

Mechanisms and Strategies to Overcome PD-1/PD-L1 Blockade Resistance in Triple-Negative Breast Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Resistance Mechanism of PD-1/PD-L1 Inhibit Therapies
2.1. Disturbed Presentation of Tumor-Specific Antigens
2.2. Immunosuppressive TME
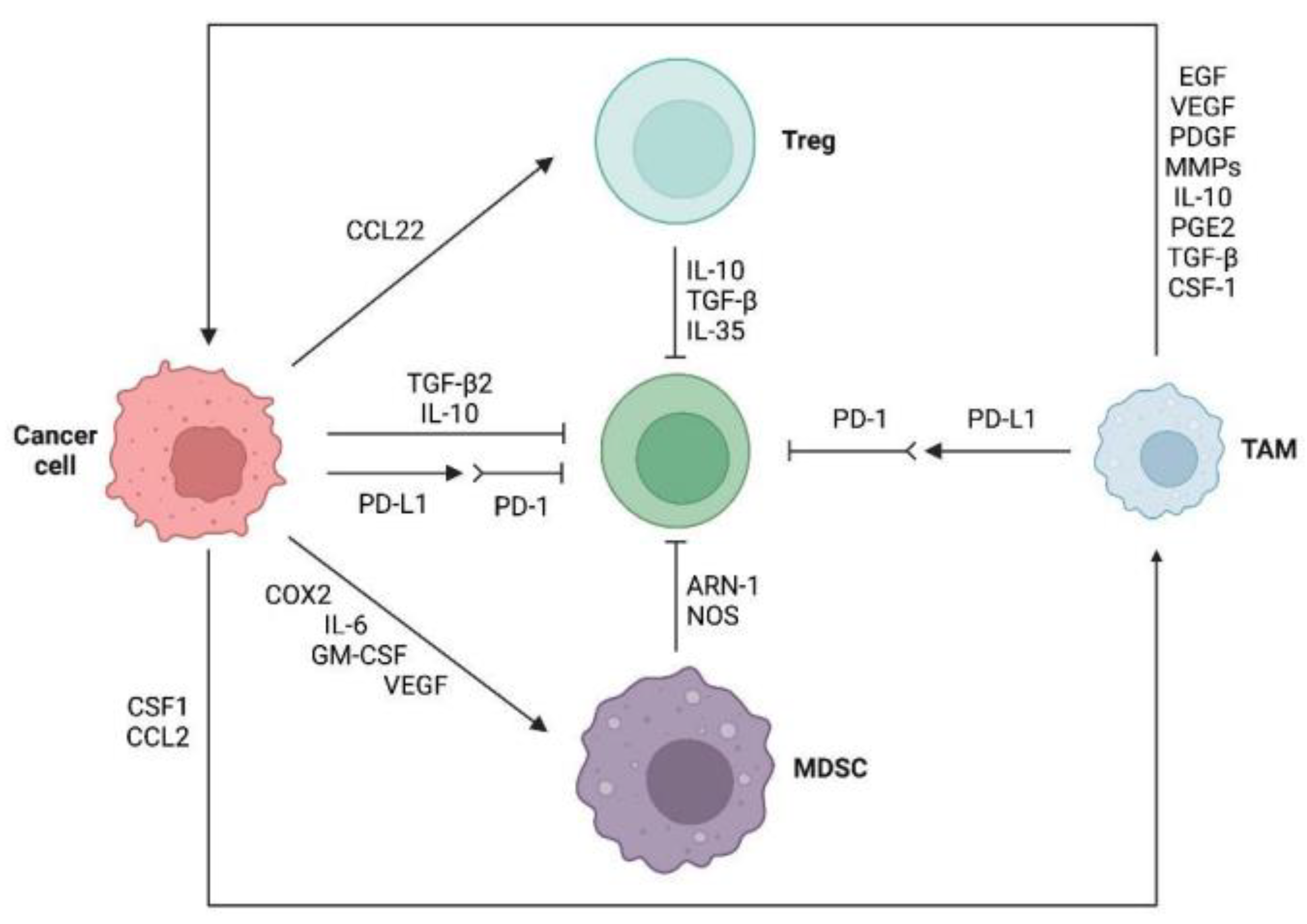
2.2.1. Immunosuppressive Cells
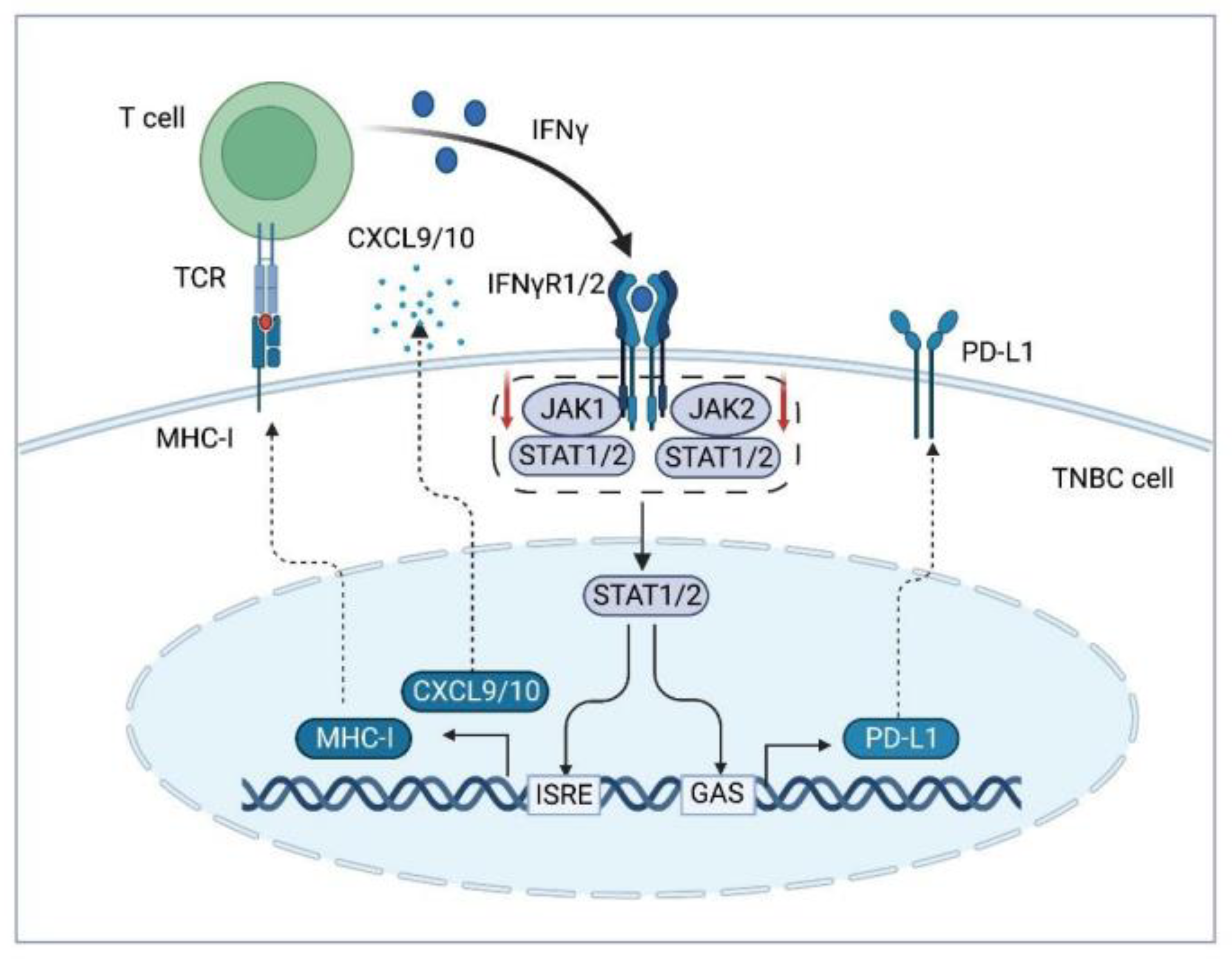
2.2.2. Cytokines
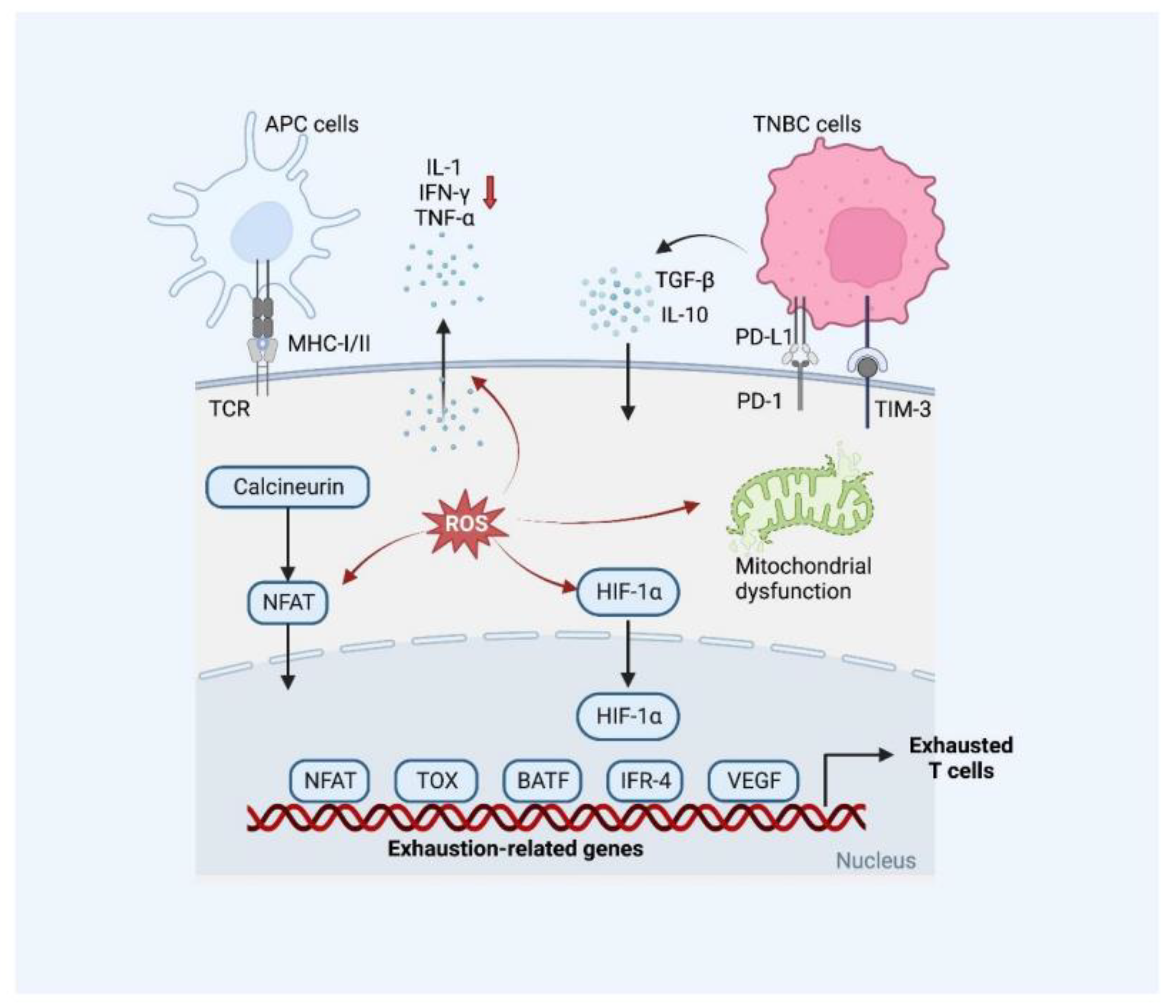
2.2.3. T Cell Exhaustion
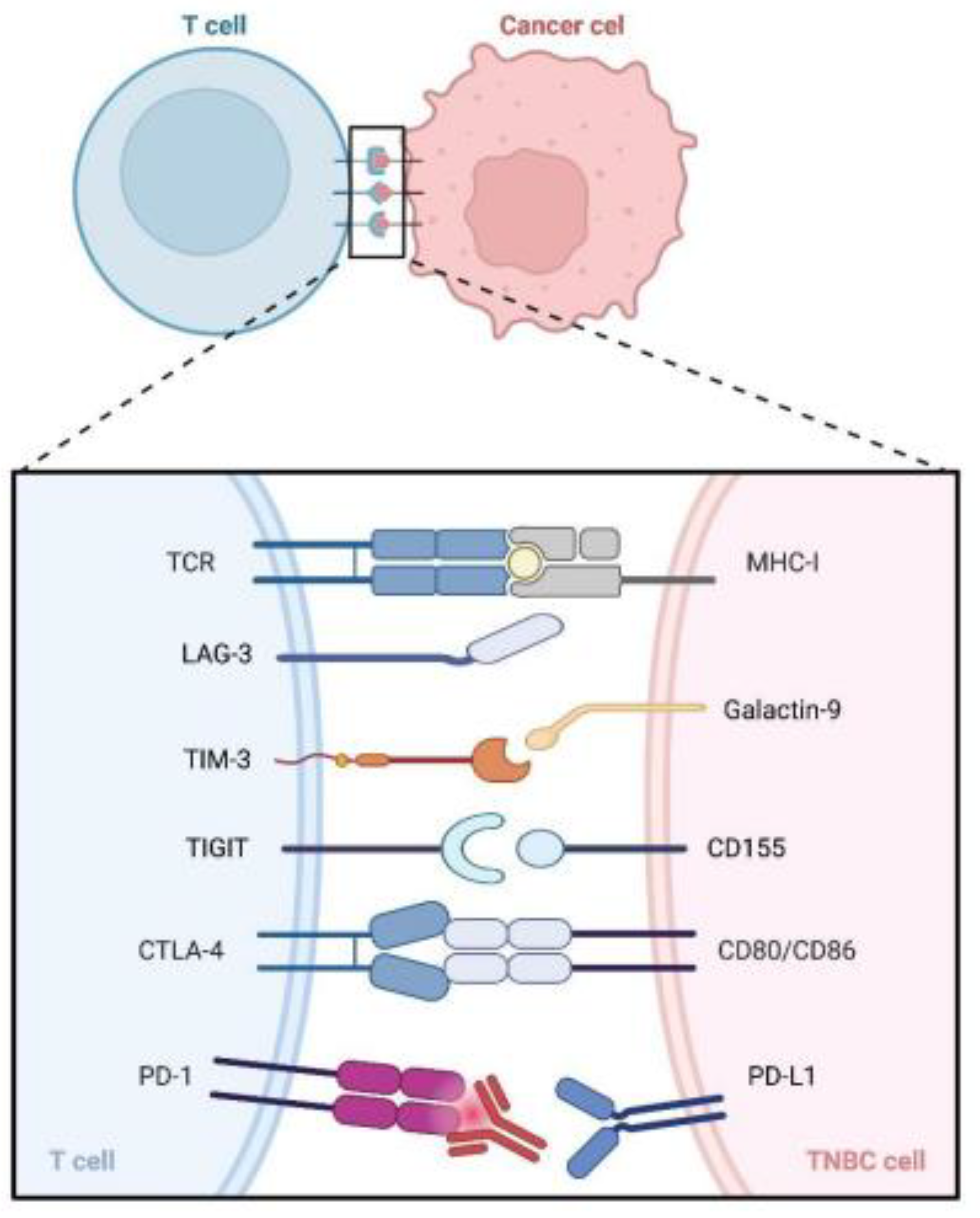
2.3. Compensatory Upregulation of Alternative Immune Checkpoints
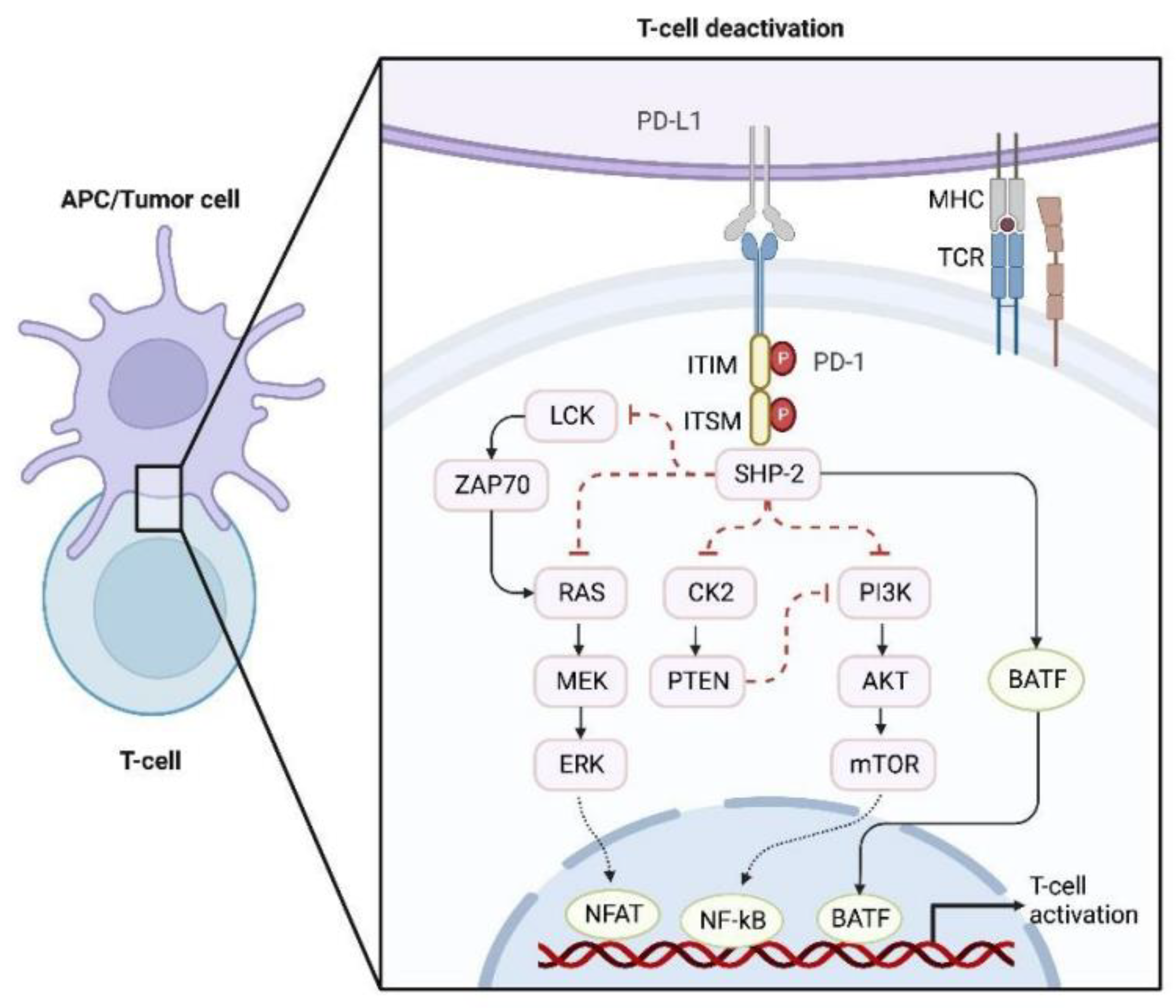
2.4. Abnormal Signaling Transduction in Tumor Cells
3. Biomarkers for Predicting the Efficacy of Anti-PD-1/PD-L1 Immunotherapy
3.1. PD-L1 Expression Level
3.2. TILs
3.3. TMB and MSI
3.4. Driver Gene Mutation
3.5. TNBC Microenvironment Phenotypes
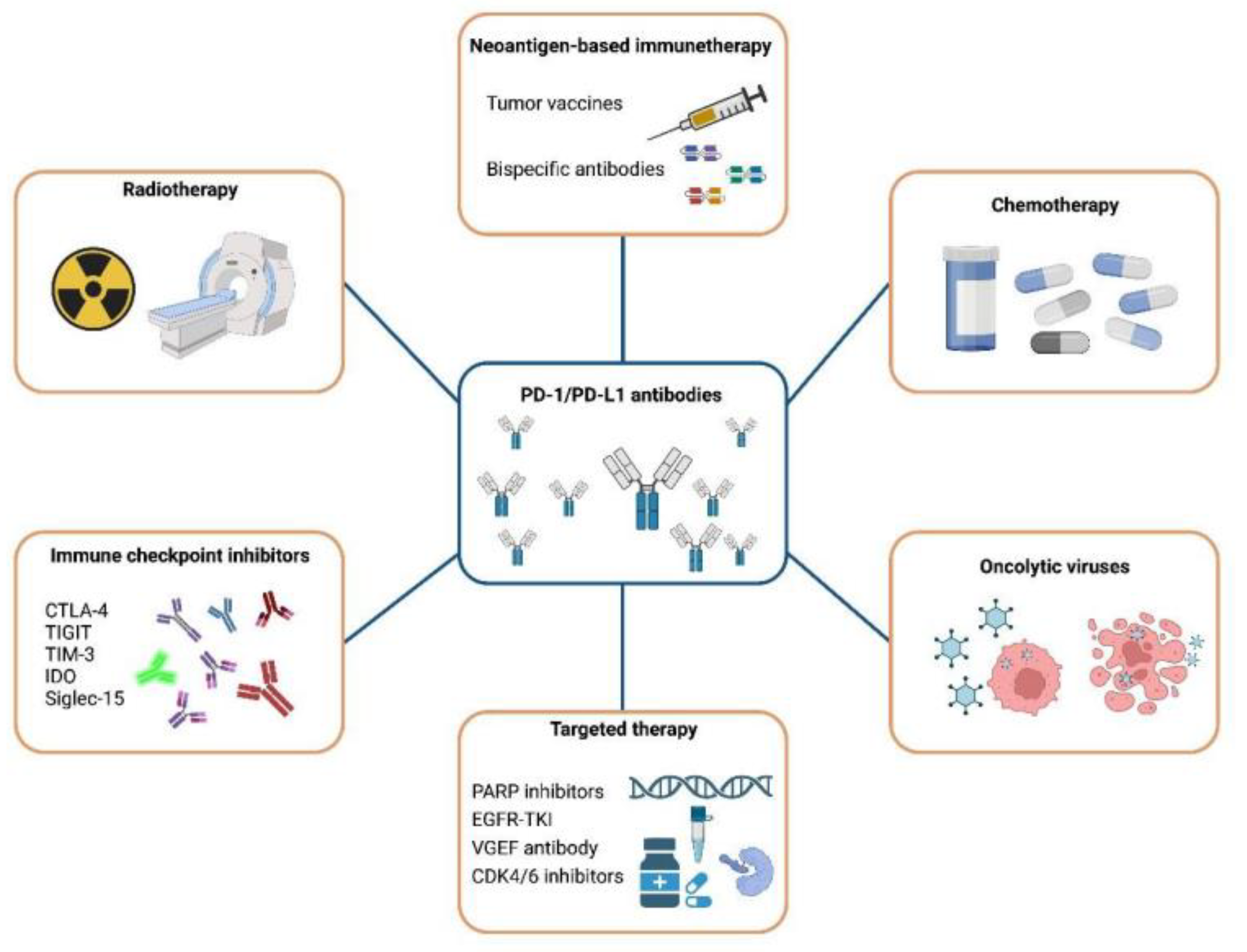
4. Therapeutic Strategies for Immune Checkpoint PD-1/PD-L1 Inhibitor Resistance
4.1. Combination with Chemotherapy
4.2. Combination with Radiotherapy
4.3. The Synergistic Effect of ICIs
4.4. Combination with Targeted Therapy
4.5. Oncolytic Viruses
4.6. Neoantigen-Based Immunotherapy
5. Conclusions and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.D.; Liu, Y.R.; Yu, Y.; et al. Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 2019, 35, 428–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.; Han, Y.; Lian, M.; Colditz, G.A.; Weber, J.D.; Ma, C.; Liu, Y. Evaluation of Racial/Ethnic Differences in Treatment and Mortality among Women with Triple-Negative Breast Cancer. JAMA Oncol. 2021, 7, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Rossi, L.; Mazzara, C.; Pagani, O. Diagnosis and Treatment of Breast Cancer in Young Women. Curr. Treat. Options Oncol. 2019, 20, 86. [Google Scholar] [CrossRef] [PubMed]
- Planes-Laine, G.; Rochigneux, P.; Bertucci, F.; Chrétien, A.S.; Viens, P.; Sabatier, R.; Gonçalves, A. PD-1/PD-L1 Targeting in Breast Cancer: The First Clinical Evidences Are Emerging. A Literature Review. Cancers 2019, 11, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartkopf, A.D.; Taran, F.A.; Wallwiener, M.; Walter, C.B.; Krämer, B.; Grischke, E.M.; Brucker, S.Y. PD-1 and PD-L1 Immune Checkpoint Blockade to Treat Breast Cancer. Breast Care 2016, 11, 385–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Neel, B.G.; Gu, H.; Pao, L. The ′Shp′ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Song, Y.; Wang, Y.; Huang, Y.; Li, Z.; Cui, Y.; Yi, M.; Xia, L.; Zhuang, W.; Wu, X.; et al. PD-1/PD-L1 blockade rescue exhausted CD8+ T cells in gastrointestinal stromal tumours via the PI3K/Akt/mTOR signalling pathway. Cell Prolif. 2019, 52, e12571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stutvoet, T.S.; Kol, A.; de Vries, E.G.; de Bruyn, M.; Fehrmann, R.S.; Terwisscha van Scheltinga, A.G.; de Jong, S. MAPK pathway activity plays a key role in PD-L1 expression of lung adenocarcinoma cells. J. Pathol. 2019, 249, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, T.; Ishikawa, T.; Okayama, T.; Oka, K.; Mizushima, K.; Yasuda, T.; Sakamoto, N.; Katada, K.; Kamada, K.; Uchiyama, K.; et al. The JAK/STAT pathway is involved in the upregulation of PD-L1 expression in pancreatic cancer cell lines. Oncol. Rep. 2017, 37, 1545–1554. [Google Scholar] [CrossRef] [Green Version]
- Shurin, M.R.; Umansky, V. Cross-talk between HIF and PD-1/PD-L1 pathways in carcinogenesis and therapy. J. Clin. Investig. 2022, 132, e159473. [Google Scholar] [CrossRef]
- Kataoka, K.; Shiraishi, Y.; Takeda, Y.; Sakata, S.; Matsumoto, M.; Nagano, S.; Maeda, T.; Nagata, Y.; Kitanaka, A.; Mizuno, S.; et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature 2016, 534, 402–406. [Google Scholar] [CrossRef]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [Green Version]
