
Divergent Roles of Mitochondria Dynamics in Pancreatic Ductal Adenocarcinoma

 , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Pancreatic Cancer and Mitochondria Alterations
1.1. Pancreatic Cancer
1.2. Pancreatic Cancer Cells Show Mitochondrial and Metabolic Defects
2. Mitochondria Dynamics
2.1. Mitochondrial Fusion
2.2. Mitochondrial Fission
2.3. Physiological Significance of Fusion/Fission Balance
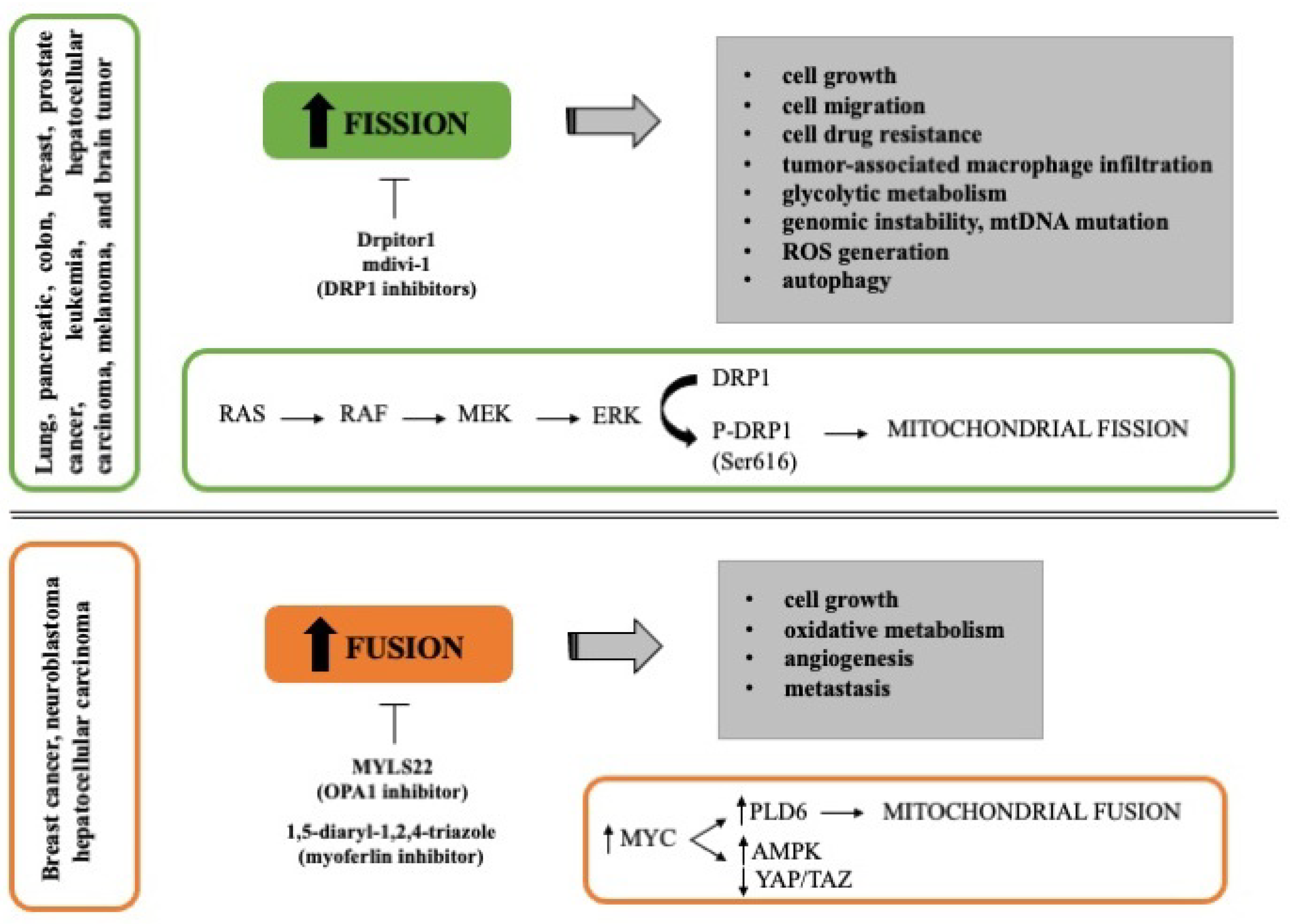
2.4. Mitochondria Dynamics and Cancer
3. Mitochondria Dynamics in Pancreatic Cancer
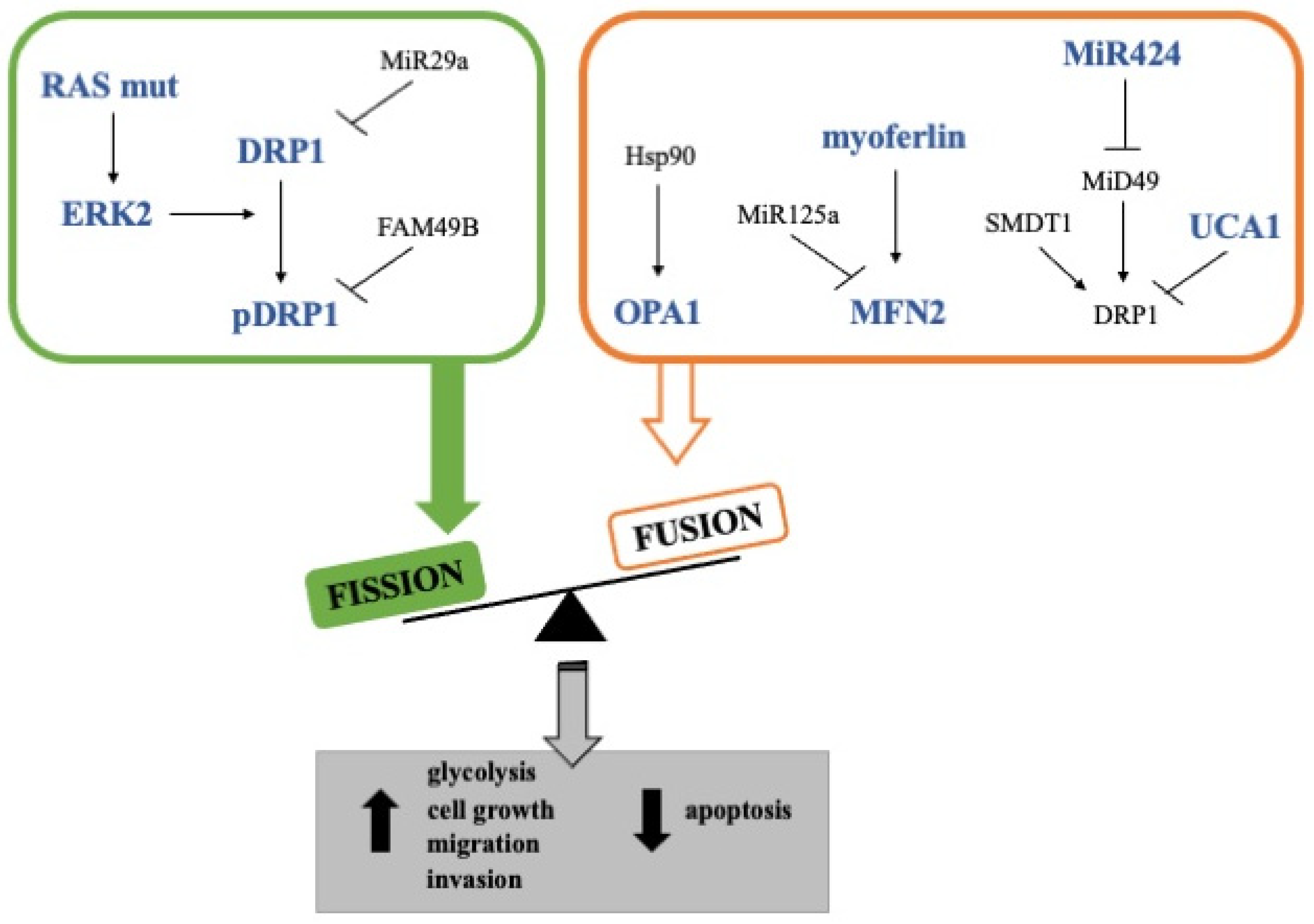
3.1. Mitochondrial Fusion in Pancreatic Cancer
3.2. Mitochondrial Fission in Pancreatic Cancer
4. Mitochondria Dynamics in Pancreatic Cancer Stem Cells
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic Cancer. Lancet 2020, 395, 73–85. [Google Scholar] [CrossRef]
- Klein, A.P. Pancreatic Cancer Epidemiology: Understanding the Role of Lifestyle and Inherited Risk Factors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 493–502. [Google Scholar] [CrossRef]
- Morani, A.C.; Hanafy, A.K.; Marcal, L.P.; Subbiah, V.; Le, O.; Bathala, T.K.; Elsayes, K.M. Imaging of Acute Abdomen in Cancer Patients. Abdom. Radiol. 2020, 45, 2287–2304. [Google Scholar] [CrossRef]
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic Ductal Adenocarcinoma: Treatment Hurdles, Tumor Microenvironment and Immunotherapy. World J. Gastrointest. Oncol. 2020, 12, 173–181. [Google Scholar] [CrossRef]
- Lambert, A.; Schwarz, L.; Borbath, I.; Henry, A.; van Laethem, J.L.; Malka, D.; Ducreux, M.; Conroy, T. An Update on Treatment Options for Pancreatic Adenocarcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1–43. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.; Yang, G.; Yang, J.; Ren, B.; Wang, H.; Chen, G.; Zhao, F.; You, L.; Wang, W.; Zhao, Y. Metabolism of Pancreatic Cancer: Paving the Way to Better Anticancer Strategies. Mol. Cancer 2020, 19, 50. [Google Scholar] [CrossRef] [Green Version]
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer Metabolism and Mitochondria: Finding Novel Mechanisms to Fight Tumours. EBioMedicine 2020, 59, 102943. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Reyes-Castellanos, G.; Masoud, R.; Carrier, A. Mitochondrial Metabolism in PDAC: From Better Knowledge to New Targeting Strategies. Biomedicines 2020, 8, 270. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.S.; Kroemer, G.; Galluzzi, L. Mitochondrial Metabolism and Cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Pascale, R.M.; Calvisi, D.F.; Simile, M.M.; Feo, C.F.; Feo, F. The Warburg Effect 97 Years after Its Discovery. Cancers 2020, 12, 2819. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 212–218. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Enríquez, S.; Hernández-Esquivel, L.; Marín-Hernández, A.; el Hafidi, M.; Gallardo-Pérez, J.C.; Hernández-Reséndiz, I.; Rodríguez-Zavala, J.S.; Pacheco-Velázquez, S.C.; Moreno-Sánchez, R. Mitochondrial Free Fatty Acid β-Oxidation Supports Oxidative Phosphorylation and Proliferation in Cancer Cells. Int. J. Biochem. Cell Biol. 2015, 65, 209–221. [Google Scholar] [CrossRef]
- Salem, A.F.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; Lisanti, M.P. Mitochondrial Biogenesis in Epithelial Cancer Cells Promotes Breast Cancer Tumor Growth and Confers Autophagy Resistance. Cell Cycle 2012, 11, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Ralph, S.J.; Rodríguez-Enríquez, S.; Neuzil, J.; Moreno-Sánchez, R. Bioenergetic Pathways in Tumor Mitochondria as Targets for Cancer Therapy and the Importance of the ROS-Induced Apoptotic Trigger. Mol. Asp. Med. 2010, 31, 29–59. [Google Scholar] [CrossRef] [Green Version]
