceccarelli, C.; Santini, D.; Taffurelli, M.; Montanaro, L.; Derenzini, M. High prevalence of retinoblastoma protein loss in triple-negative breast cancers and its association with a good prognosis in patients treated with adjuvant chemotherapy. Ann. Oncol. 2009, 20, 1818–1823. [Google Scholar] [CrossRef] [PubMed]
- Hald, Ø.H.; Olsen, L.; Gallo-Oller, G.; Elfman, L.H.M.; Løkke, C.; Kogner, P.; Sveinbjörnsson, B.; Flægstad, T.; Johnsen, J.I.; Einvik, C. Inhibitors of ribosome biogenesis repress the growth of MYCN-amplified neuroblastoma. Oncogene 2019, 38, 2800–2813. [Google Scholar] [CrossRef]
- Low, J.-Y.; Sirajuddin, P.; Moubarek, M.; Agarwal, S.; Rege, A.; Guner, G.; Liu, H.; Yang, Z.; De Marzo, A.M.; Bieberich, C.; et al. Effective targeting of RNA polymerase I in treatment-resistant prostate cancer. Prostate 2019, 79, 1837–1851. [Google Scholar] [CrossRef] [PubMed]
- Sanij, E.; Hannan, K.M.; Xuan, J.; Yan, S.; Ahern, J.E.; Trigos, A.S.; Brajanovski, N.; Son, J.; Chan, K.T.; Kondrashova, O.; et al. CX-5461 activates the DNA damage response and demonstrates therapeutic efficacy in high-grade serous ovarian cancer. Nat. Commun. 2020, 11, 2641. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Xuan, J.; Brajanovski, N.; Tancock, M.R.C.; Madhamshettiwar, P.B.; Simpson, K.J.; Ellis, S.; Kang, J.; Cullinane, C.; Sheppard, K.E.; et al. The RNA polymerase I transcription inhibitor CX-5461 cooperates with topoisomerase 1 inhibition by enhancing the DNA damage response in homologous recombination-proficient high-grade serous ovarian cancer. Br. J. Cancer 2021, 124, 616–627. [Google Scholar] [CrossRef]
- Makhale, A.; Nanayakkara, D.; Raninga, P.; Khanna, K.K.; Kalimutho, M. CX-5461 Enhances the Efficacy of APR-246 via Induction of DNA Damage and Replication Stress in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 5782. [Google Scholar] [CrossRef]
- Devlin, J.R.; Hannan, K.M.; Hein, N.; Cullinane, C.; Kusnadi, E.; Ng, P.Y.; George, A.J.; Shortt, J.; Bywater, M.J.; Poortinga, G.; et al. Combination Therapy Targeting Ribosome Biogenesis and mRNA Translation Synergistically Extends Survival in MYC-Driven Lymphoma. Cancer Discov. 2016, 6, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Rebello, R.J.; Kusnadi, E.; Cameron, D.P.; Pearson, H.B.; Lesmana, A.; Devlin, J.R.; Drygin, D.; Clark, A.K.; Porter, L.; Pedersen, J.; et al. The Dual Inhibition of RNA Pol I Transcription and PIM Kinase as a New Therapeutic Approach to Treat Advanced Prostate Cancer. Clin. Cancer Res. 2016, 22, 5539–5552. [Google Scholar] [CrossRef] [Green Version]
- Ismael, M.; Webb, R.; Ajaz, M.; Kirkby, K.J.; Coley, H.M. The Targeting of RNA Polymerase I Transcription Using CX-5461 in Combination with Radiation Enhances Tumour Cell Killing Effects in Human Solid Cancers. Cancers 2019, 11, 1429. [Google Scholar] [CrossRef] [Green Version]
- Bray, M.-A.; Gustafsdottir, S.M.; Rohban, M.H.; Singh, S.; Ljosa, V.; Sokolnicki, K.L.; Bittker, J.A.; Bodycombe, N.E.; Dancík, V.; Hasaka, T.P.; et al. A dataset of images and morphological profiles of 30 000 small-molecule treatments using the Cell Painting assay. GigaScience 2017, 6, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Bray, M.-A.; Singh, S.; Han, H.; Davis, C.T.; Borgeson, B.; Hartland, C.; Kost-Alimova, M.; Gustafsdottir, S.M.; Gibson, C.C.; Carpenter, A.E. Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat. Protoc. 2016, 11, 1757–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaker, B.; Ahmad, S.; Lee, J.; Jung, C.; Na, D. In silico methods and tools for drug discovery. Comput. Biol. Med. 2021, 137, 104851. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.J.; McCool, M.A.; Abriola, L.; Surovtseva, Y.V.; Baserga, S.J. A high-throughput assay for directly monitoring nucleolar rRNA biogenesis. Open Biol. 2022, 12, 210305. [Google Scholar] [CrossRef] [PubMed]
- Farley-Barnes, K.I.; McCann, K.L.; Ogawa, L.M.; Merkel, J.; Surovtseva, Y.V.; Baserga, S.J. Diverse Regulators of Human Ribosome Biogenesis Discovered by Changes in Nucleolar Number. Cell Rep. 2018, 22, 1923–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatopoulou, V.; Parisot, P.; De Vleeschouwer, C.; Lafontaine, D.L.J. Use of the iNo score to discriminate normal from altered nucleolar morphology, with applications in basic cell biology and potential in human disease diagnostics. Nat. Protoc. 2018, 13, 2387–2406. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, N.; Dai, L.; Nordlund, P. CETSA in integrated proteomics studies of cellular processes. Curr. Opin. Chem. Biol. 2020, 54, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Saei, A.A.; Beusch, C.M.; Chernobrovkin, A.; Sabatier, P.; Zhang, B.; Tokat, Ü.G.; Stergiou, E.; Gaetani, M.; Végvári, Á.; Zubarev, R.A. ProTargetMiner as a proteome signature library of anticancer molecules for functional discovery. Nat. Commun. 2019, 10, 5715. [Google Scholar] [CrossRef] [Green Version]
- Misiaszek, A.D.; Girbig, M.; Grötsch, H.; Baudin, F.; Murciano, B.; Lafita, A.; Müller, C.W. Cryo-EM structures of human RNA polymerase I. Nat. Struct. Mol. Biol. 2021, 28, 997–1008. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, W.; Chen, K.; Wu, Z.; Yang, H.; Xu, Y. Structure of the human RNA polymerase I elongation complex. Cell Discov. 2021, 7, 97. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zisi, A.; Bartek, J.; Lindström, M.S. Targeting Ribosome Biogenesis in Cancer: Lessons Learned and Way Forward. Cancers 2022, 14, 2126. https://doi.org/10.3390/cancers14092126
Zisi A, Bartek J, Lindström MS. Targeting Ribosome Biogenesis in Cancer: Lessons Learned and Way Forward. Cancers. 2022; 14(9):2126. https://doi.org/10.3390/cancers14092126
Chicago/Turabian StyleZisi, Asimina, Jiri Bartek, and Mikael S. Lindström. 2022. "Targeting Ribosome Biogenesis in Cancer: Lessons Learned and Way Forward" Cancers 14, no. 9: 2126. https://doi.org/10.3390/cancers14092126
APA StyleZisi, A., Bartek, J., & Lindström, M. S. (2022). Targeting Ribosome Biogenesis in Cancer: Lessons Learned and Way Forward. Cancers, 14(9), 2126. https://doi.org/10.3390/cancers14092126