- Bayraktar, R.; Van Roosbroeck, K. miR-155 in cancer drug resistance and as target for miRNA-based therapeutics. Cancer Metastasis Rev. 2018, 37, 33–44. [Google Scholar] [CrossRef]
- Gong, A.Y.; Zhou, R.; Hu, G.; Li, X.; Splinter, P.L.; O’Hara, S.P.; LaRusso, N.F.; Soukup, G.A.; Dong, H.; Chen, X.M. MicroRNA-513 regulates B7-H1 translation and is involved in IFN-gamma-induced B7-H1 expression in cholangiocytes. J. Immunol. 2009, 182, 1325–1333. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Kwapisz, D. Pembrolizumab and atezolizumab in triple-negative breast cancer. Cancer Immunol. Immunother. 2021, 70, 607–617. [Google Scholar] [CrossRef]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.; Sinn, B.V.; Blohmer, J.U.; Grischke, E.M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: Clinical results and biomarker analysis of GeparNuevo study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1279–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bense, R.D.; Sotiriou, C.; Piccart-Gebhart, M.J.; Haanen, J.; van Vugt, M.; de Vries, E.G.E.; Schröder, C.P.; Fehrmann, R.S.N. Relevance of Tumor-Infiltrating Immune Cell Composition and Functionality for Disease Outcome in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, 372–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Van Eenoo, F.; Rouas, G.; Francis, P.; Crown, J.P.; Hitre, E.; et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jiang, Z.; Gao, Y.; Wang, L.; Chen, C.; Wang, X. TP53 Mutations Promote Immunogenic Activity in Breast Cancer. J. Oncol. 2019, 2019, 5952836. [Google Scholar] [CrossRef] [Green Version]
- Andrews, L.P.; Yano, H.; Vignali, D.A.A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: Breakthroughs or backups. Nat. Immunol. 2019, 20, 1425–1434. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.K.; Vandsemb, E.N.; Herbst, R.S.; Chen, L. Adaptive immune resistance at the tumour site: Mechanisms and therapeutic opportunities. Nat. Rev. Drug Discov. 2022, 21, 529–540. [Google Scholar] [CrossRef]
- Patel, S.A.; Minn, A.J. Combination Cancer Therapy with Immune Checkpoint Blockade: Mechanisms and Strategies. Immunity 2018, 48, 417–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef] [Green Version]
- Verdegaal, E.M.; de Miranda, N.F.; Visser, M.; Harryvan, T.; van Buuren, M.M.; Andersen, R.S.; Hadrup, S.R.; van der Minne, C.E.; Schotte, R.; Spits, H.; et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 2016, 536, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Haricharan, S.; Bainbridge, M.N.; Scheet, P.; Brown, P.H. Somatic mutation load of estrogen receptor-positive breast tumors predicts overall survival: An analysis of genome sequence data. Breast Cancer Res. Treat. 2014, 146, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Barroso-Sousa, R.; Jain, E.; Cohen, O.; Kim, D.; Buendia-Buendia, J.; Winer, E.; Lin, N.; Tolaney, S.M.; Wagle, N. Prevalence and mutational determinants of high tumor mutation burden in breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Rugo, H.; Delord, J.-P.; Im, S.-A.; Ott, P.; Piha-Paul, S.; Bedard, P.; Sachdev, J.; Le Tourneau, C.; van Brummelen, E.; Varga, A.; et al. Abstract S5-07: Preliminary efficacy and safety of pembrolizumab (MK-3475) in patients with PD-L1–positive, estrogen receptor-positive (ER+)/HER2-negative advanced breast cancer enrolled in KEYNOTE-028. Cancer Res. 2016, 76, S5. [Google Scholar] [CrossRef]
- Li, Z.; Qiu, Y.; Lu, W.; Jiang, Y.; Wang, J. Immunotherapeutic interventions of Triple Negative Breast Cancer. J. Transl. Med. 2018, 16, 147. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.Z.; Sun, Y.; Jiang, X.F.; Liu, L.; Liu, L.; Sun, L.X. Promotion on NLRC5 upregulating MHC-I expression by IFN-γ in MHC-I-deficient breast cancer cells. Immunol. Res. 2019, 67, 497–504. [Google Scholar] [CrossRef]