- Yuneva, M.O.; Fan, T.W.M.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The Metabolic Profile of Tumors Depends on Both the Responsible Genetic Lesion and Tissue Type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.N.; Li, Z.; Danai, L.V.; Westermark, A.M.; Darnell, A.M.; Ferreira, R.; Gocheva, V.; Sivanand, S.; Lien, E.C.; Sapp, K.M.; et al. Dissecting Cell-Type-Specific Metabolism in Pancreatic Ductal Adenocarcinoma. eLife 2020, 9, e56782. [Google Scholar] [CrossRef]
- Chaika, N.V.; Yu, F.; Purohit, V.; Mehla, K.; Lazenby, A.J.; DiMaio, D.; Anderson, J.M.; Yeh, J.J.; Johnson, K.R.; Hollingsworth, M.A.; et al. Differential Expression of Metabolic Genes in Tumor and Stromal Components of Primary and Metastatic Loci in Pancreatic Adenocarcinoma. PLoS ONE 2012, 7, e32996. [Google Scholar] [CrossRef] [Green Version]
- Kerk, S.A.; Papagiannakopoulos, T.; Shah, Y.M.; Lyssiotis, C.A. Metabolic Networks in Mutant KRAS-Driven Tumours: Tissue Specificities and the Microenvironment. Nat. Rev. Cancer 2021, 21, 510–525. [Google Scholar] [CrossRef]
- Humpton, T.J.; Alagesan, B.; Denicola, G.M.; Lu, D.; Yordanov, G.N.; Leonhardt, C.S.; Yao, M.A.; Alagesan, P.; Zaatari, M.N.; Park, Y.; et al. Oncogenic KRAS Induces NIX-Mediated Mitophagy to Promote Pancreatic Cancer. Cancer Discov. 2019, 9, 1268–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During Autophagy Mitochondria Elongate, Are Spared from Degradation and Sustain Cell Viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Ying, J.; Wang, X.; Zhao, T.; Yoon, S.; Fang, Y.; Zheng, Q.; Liu, X.; Yu, W.; Hua, F. Mitochondrial Dynamics: A Key Role in Neurodegeneration and a Potential Target for Neurodegenerative Disease. Front. Neurosci. 2021, 15, 359. [Google Scholar] [CrossRef]
- Maycotte, P.; Marín-Hernández, A.; Goyri-Aguirre, M.; Anaya-Ruiz, M.; Reyes-Leyva, J.; Cortés-Hernández, P. Mitochondrial Dynamics and Cancer. Tumor Biol. 2017, 39, 1010428317698391. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Nunnari, J. Mitochondrial Form and Function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The Cell Biology of Mitochondrial Membrane Dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 Coordinately Regulate Mitochondrial Fusion and Are Essential for Embryonic Development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Liesa, M.; Bord-d’Água, B.; Medina-Gómez, G.; Lelliott, C.J.; Paz, J.C.; Rojo, M.; Palacín, M.; Vidal-Puig, A.; Zorzano, A. Mitochondrial Fusion Is Increased by the Nuclear Coactivator PGC-1β. PLoS ONE 2008, 3, e3613. [Google Scholar] [CrossRef] [Green Version]
- Sebastián, D.; Hernández-Alvarez, M.I.; Segalés, J.; Sorianello, E.; Muñoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) Links Mitochondrial and Endoplasmic Reticulum Function with Insulin Signaling and Is Essential for Normal Glucose Homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef] [Green Version]
- Segalés, J.; Paz, J.C.; Hernández-Alvarez, M.I.; Sala, D.; Muñoz, J.P.; Noguera, E.; Pich, S.; Palacín, M.; Enríquez, J.A.; Zorzano, A. A Form of Mitofusin 2 (Mfn2) Lacking the Transmembrane Domains and the COOH-Terminal End Stimulates Metabolism in Muscle and Liver Cells. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1208–E1221. [Google Scholar] [CrossRef] [Green Version]
- Xue, R.; Yang, J.; Jia, L.; Zhu, X.; Wu, J.; Zhu, Y.; Meng, Q. Mitofusin2, as a Protective Target in the Liver, Controls the Balance of Apoptosis and Autophagy in Acute-on-Chronic Liver Failure. Front. Pharmacol. 2019, 10, 601. [Google Scholar] [CrossRef] [Green Version]
