
Upregulation of the Mevalonate Pathway through EWSR1-FLI1/EGR2 Regulatory Axis Confers Ewing Cells Exquisite Sensitivity to Statins

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, EWSR1-FLI1, and EGR2 Invalidation
2.2. Gene Expression Profile
2.3. Chemical Compounds
2.4. High-Throughput Drug Screening and Hit Calling
2.5. Western Blot Analysis
2.6. ChIP-Seq
2.7. Ewing PDX Tumor-Dissociated Cells
2.8. Cell Proliferation in 2D and 3D Cultures (Spheroid Assay) and IC50 Determination
2.9. Cell-Cycle Analysis and Annexin V Staining
2.10. Rescue Experiments on Cell Proliferation
2.11. Intracellular Cell ROS and Lipid Peroxidation Measurement
2.12. Prenylated Protein Extraction
2.13. Xenograft of Ewing PDX Models
2.14. Statistics
3. Results
3.1. EWSR1-FLI1 and EGR2 Regulate Lipid/Cholesterol/Mevalonate Pathway
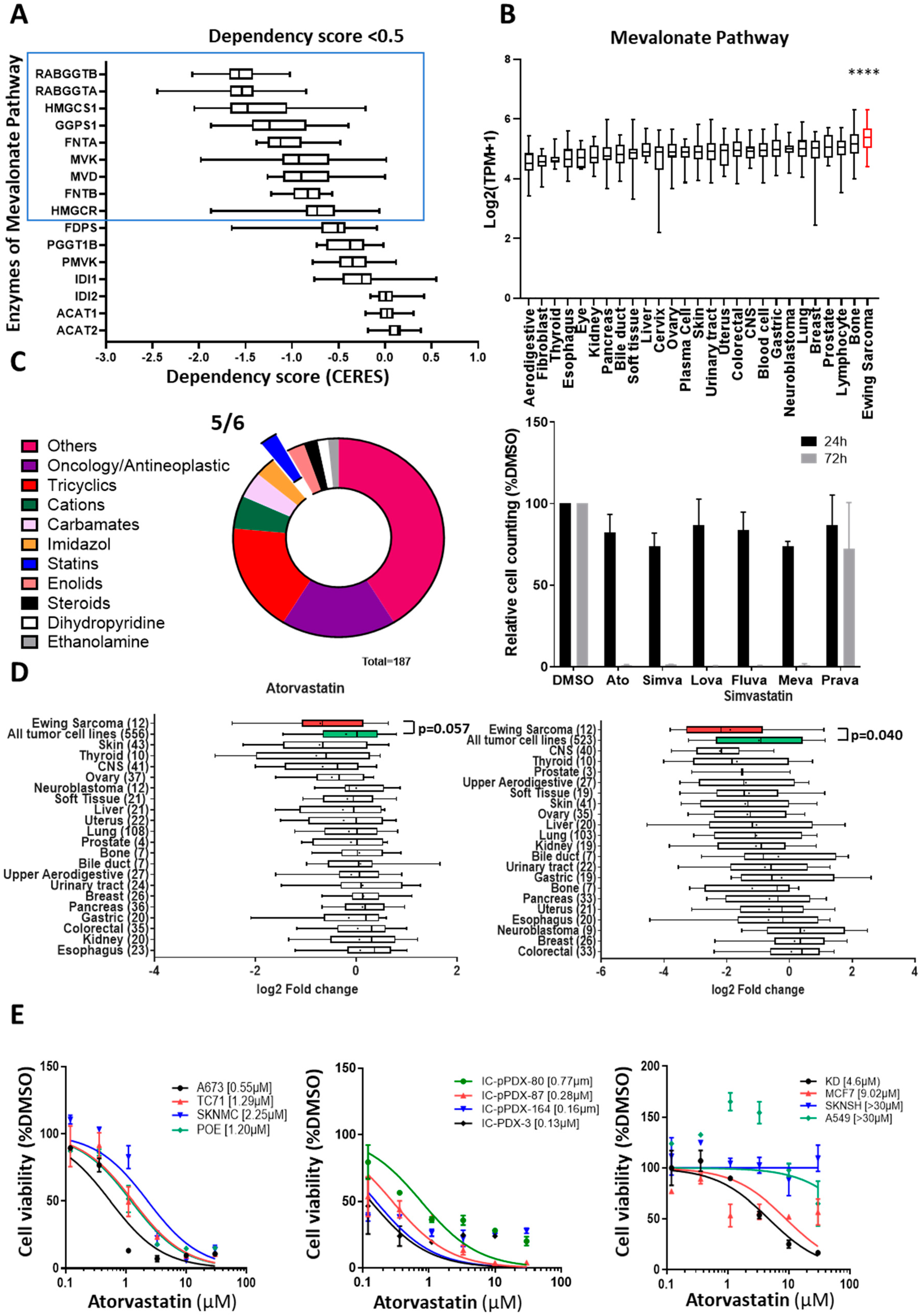
3.2. Ewing Sarcoma Cells Are Dependent on the Mevalonate Pathway
3.3. Atorvastatin Impacts Cell Proliferation of Ewing Cell Lines through Altered Anchorage on the Membrane of Small G-Proteins and Increased Intra-Cellular ROS
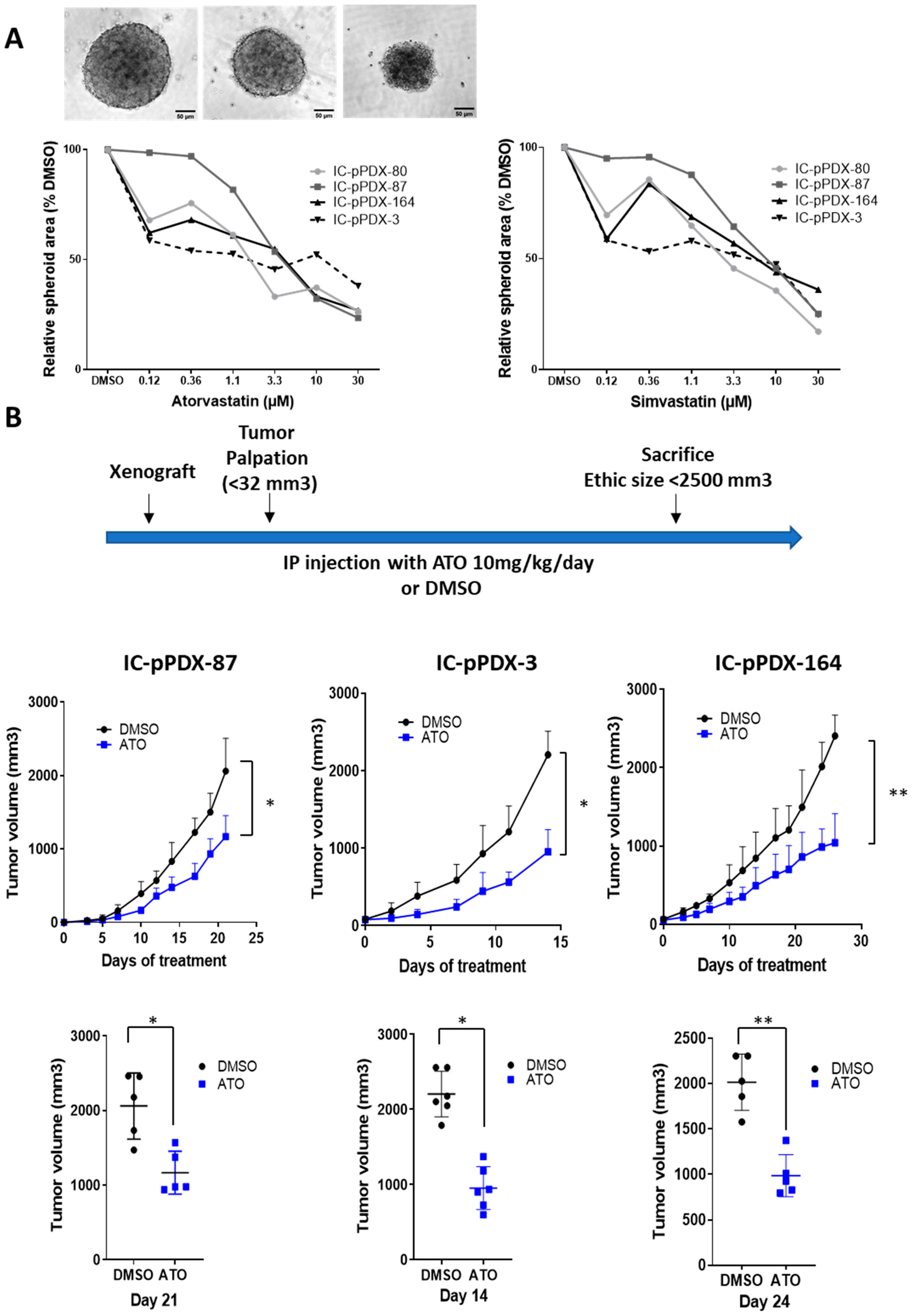
3.4. Atorvastatin Induces a Reduction in Ewing Tumor Growth In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.-C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3036–3046. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Álava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing Sarcoma. Nat. Rev. Dis. Primer 2018, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G. Gene Fusion with an ETS DNA-Binding Domain Caused by Chromosome Translocation in Human Tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Boulay, G.; Volorio, A.; Iyer, S.; Broye, L.C.; Stamenkovic, I.; Riggi, N.; Rivera, M.N. Epigenome Editing of Microsatellite Repeats Defines Tumor-Specific Enhancer Functions and Dependencies. Genes