- Vitale, M.; Rezzani, R.; Rodella, L.; Zauli, G.; Grigolato, P.; Cadei, M.; Hicklin, D.J.; Ferrone, S. HLA class I antigen and transporter associated with antigen processing (TAP1 and TAP2) down-regulation in high-grade primary breast carcinoma lesions. Cancer Res. 1998, 58, 737–742. [Google Scholar] [PubMed]
- Dusenbery, A.C.; Maniaci, J.L.; Hillerson, N.D.; Dill, E.A.; Bullock, T.N.; Mills, A.M. MHC Class I Loss in Triple-negative Breast Cancer: A Potential Barrier to PD-1/PD-L1 Checkpoint Inhibitors. Am. J. Surg. Pathol. 2021, 45, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Colaprico, A.; Silva, T.C.; Chen, J.; An, H.; Ban, Y.; Huang, H.; Wang, L.; James, J.L.; Balko, J.M.; et al. Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes. Nat. Commun. 2021, 12, 6276. [Google Scholar] [CrossRef] [PubMed]
- Tu, K.; Yu, Y.; Wang, Y.; Yang, T.; Hu, Q.; Qin, X.; Tu, J.; Yang, C.; Kong, L.; Zhang, Z. Combination of Chidamide-Mediated Epigenetic Modulation with Immunotherapy: Boosting Tumor Immunogenicity and Response to PD-1/PD-L1 Blockade. ACS Appl. Mater. Interfaces 2021, 13, 39003–39017. [Google Scholar] [CrossRef]
- Lee, J.V.; Housley, F.; Yau, C.; Nakagawa, R.; Winkler, J.; Anttila, J.M.; Munne, P.M.; Savelius, M.; Houlahan, K.E.; Van de Mark, D.; et al. Combinatorial immunotherapies overcome MYC-driven immune evasion in triple negative breast cancer. Nat. Commun. 2022, 13, 3671. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, L.; Wan, C.; Sun, Y.; Van der Jeught, K.; Zhou, Z.; Dong, T.; So, K.M.; Yu, T.; Li, Y.; et al. MAL2 drives immune evasion in breast cancer by suppressing tumor antigen presentation. J. Clin. Investig. 2021, 131, e140837. [Google Scholar] [CrossRef]
- Park, I.A.; Hwang, S.H.; Song, I.H.; Heo, S.H.; Kim, Y.A.; Bang, W.S.; Park, H.S.; Lee, M.; Gong, G.; Lee, H.J. Expression of the MHC class II in triple-negative breast cancer is associated with tumor-infiltrating lymphocytes and interferon signaling. PLoS ONE 2017, 12, e0182786. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Ericsson, P.I.; Wulfkhule, J.D.; Gallagher, R.I.; Sun, X.; Axelrod, M.L.; Sheng, Q.; Luo, N.; Gomez, H.; Sanchez, V.; Sanders, M.; et al. Tumor-Specific Major Histocompatibility-II Expression Predicts Benefit to Anti-PD-1/L1 Therapy in Patients With HER2-Negative Primary Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 5299–5306. [Google Scholar] [CrossRef]
- Johnson, D.B.; Nixon, M.J.; Wang, Y.; Wang, D.Y.; Castellanos, E.; Estrada, M.V.; Ericsson-Gonzalez, P.I.; Cote, C.H.; Salgado, R.; Sanchez, V.; et al. Tumor-specific MHC-II expression drives a unique pattern of resistance to immunotherapy via LAG-3/FCRL6 engagement. JCI Insight 2018, 3, e120360. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wu, X. Study and analysis of antitumor resistance mechanism of PD1/PD-L1 immune checkpoint blocker. Cancer Med. 2020, 9, 8086–8121. [Google Scholar] [CrossRef]
- Martín-Gayo, E.; Sierra-Filardi, E.; Corbí, A.L.; Toribio, M.L. Plasmacytoid dendritic cells resident in human thymus drive natural Treg cell development. Blood 2010, 115, 5366–5375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Du, H.; Zhan, S.; Liu, W.; Wang, Z.; Lan, J.; PuYang, L.; Wan, Y.; Qu, Q.; Wang, S.; et al. The interaction between the soluble programmed death ligand-1 (sPD-L1) and PD-1(+) regulator B cells mediates immunosuppression in triple-negative breast cancer. Front. Immunol. 2022, 13, 830606. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.X.; Jiao, Y.N.; Hao, H.F.; Xue, D.; Bai, C.C.; Han, S.Y. Taraxacum mongolicum extract inhibited malignant phenotype of triple-negative breast cancer cells in tumor-associated macrophages microenvironment through suppressing IL-10/STAT3/PD-L1 signaling pathways. J. Ethnopharmacol. 2021, 274, 113978. [Google Scholar] [CrossRef] [PubMed]