- Kawalec, M.; Boratyńska-Jasińska, A.; Beresewicz, M.; Dymkowska, D.; Zabłocki, K.; Zabłocka, B. Mitofusin 2 Deficiency Affects Energy Metabolism and Mitochondrial Biogenesis in MEF Cells. PLoS ONE 2015, 10, e0134162. [Google Scholar] [CrossRef]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 Tethers Endoplasmic Reticulum to Mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- MacVicar, T.; Langer, T. OPA1 Processing in Cell Death and Disease—the Long and Short of It. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of Mitochondrial Morphology through Proteolytic Cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The I-AAA Protease YME1L and OMA1 Cleave OPA1 to Balance Mitochondrial Fusion and Fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef]
- Lee, H.; Smith, S.B.; Yoon, Y. The Short Variant of the Mitochondrial Dynamin OPA1 Maintains Mitochondrial Energetics and Cristae Structure. J. Biol. Chem. 2017, 292, 7115–7130. [Google Scholar] [CrossRef] [Green Version]
- del Dotto, V.; Fogazza, M.; Carelli, V.; Rugolo, M.; Zanna, C. Eight Human OPA1 Isoforms, Long and Short: What Are They For? Biochim. Biophys. Acta 2018, 1859, 263–269. [Google Scholar] [CrossRef]
- del Dotto, V.; Mishra, P.; Vidoni, S.; Fogazza, M.; Maresca, A.; Caporali, L.; McCaffery, J.M.; Cappelletti, M.; Baruffini, E.; Lenaers, G.; et al. OPA1 Isoforms in the Hierarchical Organization of Mitochondrial Functions. Cell Rep. 2017, 19, 2557–2571. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Shu, L.; Huang, X.; Yu, J.; Li, L.; Gong, L.; Yang, M.; Wu, Z.; Gao, Z.; Zhao, Y.; et al. OPA1 and MICOS Regulate Mitochondrial Crista Dynamics and Formation. Cell Death Dis. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 Phosphorylation of Drp1 Promotes Mitochondrial Fission and MAPK-Driven Tumor Growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.R.; Blackstone, C. Cyclic AMP-Dependent Protein Kinase Phosphorylation of Drp1 Regulates Its GTPase Activity and Mitochondrial Morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [Green Version]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff Is an Essential Factor for Mitochondrial Recruitment of Drp1 during Mitochondrial Fission in Mammalian Cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Yamano, K.; Head, B.P.; Kawajiri, S.; Cheung, J.T.M.; Wang, C.; Cho, J.H.; Hattori, N.; Youle, R.J.; van der Bliek, A.M. Mutations in Fis1 Disrupt Orderly Disposal of Defective Mitochondria. Mol. Biol. Cell 2014, 25, 145–159. [Google Scholar] [CrossRef] [Green Version]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 Mediate Drp1 Recruitment in Mitochondrial Fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Palmer, C.S.; Elgass, K.D.; Parton, R.G.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. Adaptor Proteins MiD49 and MiD51 Can Act Independently of Mff and Fis1 in Drp1 Recruitment and Are Specific for Mitochondrial Fission. J. Biol. Chem. 2013, 288, 27584–27593. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The Dynamin-Related GTPase Drp1 Is Required for Embryonic and Brain Development in Mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, T.; Ban-Ishihara, R.; Maeda, M.; Matsunaga, Y.; Ichimura, A.; Kyogoku, S.; Aoki, H.; Katada, S.; Nakada, K.; Nomura, M.; et al. Dynamics of Mitochondrial DNA Nucleoids Regulated by Mitochondrial Fission Is Essential for Maintenance of Homogeneously Active Mitochondria during Neonatal Heart Development. Mol. Cell. Biol. 2015, 35, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A Hyperfused Mitochondrial State Achieved at G1-S Regulates Cyclin E Buildup and Entry into S Phase. Proc. Natl. Acad. Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheridan, C.; Martin, S.J. Mitochondrial Fission/Fusion Dynamics and Apoptosis. Mitochondrion 2010, 10, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, P.; Liu, B.; Zhao, J.; Pang, Q.; Agrawal, S.G.; Jia, L.; Liu, F.T. Dynamin-Related Protein Drp1 Is Required for Bax Translocation to Mitochondria in Response to Irradiation-Induced Apoptosis. Oncotarget 2015, 6, 22598–22612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasiak, S.; Zunino, R.; McBride, H.M. Bax/Bak Promote Sumoylation of DRP1 and Its Stable Association with Mitochondria during Apoptotic Cell Death. J. Cell Biol. 2007, 177, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Arnoult, D.; Grodet, A.; Lee, Y.J.; Estaquier, J.; Blackstone, C. Release of OPA1 during Apoptosis Participates in the Rapid and Complete Release of Cytochrome c and Subsequent Mitochondrial Fragmentation. J. Biol. Chem. 2005, 280, 35742–35750. [Google Scholar] [CrossRef] [Green Version]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabò, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The Opa1-Dependent Mitochondrial Cristae Remodeling Pathway Controls Atrophic, Apoptotic, and Ischemic Tissue Damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef] [Green Version]
- Prieto, J.; León, M.; Ponsoda, X.; Sendra, R.; Bort, R.; Ferrer-Lorente, R.; Raya, A.; López-Garciá, C.; Torres, J. Early ERK1/2 Activation Promotes DRP1-Dependent Mitochondrial Fission Necessary for Cell Reprogramming. Nat. Commun. 2016, 7, 11124. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, J.P.; Ivanova, S.; Sánchez-Wandelmer, J.; Martínez-Cristóbal, P.; Noguera, E.; Sancho, A.; Díaz-Ramos, A.; Hernández-Alvarez, M.I.; Sebastián, D.; Mauvezin, C.; et al. Mfn2 Modulates the UPR and Mitochondrial Function via Repression of PERK. EMBO J. 2013, 32, 2348–2361. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Wang, L.; Jia, R. The Role of Mitochondrial Dynamics in Human Cancers. Am. J. Cancer Res. 2020, 10, 1278–1293. [Google Scholar]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.H. Specific Mitochondrial Functions in Separate Sub-Cellular Domains of Pancreatic Acinar Cells. Pflug. Arch. Eur. J. Physiol. 2012, 464, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Tinel, H.; Cancela, J.M.; Mogami, H.; Gerasimenko, J.V.; Gerasimenko, O.V.; Tepikin, A.V.; Petersen, O.H. Active Mitochondria Surrounding the Pancreatic Acinar Granule Region Prevent Spreading of Inositol Trisphosphate-Evoked Local Cytosolic Ca2+ Signals. EMBO J. 1999, 18, 4999–5008. [Google Scholar] [CrossRef]
- Yoon, Y.; Galloway, C.A.; Jhun, B.S.; Yu, T. Mitochondrial Dynamics in Diabetes. Antioxid. Redox Signal. 2011, 14, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Wiederkehr, A.; Kirkpatrick, C.; Mattenberger, Y.; Martinou, J.C.; Marchetti, P.; Demaurex, N.; Wollheim, C.B. Selective Actions of Mitochondrial Fission/Fusion Genes on Metabolism-Secretion Coupling in Insulin-Releasing Cells. J. Biol. Chem. 2008, 283, 33347–33356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Mitochondrial Dynamics and Metastasis. Cell. Mol. Life Sci. 2019, 76, 827–835. [Google Scholar] [CrossRef]
- von Eyss, B.; Jaenicke, L.A.; Kortlever, R.M.; Royla, N.; Wiese, K.E.; Letschert, S.; McDuffus, L.A.; Sauer, M.; Rosenwald, A.; Evan, G.I.; et al. A MYC-Driven Change in Mitochondrial Dynamics Limits YAP/TAZ Function in Mammary Epithelial Cells and Breast Cancer. Cancer Cell 2015, 28, 743–757. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Kim, B.; Cho, U.; Park, I.S.; Kim, S.I.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Mitochondrial Fission Causes Cisplatin Resistance under Hypoxic Conditions via ROS in Ovarian Cancer Cells. Oncogene 2019, 38, 7089–7105. [Google Scholar] [CrossRef]