Dev. 2018, 32, 1008–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangwal, K.; Sankar, S.; Hollenhorst, P.C.; Kinsey, M.; Haroldsen, S.C.; Shah, A.A.; Boucher, K.M.; Watkins, W.S.; Jorde, L.B.; Graves, B.J.; et al. Microsatellites as EWS/FLI Response Elements in Ewing’s Sarcoma. Proc. Natl. Acad. Sci. USA 2008, 105, 10149–10154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillon, N.; Tirode, F.; Boeva, V.; Zynovyev, A.; Barillot, E.; Delattre, O. The Oncogenic EWS-FLI1 Protein Binds in Vivo GGAA Microsatellite Sequences with Potential Transcriptional Activation Function. PLoS ONE 2009, 4, e4932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheffield, N.C.; Pierron, G.; Klughammer, J.; Datlinger, P.; Schönegger, A.; Schuster, M.; Hadler, J.; Surdez, D.; Guillemot, D.; Lapouble, E.; et al. DNA Methylation Heterogeneity Defines a Disease Spectrum in Ewing Sarcoma. Nat. Med. 2017, 23, 386–395. [Google Scholar] [CrossRef]
- Tomazou, E.M.; Sheffield, N.C.; Schmidl, C.; Schuster, M.; Schönegger, A.; Datlinger, P.; Kubicek, S.; Bock, C.; Kovar, H. Epigenome Mapping Reveals Distinct Modes of Gene Regulation and Widespread Enhancer Reprogramming by the Oncogenic Fusion Protein EWS-FLI1. Cell Rep. 2015, 10, 1082–1095. [Google Scholar] [CrossRef] [Green Version]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The Genomic Landscape of Pediatric Ewing Sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [Green Version]
- Tirode, F.; Surdez, D.; Ma, X.; Parker, M.; Le Deley, M.C.; Bahrami, A.; Zhang, Z.; Lapouble, E.; Grossetête-Lalami, S.; Rusch, M.; et al. Genomic Landscape of Ewing Sarcoma Defines an Aggressive Subtype with Co-Association of STAG2 and TP53 Mutations. Cancer Discov. 2014, 4, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Machiela, M.J.; Grünewald, T.G.P.; Surdez, D.; Reynaud, S.; Mirabeau, O.; Karlins, E.; Rubio, R.A.; Zaidi, S.; Grossetete-Lalami, S.; Ballet, S.; et al. Genome-Wide Association Study Identifies Multiple New Loci Associated with Ewing Sarcoma Susceptibility. Nat. Commun. 2018, 9, 3184. [Google Scholar] [CrossRef] [PubMed]