- Kondou, R.; Iizuka, A.; Nonomura, C.; Miyata, H.; Ashizawa, T.; Nagashima, T.; Ohshima, K.; Urakami, K.; Kusuhara, M.; Yamaguchi, K.; et al. Classification of tumor microenvironment immune types based on immune response-associated gene expression. Int. J. Oncol. 2019, 54, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Oshi, M.; Asaoka, M.; Tokumaru, Y.; Angarita, F.A.; Yan, L.; Matsuyama, R.; Zsiros, E.; Ishikawa, T.; Endo, I.; Takabe, K. Abundance of Regulatory T Cell (Treg) as a Predictive Biomarker for Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer. Cancers 2020, 12, 3038. [Google Scholar] [CrossRef]
- Bohling, S.D.; Allison, K.H. Immunosuppressive regulatory T cells are associated with aggressive breast cancer phenotypes: A potential therapeutic target. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 2008, 21, 1527–1532. [Google Scholar] [CrossRef] [Green Version]
- Ohara, M.; Yamaguchi, Y.; Matsuura, K.; Murakami, S.; Arihiro, K.; Okada, M. Possible involvement of regulatory T cells in tumor onset and progression in primary breast cancer. Cancer Immunol. Immunother. 2009, 58, 441–447. [Google Scholar] [CrossRef] [Green Version]
- Chinen, T.; Kannan, A.K.; Levine, A.G.; Fan, X.; Klein, U.; Zheng, Y.; Gasteiger, G.; Feng, Y.; Fontenot, J.D.; Rudensky, A.Y. An essential role for the IL-2 receptor in T(reg) cell function. Nat. Immunol. 2016, 17, 1322–1333. [Google Scholar] [CrossRef]
- Taylor, A.; Verhagen, J.; Blaser, K.; Akdis, M.; Akdis, C.A. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: The role of T regulatory cells. Immunology 2006, 117, 433–442. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Cheng, X.; Yang, H.; Lian, S.; Jiang, Y.; Liang, J.; Chen, X.; Mo, S.; Shi, Y.; Zhao, S.; et al. BCL-2 expression promotes immunosuppression in chronic lymphocytic leukemia by enhancing regulatory T cell differentiation and cytotoxic T cell exhaustion. Mol. Cancer 2022, 21, 59. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Kumar, V.; Ramachandran, I.R.; Youn, J.I.; Celis, E.; Finnberg, N.; El-Deiry, W.S.; Winograd, R.; Vonderheide, R.H.; English, N.R.; et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J. Clin. Investig. 2014, 124, 2626–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018, 28, 87–103.e106. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Wilkes, D.W.; Samuel, N.; Blanco, M.A.; Nayak, A.; Alicea-Torres, K.; Gluck, C.; Sinha, S.; Gabrilovich, D.; Chakrabarti, R. ΔNp63-driven recruitment of myeloid-derived suppressor cells promotes metastasis in triple-negative breast cancer. J. Clin. Investig. 2018, 128, 5095–5109. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.; Luo, F.; Sun, B.; Liu, W.; Shi, Q.; Cheng, S.Y.; Chen, C.; Chen, G.; Li, Y.; Feng, H. KAT6A Acetylation of SMAD3 Regulates Myeloid-Derived Suppressor Cell Recruitment, Metastasis, and Immunotherapy in Triple-Negative Breast Cancer. Adv. Sci. 2022, 9, e2105793. [Google Scholar] [CrossRef]
- Kajihara, N.; Kobayashi, T.; Otsuka, R.; Nio-Kobayashi, J.; Oshino, T.; Takahashi, M.; Imanishi, S.; Hashimoto, A.; Wada, H.; Seino, K.I. Tumor-derived interleukin-34 creates an immunosuppressive and chemoresistant tumor microenvironment by modulating myeloid-derived suppressor cells in triple-negative breast cancer. Cancer Immunol. Immunother. 2022. Available online: https://link.springer.com/article/10.1007/s00262-022-03293-3 (accessed on 14 September 2022). [CrossRef]
- Parihar, R.; Rivas, C.; Huynh, M.; Omer, B.; Lapteva, N.; Metelitsa, L.S.; Gottschalk, S.M.; Rooney, C.M. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors. Cancer Immunol. Res. 2019, 7, 363–375. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Z.T.; Wang, Q. Effect of Expression Level Changes of M-MDSC to Related Immune Function in Patients with Primary ITP. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2021, 29, 581–585. [Google Scholar] [CrossRef]