- Bao, D.; Zhao, J.; Zhou, X.; Yang, Q.; Chen, Y.; Zhu, J.; Yuan, P.; Yang, J.; Qin, T.; Wan, S.; et al. Mitochondrial Fission-Induced MtDNA Stress Promotes Tumor-Associated Macrophage Infiltration and HCC Progression. Oncogene 2019, 38, 5007–5020. [Google Scholar] [CrossRef] [Green Version]
- Serasinghe, M.N.; Wieder, S.Y.; Renault, T.T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; Sesaki, H.; et al. Mitochondrial Division Is Requisite to RAS-Induced Transformation and Targeted by Oncogenic MAPK Pathway Inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial Dynamics Regulates Migration and Invasion of Breast Cancer Cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Wang, G.; Chwa, J.; Oh, M.E.; Abeywardana, T.; Yang, Y.; Wang, Q.A.; Jiang, L. Mitochondrial Division Inhibitor (Mdivi-1) Decreases Oxidative Metabolism in Cancer. Br. J. Cancer 2020, 122, 1288–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Dasgupta, A.; Chen, K.H.; Neuber-Hess, M.; Patel, J.; Hurst, T.E.; Mewburn, J.D.; Lima, P.D.A.; Alizadeh, E.; Martin, A.; et al. Identification of Novel Dynamin-Related Protein 1 (Drp1) GTPase Inhibitors: Therapeutic Potential of Drpitor1 and Drpitor1a in Cancer and Cardiac Ischemia-Reperfusion Injury. FASEB J. 2020, 34, 1447–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Nguyen, N.D.; Huang, Y.; Lin, D.; Fujimoto, T.N.; Molkentine, J.M.; Deorukhkar, A.; Kang, Y.; Anthony San Lucas, F.; Fernandes, C.J.; et al. Mitochondrial Fusion Exploits a Therapeutic Vulnerability of Pancreatic Cancer. JCI Insight 2019, 4, e126915. [Google Scholar] [CrossRef]
- Kong, B.; Wang, Q.; Fung, E.; Xue, K.; Tsang, B.K. P53 Is Required for Cisplatin-Induced Processing of the Mitochondrial Fusion Protein L-Opa1 That Is Mediated by the Mitochondrial Metallopeptidase Oma1 in Gynecologic Cancers. J. Biol. Chem. 2014, 289, 27134–27145. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Wang, L.; Wang, Y.; Zhang, S.; Zhou, G.; Lieshout, R.; Ma, B.; Liu, J.; Qu, C.; Verstegen, M.M.A.; et al. Mitochondrial Fusion Via OPA1 and MFN1 Supports Liver Tumor Cell Metabolism and Growth. Cells 2020, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.H.; Wang, R.; Wang, Y.; Kung, C.P.; Weber, J.D.; Patti, G.J. Mitochondrial Fusion Supports Increased Oxidative Phosphorylation during Cell Proliferation. eLife 2019, 8, 27134–27145. [Google Scholar] [CrossRef]
- Herkenne, S.; Ek, O.; Zamberlan, M.; Pellattiero, A.; Chergova, M.; Chivite, I.; Novotná, E.; Rigoni, G.; Fonseca, T.B.; Samardzic, D.; et al. Developmental and Tumor Angiogenesis Requires the Mitochondria-Shaping Protein Opa1. Cell Metab. 2020, 31, 987–1003.e8. [Google Scholar] [CrossRef]
- Casinelli, G.; LaRosa, J.; Sharma, M.; Cherok, E.; Banerjee, S.; Branca, M.; Edmunds, L.; Wang, Y.; Sims-Lucas, S.; Churley, L.; et al. N-Myc Overexpression Increases Cisplatin Resistance in Neuroblastoma via Deregulation of Mitochondrial Dynamics. Cell Death Discov. 2016, 2, 16082. [Google Scholar] [CrossRef] [Green Version]
- Rademaker, G.; Hennequière, V.; Brohée, L.; Nokin, M.J.; Lovinfosse, P.; Durieux, F.; Gofflot, S.; Bellier, J.; Costanza, B.; Herfs, M.; et al. Myoferlin Controls Mitochondrial Structure and Activity in Pancreatic Ductal Adenocarcinoma, and Affects Tumor Aggressiveness. Oncogene 2018, 37, 4398–4412. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Zhou, L.; Yin, W.; Bai, J.; Liu, R. MiR-125a Induces Apoptosis, Metabolism Disorder and Migration Impairment in Pancreatic Cancer Cells by Targeting Mfn2-Related Mitochondrial Fission. Int. J. Oncol. 2018, 53, 124–136. [Google Scholar] [CrossRef]