- Postel-Vinay, S.; Véron, A.S.; Tirode, F.; Pierron, G.; Reynaud, S.; Kovar, H.; Oberlin, O.; Lapouble, E.; Ballet, S.; Lucchesi, C.; et al. Common Variants near TARDBP and EGR2 Are Associated with Susceptibility to Ewing Sarcoma. Nat. Genet. 2012, 44, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, T.G.P.; Bernard, V.; Gilardi-Hebenstreit, P.; Raynal, V.; Surdez, D.; Aynaud, M.-M.; Mirabeau, O.; Cidre-Aranaz, F.; Tirode, F.; Zaidi, S.; et al. Chimeric EWSR1-FLI1 Regulates the Ewing Sarcoma Susceptibility Gene EGR2 via a GGAA Microsatellite. Nat. Genet. 2015, 47, 1073–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donovan, K.J.; Tourtellotte, W.G.; Millbrandt, J.; Baraban, J.M. The EGR Family of Transcription-Regulatory Factors: Progress at the Interface of Molecular and Systems Neuroscience. Trends Neurosci. 1999, 22, 167–173. [Google Scholar] [CrossRef]
- Nagarajan, R.; Svaren, J.; Le, N.; Araki, T.; Watson, M.; Milbrandt, J. EGR2 Mutations in Inherited Neuropathies Dominant-Negatively Inhibit Myelin Gene Expression. Neuron 2001, 30, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Topilko, P.; Schneider-Maunoury, S.; Levi, G.; Baron-Van Evercooren, A.; Chennoufi, A.B.; Seitanidou, T.; Babinet, C.; Charnay, P. Krox-20 Controls Myelination in the Peripheral Nervous System. Nature 1994, 371, 796–799. [Google Scholar] [CrossRef]
- Leblanc, S.E.; Srinivasan, R.; Ferri, C.; Mager, G.M.; Gillian-Daniel, A.L.; Wrabetz, L.; Svaren, J. Regulation of Cholesterol/Lipid Biosynthetic Genes by Egr2/Krox20 during Peripheral Nerve Myelination. J. Neurochem. 2005, 93, 737–748. [Google Scholar] [CrossRef]