- Rico, L.G.; Aguilar Hernández, A.; Ward, M.D.; Bradford, J.A.; Juncà, J.; Rosell, R.; Petriz, J. Unmasking the expression of PD-L1 in Myeloid Derived Suppressor Cells: A case study in lung cancer to discover new drugs with specific on-target efficacy. Transl. Oncol. 2021, 14, 100969. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, X.; Sun, Q.; Jiang, Y.; Zhang, W.; Luo, J.; Li, Y. Synergic effect of PD-1 blockade and endostar on the PI3K/AKT/mTOR-mediated autophagy and angiogenesis in Lewis lung carcinoma mouse model. Biomed. Pharmacother. 2020, 125, 109746. [Google Scholar] [CrossRef] [PubMed]
- Ngambenjawong, C.; Gustafson, H.H.; Pun, S.H. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 2017, 114, 206–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Lin, K.; Li, X.; Yuan, X.; Xu, P.; Ni, P.; Xu, D. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front. Immunol. 2020, 11, 1731. [Google Scholar] [CrossRef] [PubMed]
- Sami, E.; Paul, B.T.; Koziol, J.A.; ElShamy, W.M. The Immunosuppressive Microenvironment in BRCA1-IRIS-Overexpressing TNBC Tumors Is Induced by Bidirectional Interaction with Tumor-Associated Macrophages. Cancer Res. 2020, 80, 1102–1117. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Le, K.; Xu, M.; Zhou, J.; Xiao, Y.; Yang, W.; Jiang, Y.; Xi, Z.; Huang, T. Combined MEK inhibition and tumor-associated macrophages depletion suppresses tumor growth in a triple-negative breast cancer mouse model. Int. Immunopharmacol. 2019, 76, 105864. [Google Scholar] [CrossRef]
- Santoni, M.; Romagnoli, E.; Saladino, T.; Foghini, L.; Guarino, S.; Capponi, M.; Giannini, M.; Cognigni, P.D.; Ferrara, G.; Battelli, N. Triple negative breast cancer: Key role of Tumor-Associated Macrophages in regulating the activity of anti-PD-1/PD-L1 agents. Biochim. Et Biophys. Acta. Rev. Cancer. 2018, 1869, 78–84. [Google Scholar] [CrossRef]
- Li, M.; Li, M.; Yang, Y.; Liu, Y.; Xie, H.; Yu, Q.; Tian, L.; Tang, X.; Ren, K.; Li, J.; et al. Remodeling tumor immune microenvironment via targeted blockade of PI3K-γ and CSF-1/CSF-1R pathways in tumor associated macrophages for pancreatic cancer therapy. J. Control. Release Off. J. Control. Release Soc. 2020, 321, 23–35. [Google Scholar] [CrossRef]
- Lin, R.L.; Zhao, L.J. Mechanistic basis and clinical relevance of the role of transforming growth factor-β in cancer. Cancer Biol. Med. 2015, 12, 385–393. [Google Scholar] [CrossRef]
- Panagi, M.; Voutouri, C.; Mpekris, F.; Papageorgis, P.; Martin, M.R.; Martin, J.D.; Demetriou, P.; Pierides, C.; Polydorou, C.; Stylianou, A.; et al. TGF-β inhibition combined with cytotoxic nanomedicine normalizes triple negative breast cancer microenvironment towards anti-tumor immunity. Theranostics 2020, 10, 1910–1922. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef]
- Lee, E.; Fertig, E.J.; Jin, K.; Sukumar, S.; Pandey, N.B.; Popel, A.S. Breast cancer cells condition lymphatic endothelial cells within pre-metastatic niches to promote metastasis. Nat. Commun. 2014, 5, 4715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudis, C.A.; Gianni, L. Triple-negative breast cancer: An unmet medical need. Oncologist 2011, 16 (Suppl. 1), 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wawrocki, S.; Druszczynska, M.; Kowalewicz-Kulbat, M.; Rudnicka, W. Interleukin 18 (IL-18) as a target for immune intervention. Acta Biochim. Pol. 2016, 63, 59–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbi, M.; Carbotti, G.; Ferrini, S. Context-dependent role of IL-18 in cancer biology and counter-regulation by IL-18BP. J. Leukoc. Biol. 2015, 97, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.W.; Chen, P.C.; Lin, M.H.; Ger, T.R.; Chiu, H.W.; Lin, Y.F. GBP5 Repression Suppresses the Metastatic Potential and PD-L1 Expression in Triple-Negative Breast Cancer. Biomedicines 2021, 9, 371. [Google Scholar] [CrossRef]
- Singh, S.; Kumar, S.; Srivastava, R.K.; Nandi, A.; Thacker, G.; Murali, H.; Kim, S.; Baldeon, M.; Tobias, J.; Blanco, M.A.; et al. Loss of ELF5-FBXW7 stabilizes IFNGR1 to promote the growth and metastasis of triple-negative breast cancer through interferon-γ signalling. Nat. Cell Biol. 2020, 22, 591–602. [Google Scholar] [CrossRef]