- Teng, B.W.; Feng, T.; Li, W.; Wang, Z. Abnormal Expression of LncRNA UCA1 Disturbed Cell Apoptosis through Mediating Mitochondrial Dynamics in PDAC. Neoplasma 2021, 68, 334–341. [Google Scholar] [CrossRef]
- Herkenne, S.; Scorrano, L. OPA1, a New Mitochondrial Target in Cancer Therapy. Aging 2020, 12, 20931–20933. [Google Scholar] [CrossRef]
- Liang, J.; Yang, Y.; Bai, L.; Li, F.; Li, E. DRP1 Upregulation Promotes Pancreatic Cancer Growth and Metastasis through Increased Aerobic Glycolysis. J. Gastroenterol. Hepatol. 2020, 35, 885–895. [Google Scholar] [CrossRef]
- Nagdas, S.; Kashatus, J.A.; Nascimento, A.; Hussain, S.S.; Trainor, R.E.; Pollock, S.R.; Adair, S.J.; Michaels, A.D.; Sesaki, H.; Stelow, E.B.; et al. Drp1 Promotes KRas-Driven Metabolic Changes to Drive Pancreatic Tumor Growth. Cell Rep. 2019, 28, 1845–1859.e5. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Liang, J.; Li, L.; Li, E. Downregulation of MiD49 Contributes to Tumor Growth and Metastasis of Human Pancreatic Cancer. Oncol. Rep. 2020, 43, 1208–1220. [Google Scholar] [CrossRef] [Green Version]
- Xie, K.F.; Guo, D.D.; Luo, X.J. SMDT1-Driven Change in Mitochondrial Dynamics Mediate Cell Apoptosis in PDAC. Biochem. Biophys. Res. Commun. 2019, 511, 323–329. [Google Scholar] [CrossRef]
- Lin, Z.; Lin, X.; Chen, J.; Huang, G.; Chen, T.; Zheng, L. Mitofusin-2 Is a Novel Anti-Angiogenic Factor in Pancreatic Cancer. J. Gastrointest. Oncol. 2021, 12, 484–495. [Google Scholar] [CrossRef]
- Xue, R.; Meng, Q.; Lu, D.; Liu, X.; Wang, Y.; Hao, J. Mitofusin2 Induces Cell Autophagy of Pancreatic Cancer through Inhibiting the PI3K/Akt/MTOR Signaling Pathway. Oxidative Med. Cell. Longev. 2018, 2018, 2798070. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Zhang, Z.; Wakabayashi, N.; Wakabayashi, J.; Tamura, Y.; Song, W.J.; Sereda, S.; Clerc, P.; Polster, B.M.; Aja, S.M.; Pletnikov, M.V.; et al. The Dynamin-Related GTPase Opa1 Is Required for Glucose-Stimulated ATP Production in Pancreatic Beta Cells. Mol. Biol. Cell 2011, 22, 2235–2245. [Google Scholar] [CrossRef]
- Li, J.; Huang, Q.; Long, X.; Guo, X.; Sun, X.; Jin, X.; Li, Z.; Ren, T.; Yuan, P.; Huang, X.; et al. Mitochondrial Elongation-Mediated Glucose Metabolism Reprogramming Is Essential for Tumour Cell Survival during Energy Stress. Oncogene 2017, 36, 4901–4912. [Google Scholar] [CrossRef]
- Chattaragada, M.S.; Riganti, C.; Sassoe, M.; Principe, M.; Santamorena, M.M.; Roux, C.; Curcio, C.; Evangelista, A.; Allavena, P.; Salvia, R.; et al. FAM49B, a Novel Regulator of Mitochondrial Function and Integrity That Suppresses Tumor Metastasis. Oncogene 2018, 37, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Wee, Y.; Liu, Y.; Lu, J.; Li, X.; Zhao, M. Identification of Novel Prognosis-Related Genes Associated with Cancer Using Integrative Network Analysis. Sci. Rep. 2018, 8, 3233. [Google Scholar] [CrossRef]
- Hu, L.P.; Zhou, K.X.; Huo, Y.M.; Liu, D.J.; Li, Q.; Yang, M.W.; Huang, P.Q.; Xu, C.J.; Tian, G.A.; Yao, L.L.; et al. Single-Cell RNA Sequencing Reveals That Targeting HSP90 Suppresses PDAC Progression by Restraining Mitochondrial Bioenergetics. Oncogenesis 2021, 10, 22. [Google Scholar] [CrossRef]
- Ohba, Y.; MacVicar, T.; Langer, T. Regulation of Mitochondrial Plasticity by the I-AAA Protease YME1L. Biol. Chem. 2020, 401, 877–890. [Google Scholar] [CrossRef]
- MacVicar, T.; Ohba, Y.; Nolte, H.; Mayer, F.C.; Tatsuta, T.; Sprenger, H.G.; Lindner, B.; Zhao, Y.; Li, J.; Bruns, C.; et al. Lipid Signalling Drives Proteolytic Rewiring of Mitochondria by YME1L. Nature 2019, 575, 361–365. [Google Scholar] [CrossRef]