- Carrillo, J.; Garcia-Aragoncillo, E.; Azorin, D.; Agra, N.; Sastre, A.; Gonzalez-Mediero, I.; Garcia-Miguel, P.; Pestana, A.; Gallego, S.; Segura, D.; et al. Cholecystokinin Down-Regulation by RNA Interference Impairs Ewing Tumor Growth. Clin. Cancer Res. 2007, 13, 2429–2440. [Google Scholar] [CrossRef] [Green Version]
- Sole, A.; Grossetête, S.; Heintzé, M.; Babin, L.; Zaïdi, S.; Revy, P.; Renouf, B.; De Cian, A.; Giovannangeli, C.; Pierre-Eugène, C.; et al. Unraveling Ewing Sarcoma Tumorigenesis Originating from Patient-Derived Mesenchymal Stem Cells. Cancer Res. 2021, 81, 4994–5006. [Google Scholar] [CrossRef]
- Chauvin, C.; Leruste, A.; Tauziede-Espariat, A.; Andrianteranagna, M.; Surdez, D.; Lescure, A.; Han, Z.-Y.; Anthony, E.; Richer, W.; Baulande, S.; et al. High-Throughput Drug Screening Identifies Pazopanib and Clofilium Tosylate as Promising Treatments for Malignant Rhabdoid Tumors. Cell Rep. 2017, 21, 1737–1745. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-Based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aynaud, M.-M.; Mirabeau, O.; Gruel, N.; Grossetête, S.; Boeva, V.; Durand, S.; Surdez, D.; Saulnier, O.; Zaïdi, S.; Gribkova, S.; et al. Transcriptional Programs Define Intratumoral Heterogeneity of Ewing Sarcoma at Single-Cell Resolution. Cell Rep. 2020, 30, 1767–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, Y.; Mistica, A.M.A.; Kitamoto, T.; Schätzl, H.M. Critical Significance of the Region between Helix 1 and 2 for Efficient Dominant-Negative Inhibition by Conversion-Incompetent Prion Protein. PLoS Pathog. 2013, 9, e1003466. [Google Scholar] [CrossRef] [Green Version]
- DepMap, Broad (2022): DepMap 22Q1 Public. Figshare. Dataset. Available online: https://figshare.com/articles/dataset/DepMap_22Q1_Public/19139906 (accessed on 6 January 2022).