- Ma, S.; Zhao, Y.; Lee, W.C.; Ong, L.T.; Lee, P.L.; Jiang, Z.; Oguz, G.; Niu, Z.; Liu, M.; Goh, J.Y.; et al. Hypoxia induces HIF1α-dependent epigenetic vulnerability in triple negative breast cancer to confer immune effector dysfunction and resistance to anti-PD-1 immunotherapy. Nat. Commun. 2022, 13, 4118. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Ngiow, S.F.; Young, A.; Jacquelot, N.; Yamazaki, T.; Enot, D.; Zitvogel, L.; Smyth, M.J. A Threshold Level of Intratumor CD8+ T-cell PD1 Expression Dictates Therapeutic Response to Anti-PD1. Cancer Res. 2015, 75, 3800–3811. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, S.D.; Shin, H.; Freeman, G.J.; Wherry, E.J. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc. Natl. Acad. Sci. USA 2008, 105, 15016–15021. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schreiner, J.; Müller, P.; Herzig, P.; Roller, A.; Belousov, A.; Umana, P.; Pisa, P.; Klein, C.; Bacac, M.; et al. Progression of Lung Cancer Is Associated with Increased Dysfunction of T Cells Defined by Coexpression of Multiple Inhibitory Receptors. Cancer Immunol. Res. 2015, 3, 1344–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curdy, N.; Lanvin, O.; Laurent, C.; Fournié, J.J.; Franchini, D.M. Regulatory Mechanisms of Inhibitory Immune Checkpoint Receptors Expression. Trends Cell Biol. 2019, 29, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef] [PubMed]
- de Mingo Pulido, Á.; Gardner, A.; Hiebler, S.; Soliman, H.; Rugo, H.S.; Krummel, M.F.; Coussens, L.M.; Ruffell, B. TIM-3 Regulates CD103(+) Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 2018, 33, 60–74.e66. [Google Scholar] [CrossRef] [Green Version]
- Sabatos-Peyton, C.A.; Nevin, J.; Brock, A.; Venable, J.D.; Tan, D.J.; Kassam, N.; Xu, F.; Taraszka, J.; Wesemann, L.; Pertel, T.; et al. Blockade of Tim-3 binding to phosphatidylserine and CEACAM1 is a shared feature of anti-Tim-3 antibodies that have functional efficacy. Oncoimmunology 2018, 7, e1385690. [Google Scholar] [CrossRef] [Green Version]
- Saleh, R.; Toor, S.M.; Khalaf, S.; Elkord, E. Breast Cancer Cells and PD-1/PD-L1 Blockade Upregulate the Expression of PD-1, CTLA-4, TIM-3 and LAG-3 Immune Checkpoints in CD4(+) T Cells. Vaccines 2019, 7, 149. [Google Scholar] [CrossRef] [Green Version]
- Corti, C.; Nicolò, E.; Curigliano, G. Novel immune targets for the treatment of triple-negative breast cancer. Expert Opin. Ther. Targets 2021, 25, 815–834. [Google Scholar] [CrossRef]
- Workman, C.J.; Rice, D.S.; Dugger, K.J.; Kurschner, C.; Vignali, D.A. Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3). Eur. J. Immunol. 2002, 32, 2255–2263. [Google Scholar] [CrossRef]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stovgaard, E.S.; Kümler, I.; List-Jensen, K.; Roslind, A.; Christensen, I.J.; Høgdall, E.; Nielsen, D.; Balslev, E. Prognostic and Clinicopathologic Associations of LAG-3 Expression in Triple-negative Breast Cancer. Appl. Immunohistochem. Mol. Morphol. AIMM 2022, 30, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Heimes, A.S.; Almstedt, K.; Krajnak, S.; Runkel, A.; Droste, A.; Schwab, R.; Stewen, K.; Lebrecht, A.; Battista, M.J.; Brenner, W.; et al. Prognostic Impact of LAG-3 mRNA Expression in Early Breast Cancer. Biomedicines 2022, 10, 2656. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Tan, D.S.W.; Martín, M.; Ochoa-de-Olza, M.; Sarantopoulos, J.; Carvajal, R.D.; Kyi, C.; Esaki, T.; Prawira, A.; Akerley, W.; et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) ± anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. J. Immunother. Cancer 2022, 10, e003776. [Google Scholar] [CrossRef] [PubMed]