- Rademaker, G.; Costanza, B.; Anania, S.; Agirman, F.; Maloujahmoum, N.; di Valentin, E.; Goval, J.J.; Bellahcène, A.; Castronovo, V.; Peulen, O. Myoferlin Contributes to the Metastatic Phenotype of Pancreatic Cancer Cells by Enhancing Their Migratory Capacity through the Control of Oxidative Phosphorylation. Cancers 2019, 11, 853. [Google Scholar] [CrossRef] [Green Version]
- Anania, S.; Peiffer, R.; Rademaker, G.; Hego, A.; Thiry, M.; Deldicque, L.; Francaux, M.; Maloujahmoum, N.; Agirman, F.; Bellahcène, A.; et al. Myoferlin Is a yet Unknown Interactor of the Mitochondrial Dynamics’ Machinery in Pancreas Cancer Cells. Cancers 2020, 12, 1643. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.; Shao, T.; Pei, H.; Guo, W.; Mi, D.; Krimm, I.; Zhang, Y.; Wang, P.; Wang, X.; et al. Modification and Biological Evaluation of a Series of 1,5-Diaryl-1,2,4-Triazole Compounds as Novel Agents against Pancreatic Cancer Metastasis through Targeting Myoferlin. J. Med. Chem. 2019, 62, 4949–4966. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Sudo, H.; Kawauchi, J.; Takizawa, S.; Kondou, S.; Nobumasa, H.; Ochiai, A. MicroRNA Markers for the Diagnosis of Pancreatic and Biliary-Tract Cancers. PLoS ONE 2015, 10, e0118220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, B.; Ahmad, A.; Azmi, A.S.; Ali, S.; Sarkar, F.H. Overview of Cancer Stem Cells (CSCS) and Mechanisms of Their Regulation: Implications for Cancer Therapy. Curr. Protoc. Pharmacol. 2013, 61, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Ambrosini, G.; Dalla Pozza, E.; Fanelli, G.; di Carlo, C.; Vettori, A.; Cannino, G.; Cavallini, C.; Carmona-Carmona, C.A.; Brandi, J.; Rinalducci, S.; et al. Progressively De-Differentiated Pancreatic Cancer Cells Shift from Glycolysis to Oxidative Metabolism and Gain a Quiescent Stem State. Cells 2020, 9, 1572. [Google Scholar] [CrossRef]
- Katajisto, P.; Döhla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.; Weinberg, R.A.; Sabatini, D.M. Asymmetric Apportioning of Aged Mitochondria between Daughter Cells Is Required for Stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef] [Green Version]
- Jagust, P.; Alcalá, S.; Jr, B.S.; Heeschen, C.; Sancho, P. Glutathione Metabolism Is Essential for Self-Renewal and Chemoresistance of Pancreatic Cancer Stem Cells. World J. Stem Cells 2020, 12, 1410–1428. [Google Scholar] [CrossRef]
- Courtois, S.; de Luxán-Delgado, B.; Penin-Peyta, L.; Royo-García, A.; Parejo-Alonso, B.; Jagust, P.; Alcalá, S.; Rubiolo, J.A.; Sánchez, L.; Sainz, B.; et al. Inhibition of Mitochondrial Dynamics Preferentially Targets Pancreatic Cancer Cells with Enhanced Tumorigenic and Invasive Potential. Cancers 2021, 13, 698. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin Targets the Metabolic Achilles Heel of Human Pancreatic Cancer Stem Cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Ricciardiello, F.; Yang, G.; Qiu, J.; Huang, H.; Xiao, J.; Cao, Z.; Zhao, F.; Liu, Y.; Luo, W.; et al. The Role of Mitochondria in the Chemoresistance of Pancreatic Cancer Cells. Cells 2021, 10, 497. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Mitochondrial Fusion | |||||
| Tumor Type | Players | Functional Effect | Method | Results | Reference |
| PDAC | OPA1 | Pro-tumor | Inhibition of the interaction between Hsp90 and OPA1 (loss of myoferlin) | Reduction in mitochondrial cristae amount, energy production, cell proliferation, and induction of autophagy | Rademaker et al., 2018 [81] |