- Dempster, J.M.; Rossen, J.; Kazachkova, M.; Pan, J.; Kugener, G.; Root, D.E.; Tsherniak, A. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv 2019, 720243. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational Correction of Copy Number Effect Improves Specificity of CRISPR–Cas9 Essentiality Screens in Cancer Cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: A CRISPR Cell Population Dynamics Model. bioRxiv 2021. [Google Scholar] [CrossRef]
- Pacini, C.; Dempster, J.M.; Boyle, I.; Gonçalves, E.; Najgebauer, H.; Karakoc, E.; van der Meer, D.; Barthorpe, A.; Lightfoot, H.; Jaaks, P.; et al. Integrated Cross-Study Datasets of Genetic Dependencies in Cancer. Nat. Commun. 2021, 12, 1661. [Google Scholar] [CrossRef]
- Baldauf, M.C.; Orth, M.F.; Dallmayer, M.; Marchetto, A.; Gerke, J.S.; Rubio, R.A.; Kiran, M.M.; Musa, J.; Knott, M.M.L.; Ohmura, S.; et al. Robust Diagnosis of Ewing Sarcoma by Immunohistochemical Detection of Super-Enhancer-Driven EWSR1-ETS Targets. Oncotarget 2017, 9, 1587–1601. [Google Scholar] [CrossRef] [Green Version]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Non-Oncology Drugs Are a Source of Previously Unappreciated Anti-Cancer Activity. bioRxiv 2019, 730119. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-Generation Characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-G.; Islam, R.; Cho, J.Y.; Jeong, H.; Cap, K.-C.; Park, Y.; Hossain, A.J.; Park, J.-B. Regulation of RhoA GTPase and Various Transcription Factors in the RhoA Pathway. J. Cell. Physiol. 2018, 233, 6381–6392. [Google Scholar] [CrossRef] [PubMed]
- Tebar, F.; Chavero, A.; Agell, N.; Lu, A.; Rentero, C.; Enrich, C.; Grewal, T. Pleiotropic Roles of Calmodulin in the Regulation of KRas and Rac1 GTPases: Functional Diversity in Health and Disease. Int. J. Mol. Sci. 2020, 21, 3680. [Google Scholar] [CrossRef]
- Larosa, V.; Remacle, C. Insights into the Respiratory Chain and Oxidative Stress. Biosci. Rep. 2018, 38, BSR20171492. [Google Scholar] [CrossRef] [Green Version]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations with Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, T.G.P.; Diebold, I.; Esposito, I.; Plehm, S.; Hauer, K.; Thiel, U.; da Silva-Buttkus, P.; Neff, F.; Unland, R.; Müller-Tidow, C.; et al. STEAP1 Is Associated with the Invasive and Oxidative Stress Phenotype of Ewing Tumors. Mol. Cancer Res. MCR 2012, 10, 52–65. [Google Scholar] [CrossRef] [Green Version]
- Magwere, T.; Myatt, S.S.; Burchill, S.A. Manipulation of Oxidative Stress to Induce Cell Death in Ewing’s Sarcoma Family of Tumours. Eur. J. Cancer 2008, 44, 2276–2287. [Google Scholar] [CrossRef]
- Marchetto, A.; Ohmura, S.; Orth, M.F.; Knott, M.M.L.; Colombo, M.V.; Arrigoni, C.; Bardinet, V.; Saucier, D.; Wehweck, F.S.; Li, J.; et al. Oncogenic Hijacking of a Developmental Transcription Factor Evokes Vulnerability toward Oxidative Stress in Ewing Sarcoma. Nat. Commun. 2020, 11, 2423. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A Simple Practice Guide for Dose Conversion between Animals and Human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Gao, Y.; Xu, P.; Li, K.; Xu, X.; Gao, J.; Qi, Y.; Xu, J.; Yang, Y.; Song, W.; et al. Atorvastatin Inhibits Breast Cancer Cells by Downregulating PTEN/AKT Pathway via Promoting Ras Homolog Family Member B (RhoB). BioMed Res. Int. 2019, 2019, 3235021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, K.A.; Dal-Cim, T.; Lopes, F.G.; Ludka, F.K.; Nedel, C.B.; Tasca, C.I. Atorvastatin Promotes Cytotoxicity and Reduces Migration and Proliferation of Human A172 Glioma Cells. Mol. Neurobiol. 2018, 55, 1509–1523. [Google Scholar] [CrossRef] [PubMed]