- Stamm, H.; Oliveira-Ferrer, L.; Grossjohann, E.M.; Muschhammer, J.; Thaden, V.; Brauneck, F.; Kischel, R.; Müller, V.; Bokemeyer, C.; Fiedler, W.; et al. Targeting the TIGIT-PVR immune checkpoint axis as novel therapeutic option in breast cancer. Oncoimmunology 2019, 8, e1674605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauken, K.E.; Wherry, E.J. TIGIT and CD226: Tipping the balance between costimulatory and coinhibitory molecules to augment the cancer immunotherapy toolkit. Cancer Cell 2014, 26, 785–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD96: New checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef]
- Yang, L.; Li, A.; Lei, Q.; Zhang, Y. Tumor-intrinsic signaling pathways: Key roles in the regulation of the immunosuppressive tumor microenvironment. J. Hematol. Oncol. 2019, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Peng, W.; Xu, C.; Lou, Y.; Zhang, M.; Wargo, J.A.; Chen, J.Q.; Li, H.S.; Watowich, S.S.; Yang, Y.; et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barroso-Sousa, R.; Keenan, T.E.; Pernas, S.; Exman, P.; Jain, E.; Garrido-Castro, A.C.; Hughes, M.; Bychkovsky, B.; Umeton, R.; Files, J.L.; et al. Tumor Mutational Burden and PTEN Alterations as Molecular Correlates of Response to PD-1/L1 Blockade in Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 2565–2572. [Google Scholar] [CrossRef] [PubMed]
- Atefi, M.; Avramis, E.; Lassen, A.; Wong, D.J.; Robert, L.; Foulad, D.; Cerniglia, M.; Titz, B.; Chodon, T.; Graeber, T.G.; et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 3446–3457. [Google Scholar] [CrossRef] [Green Version]
- Keniry, M.; Parsons, R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene 2008, 27, 5477–5485. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Castagnoli, L.; Cancila, V.; Cordoba-Romero, S.L.; Faraci, S.; Talarico, G.; Belmonte, B.; Iorio, M.V.; Milani, M.; Volpari, T.; Chiodoni, C.; et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 2019, 38, 4047–4060. [Google Scholar] [CrossRef]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.H.; Kuntz, S.M.; Leibovich, B.C.; Dong, H.; Lohse, C.M.; Webster, W.S.; Sengupta, S.; Frank, I.; Parker, A.S.; Zincke, H.; et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res. 2006, 66, 3381–3385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hino, R.; Kabashima, K.; Kato, Y.; Yagi, H.; Nakamura, M.; Honjo, T.; Okazaki, T.; Tokura, Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 2010, 116, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wei, S.; Hurt, E.M.; Green, M.D.; Zhao, L.; Vatan, L.; Szeliga, W.; Herbst, R.; Harms, P.W.; Fecher, L.A.; et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Investig. 2018, 128, 805–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rugo, H.S.; Loi, S.; Adams, S.; Schmid, P.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Winer, E.P.; Kockx, M.M.; et al. PD-L1 Immunohistochemistry Assay Comparison in Atezolizumab Plus nab-Paclitaxel-Treated Advanced Triple-Negative Breast Cancer. J. Natl. Cancer Inst. 2021, 113, 1733–1743. [Google Scholar] [CrossRef]
- Büttner, R.; Gosney, J.R.; Skov, B.G.; Adam, J.; Motoi, N.; Bloom, K.J.; Dietel, M.; Longshore, J.W.; López-Ríos, F.; Penault-Llorca, F.; et al. Programmed Death-Ligand 1 Immunohistochemistry Testing: A Review of Analytical Assays and Clinical Implementation in Non-Small-Cell Lung Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 3867–3876. [Google Scholar] [CrossRef]
- Chae, Y.K.; Pan, A.; Davis, A.A.; Raparia, K.; Mohindra, N.A.; Matsangou, M.; Giles, F.J. Biomarkers for PD-1/PD-L1 Blockade Therapy in Non-Small-cell Lung Cancer: Is PD-L1 Expression a Good Marker for Patient Selection? Clin. Lung Cancer 2016, 17, 350–361. [Google Scholar] [CrossRef]
- Zouein, J.; Kesrouani, C.; Kourie, H.R. PD-L1 expression as a predictive biomarker for immune checkpoint inhibitors: Between a dream and a nightmare. Immunotherapy 2021, 13, 1053–1065. [Google Scholar] [CrossRef]