| PDAC | MFN2 | Anti-tumor | Overexpression of MFN2 | Improvement of survival in preclinical models, by the promotion of autophagy and the reduction in mitochondrial mass, OCR, and ATP production | Yu et al., 2019 [75] |
| PDAC | fusion | Anti-tumor | Pharmacological induction of mitochondrial fusion by leflunomide | Improvement of survival in mouse models | Yu et al., 2019 [75] |
| PDAC | MFN2 | Anti-tumor | Downregulation of MFN2 by miR125-a | Increased fission as a tumor suppressor process | Pan et al., 2018 [82] |
| PDAC | UCA1 | Pro-tumor | UCA1 knockdown | Decreased cell viability and induced apoptosis and mitochondria fragmentation | Teng et al., 2021 [83] |
| PDAC | Myoferlin | Pro-tumor | Decreased levels of Myoferlin | Reduced cell proliferation and induced aoutophagy | Rademaker et al., 2018 [81] |
| Liver cancer | OPA1 - MFN1 | Pro-tumor | Knockdown of OPA1 or MFN1 | Inhibition of the tumor formation in vivo in mice | Li et al., 2020 [77] |
| Several cancer types | OPA1 | Pro-tumor | Deletion of endothelial OPA1 | Decrease of tumor angiogenesis, growth, and metastasis | Herkenne et al., 2020 [79] |
| Several cancer types | OPA1 | Pro-tumor | Inhibition of OPA1 by MYLS22 | Decrease of tumor angiogenesis, growth, and metastasis | Herkenne and Scorrano, 2020 [84] |
| Several cancer types | MFN2 | Pro-tumor | Deletion of MFN2 | Reduction of cell proliferation | Yao et al., 2019 [78] |
| Mitochondrial Fission | |||||
| Tumor Type | Players | Functional Effect | Method | Results | Reference |
| Pancreatic cancer/PDAC | DRP1 | Pro-tumor | DRP1 knockdown | Inhibition of fragmented mitochondria phenotype, and tumor cell growth in vitro and in mouse xenograft | Liang et al., 2020 [85] |
| Pancreatic cancer/PDAC | DRP1 | Pro-tumor | Inhibition of DRP1 by synthetic miR-29a | Reduction of cell growth in vitro | Liang et al., 2020 [85] |
| PDAC | DRP1 | Pro-tumor | DRP1 knockdown | Decrease in Hexokinase II expression and glycolytic flux | Nagdas et al., 2019 [86] |
| PDAC | MiD49 | Anti-tumor | Overexpression of MiD49 | Suppression of PDAC growth and metastasis both in vitro and in vivo | Bai et al., 2020 [87] |
| PDAC | SMDT1 | Anti-tumor | SMDT1 overexpression | Decrease of proliferation rates of PDAC cell lines | Xie et al., 2019 [88] |
| Breast cancer | DRP1 | Pro-tumor | Silencing of DRP1 | Reduction of cell migration and invasion | Zhao et al., 2013 [72] |
| Breast and lung cancer | DRP1 | Pro-tumor | Inhibition of DRP1 by Drpitor1 | Damage of oxidative metabolism and induction of cell death | Wu et al., 2020 [74] |
| Several cancer types | DRP1 | Pro-tumor | Inhibition of DRP1 by mdivi-1 | Damage of oxidative metabolism and induction of cell death | Dai et al., 2020 [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carmona-Carmona, C.A.; Dalla Pozza, E.; Ambrosini, G.; Errico, A.; Dando, I. Divergent Roles of Mitochondria Dynamics in Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 2155. https://doi.org/10.3390/cancers14092155
Carmona-Carmona CA, Dalla Pozza E, Ambrosini G, Errico A, Dando I. Divergent Roles of Mitochondria Dynamics in Pancreatic Ductal Adenocarcinoma. Cancers. 2022; 14(9):2155. https://doi.org/10.3390/cancers14092155
Chicago/Turabian StyleCarmona-Carmona, Cristian Andres, Elisa Dalla Pozza, Giulia Ambrosini, Andrea Errico, and Ilaria Dando. 2022. "Divergent Roles of Mitochondria Dynamics in Pancreatic Ductal Adenocarcinoma" Cancers 14, no. 9: 2155. https://doi.org/10.3390/cancers14092155
APA StyleCarmona-Carmona, C. A., Dalla Pozza, E., Ambrosini, G., Errico, A., & Dando, I. (2022). Divergent Roles of Mitochondria Dynamics in Pancreatic Ductal Adenocarcinoma. Cancers, 14(9), 2155. https://doi.org/10.3390/cancers14092155