- Gbelcová, H.; Lenícek, M.; Zelenka, J.; Knejzlík, Z.; Dvoráková, G.; Zadinová, M.; Poucková, P.; Kudla, M.; Balaz, P.; Ruml, T.; et al. Differences in Antitumor Effects of Various Statins on Human Pancreatic Cancer. Int. J. Cancer 2008, 122, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Amiri, S.; Pecic, S.; Machaj, F.; Rosik, J.; Łos, M.J.; Alizadeh, J.; Mahdian, R.; da Silva Rosa, S.C.; Schaafsma, D.; et al. Pleiotropic Effects of Statins: A Focus on Cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165968. [Google Scholar] [CrossRef]
- Longo, J.; van Leeuwen, J.E.; Elbaz, M.; Branchard, E.; Penn, L.Z. Statins as Anticancer Agents in the Era of Precision Medicine. Clin. Cancer Res. 2020, 26, 5791–5800. [Google Scholar] [CrossRef]
- Kubo, T.; Shimose, S.; Matsuo, T.; Tanaka, K.; Yasunaga, Y.; Sakai, A.; Ochi, M. Inhibitory Effects of a New Bisphosphonate, Minodronate, on Proliferation and Invasion of a Variety of Malignant Bone Tumor Cells. J. Orthop. Res. 2006, 24, 1138–1144. [Google Scholar] [CrossRef] [Green Version]
- Odri, G.; Kim, P.-P.; Lamoureux, F.; Charrier, C.; Battaglia, S.; Amiaud, J.; Heymann, D.; Gouin, F.; Redini, F. Zoledronic Acid Inhibits Pulmonary Metastasis Dissemination in a Preclinical Model of Ewing’s Sarcoma via Inhibition of Cell Migration. BMC Cancer 2014, 14, 169. [Google Scholar] [CrossRef] [Green Version]
- Sonnemann, J.; Eckervogt, V.; Truckenbrod, B.; Boos, J.; Winkelmann, W.; van Valen, F. The Bisphosphonate Pamidronate Is a Potent Inhibitor of Ewing’s Sarcoma Cell Growth in Vitro. Anticancer Drugs 2003, 14, 767–771. [Google Scholar] [CrossRef]
- Zhou, Z.; Guan, H.; Duan, X.; Kleinerman, E.S. Zoledronic Acid Inhibits Primary Bone Tumor Growth in Ewing Sarcoma. Cancer 2005, 104, 1713–1720. [Google Scholar] [CrossRef]
- Agarwal, B.; Halmos, B.; Feoktistov, A.S.; Protiva, P.; Ramey, W.G.; Chen, M.; Pothoulakis, C.; Lamont, J.T.; Holt, P.R. Mechanism of Lovastatin-Induced Apoptosis in Intestinal Epithelial Cells. Carcinogenesis 2002, 23, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Zheng, X.; Lytle, R.A.; Higashikubo, R.; Rich, K.M. Lovastatin-Induced up-Regulation of the BH3-Only Protein, Bim, and Cell Death in Glioblastoma Cells. J. Neurochem. 2004, 89, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Shellman, Y.G.; Ribble, D.; Miller, L.; Gendall, J.; Vanbuskirk, K.; Kelly, D.; Norris, D.A.; Dellavalle, R.P. Lovastatin-Induced Apoptosis in Human Melanoma Cell Lines. Melanoma Res. 2005, 15, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Tan, M.M.; Wong, W.W.; Dimitroulakos, J.; Minden, M.D.; Penn, L.Z. Blocking Protein Geranylgeranylation Is Essential for Lovastatin-Induced Apoptosis of Human Acute Myeloid Leukemia Cells. Leukemia 2001, 15, 1398–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin. Cancer Res. 2015, 21, 1819–1827. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the Undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Mi, W.; Lin, Q.; Childress, C.; Sudol, M.; Robishaw, J.; Berlot, C.H.; Shabahang, M.; Yang, W. Geranylgeranylation Signals to the Hippo Pathway for Breast Cancer Cell Proliferation and Migration. Oncogene 2015, 34, 3095–3106. [Google Scholar] [CrossRef]
- Kovar, H.; Bierbaumer, L.; Radic-Sarikas, B. The YAP/TAZ Pathway in Osteogenesis and Bone Sarcoma Pathogenesis. Cells 2020, 9, 972. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic Control of YAP and TAZ by the Mevalonate Pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, T.; Dou, Y.; Zhang, S.; Liu, H.; Khishignyam, T.; Li, X.; Zuo, D.; Zhang, Z.; Jin, M.; et al. Atorvastatin Exerts Antileukemia Activity via Inhibiting Mevalonate-YAP Axis in K562 and HL60 Cells. Front. Oncol. 2019, 9, 1032. [Google Scholar] [CrossRef]
- McGregor, G.H.; Campbell, A.D.; Fey, S.K.; Tumanov, S.; Sumpton, D.; Blanco, G.R.; Mackay, G.; Nixon, C.; Vazquez, A.; Sansom, O.J.; et al. Targeting the Metabolic Response to Statin-Mediated Oxidative Stress Produces a Synergistic Antitumor Response. Cancer Res. 2020, 80, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, T.; Ragu, S.; Magdalou, I.; Machon, C.; Dardillac, E.; Técher, H.; Guitton, J.; Debatisse, M.; Lopez, B.S. Slow Replication Fork Velocity of Homologous Recombination-Defective Cells Results from Endogenous Oxidative Stress. PLoS Genet. 2016, 12, e1006007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS-FLI1 Increases Transcription to Cause R-Loops and Block BRCA1 Repair in Ewing Sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.C.; Hoegel, T.J.; Lynch, M.L.; Woloszynska, A.; Melendy, T.; Ohm, J.E. Exploiting Replication Stress as a Novel Therapeutic Intervention. Mol. Cancer Res. MCR 2020, 19, 192–206. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ewing Cell Lines | Short Term Culture of Ewing PDX | Other Cell Lines | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (µM) | A673 | TC71 | SKNMC | POE | IC-pPDX-80 | IC-pPDX-87 | IC-pPDX-164 | IC-pPDX-3 | KD | MCF7 | SKNSH | A549 |
| RT | BC | NB | Lung | |||||||||
| Atorvatatin | 0.55 | 1.29 | 2.25 | 1.2 | 0.75 | 0.3 | 0.2 | 0.13 | 4.6 | 9.02 | no effect | 124.6 |
| Simvastatin | 0.34 | 1.03 | 2.6 | 0.2 | 1.08 | 0.39 | 0.24 | 0.11 | 1.51 | 12.62 | 62.55 | 17.54 |
| Lovastatin | 1.06 | 3.26 | 3.98 | 0.69 | 0.83 | 1.18 | 0.55 | 5.03 | 7.39 | 43.13 | 26.05 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buchou, C.; Laud-Duval, K.; van der Ent, W.; Grossetête, S.; Zaidi, S.; Gentric, G.; Corbé, M.; Müller, K.; Del Nery, E.; Surdez, D.; et al. Upregulation of the Mevalonate Pathway through EWSR1-FLI1/EGR2 Regulatory Axis Confers Ewing Cells Exquisite Sensitivity to Statins. Cancers 2022, 14, 2327. https://doi.org/10.3390/cancers14092327
Buchou C, Laud-Duval K, van der Ent W, Grossetête S, Zaidi S, Gentric G, Corbé M, Müller K, Del Nery E, Surdez D, et al. Upregulation of the Mevalonate Pathway through EWSR1-FLI1/EGR2 Regulatory Axis Confers Ewing Cells Exquisite Sensitivity to Statins. Cancers. 2022; 14(9):2327. https://doi.org/10.3390/cancers14092327
Chicago/Turabian StyleBuchou, Charlie, Karine Laud-Duval, Wietske van der Ent, Sandrine Grossetête, Sakina Zaidi, Géraldine Gentric, Maxime Corbé, Kévin Müller, Elaine Del Nery, Didier Surdez, and et al. 2022. "Upregulation of the Mevalonate Pathway through EWSR1-FLI1/EGR2 Regulatory Axis Confers Ewing Cells Exquisite Sensitivity to Statins" Cancers 14, no. 9: 2327. https://doi.org/10.3390/cancers14092327
APA StyleBuchou, C., Laud-Duval, K., van der Ent, W., Grossetête, S., Zaidi, S., Gentric, G., Corbé, M., Müller, K., Del Nery, E., Surdez, D., & Delattre, O. (2022). Upregulation of the Mevalonate Pathway through EWSR1-FLI1/EGR2 Regulatory Axis Confers Ewing Cells Exquisite Sensitivity to Statins. Cancers, 14(9), 2327. https://doi.org/10.3390/cancers14092327