- Arora, S.; Velichinskii, R.; Lesh, R.W.; Ali, U.; Kubiak, M.; Bansal, P.; Borghaei, H.; Edelman, M.J.; Boumber, Y. Existing and Emerging Biomarkers for Immune Checkpoint Immunotherapy in Solid Tumors. Adv. Ther. 2019, 36, 2638–2678. [Google Scholar] [CrossRef]
- Hamada, T.; Soong, T.R.; Masugi, Y.; Kosumi, K.; Nowak, J.A.; da Silva, A.; Mu, X.J.; Twombly, T.S.; Koh, H.; Yang, J.; et al. TIME (Tumor Immunity in the MicroEnvironment) classification based on tumor CD274 (PD-L1) expression status and tumor-infiltrating lymphocytes in colorectal carcinomas. Oncoimmunology 2018, 7, e1442999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, E.M.; Al-Foheidi, M.E.; Al-Mansour, M.M.; Kazkaz, G.A. The prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancer: A meta-analysis. Breast Cancer Res. Treat. 2014, 148, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Molinero, L.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Diéras, V.; Iwata, H.; Barrios, C.H.; Nechaeva, M.; Nguyen-Duc, A.; et al. Atezolizumab and nab-Paclitaxel in Advanced Triple-Negative Breast Cancer: Biomarker Evaluation of the IMpassion130 Study. J. Natl. Cancer Inst. 2021, 113, 1005–1016. [Google Scholar] [CrossRef]
- Schmid, P.; Salgado, R.; Park, Y.H.; Muñoz-Couselo, E.; Kim, S.B.; Sohn, J.; Im, S.A.; Foukakis, T.; Kuemmel, S.; Dent, R.; et al. Pembrolizumab plus chemotherapy as neoadjuvant treatment of high-risk, early-stage triple-negative breast cancer: Results from the phase 1b open-label, multicohort KEYNOTE-173 study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 569–581. [Google Scholar] [CrossRef]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019, 4, e126908. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Voutsadakis, I.A. High Tumor Mutation Burden and Other Immunotherapy Response Predictors in Breast Cancers: Associations and Therapeutic Opportunities. Target. Oncol. 2020, 15, 127–138. [Google Scholar] [CrossRef]
- Subbiah, V.; Solit, D.B.; Chan, T.A.; Kurzrock, R. The FDA approval of pembrolizumab for adult and pediatric patients with tumor mutational burden (TMB) ≥10: A decision centered on empowering patients and their physicians. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Zhang, J.; Luo, P. Crosstalk Between the MSI Status and Tumor Microenvironment in Colorectal Cancer. Front. Immunol. 2020, 11, 2039. [Google Scholar] [CrossRef] [PubMed]
- Pellat, A.; Netter, J.; Perkins, G.; Cohen, R.; Coulet, F.; Parc, Y.; Svrcek, M.; Duval, A.; André, T. Lynch syndrome: What is new? Bull. Du Cancer 2019, 106, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Baretti, M.; Le, D.T. DNA mismatch repair in cancer. Pharmacol. Ther. 2018, 189, 45–62. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef]
- Horimoto, Y.; Thinzar Hlaing, M.; Saeki, H.; Kitano, S.; Nakai, K.; Sasaki, R.; Kurisaki-Arakawa, A.; Arakawa, A.; Otsuji, N.; Matsuoka, S.; et al. Microsatellite instability and mismatch repair protein expressions in lymphocyte-predominant breast cancer. Cancer Sci. 2020, 111, 2647–2654. [Google Scholar] [CrossRef]
- Xu, Y.; Fu, Y.; Zhu, B.; Wang, J.; Zhang, B. Predictive Biomarkers of Immune Checkpoint Inhibitors-Related Toxicities. Front. Immunol. 2020, 11, 2023. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Wu, L.; Li, X.; Zhang, X.; Zhang, G.; Xu, S.; Sun, S.; Jiao, S. Diversity of Dominant Peripheral T Cell Receptor Clone and Soluble Immune Checkpoint Proteins Associated With Clinical Outcomes Following Immune Checkpoint Inhibitor Treatment in Advanced Cancers. Front. Immunol. 2021, 12, 649343. [Google Scholar] [CrossRef]
- Kurata, K.; Kubo, M.; Kai, M.; Mori, H.; Kawaji, H.; Kaneshiro, K.; Yamada, M.; Nishimura, R.; Osako, T.; Arima, N.; et al. Microsatellite instability in Japanese female patients with triple-negative breast cancer. Breast Cancer 2020, 27, 490–498. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef]
- Zimmerli, D.; Brambillasca, C.S.; Talens, F.; Bhin, J.; Linstra, R.; Romanens, L.; Bhattacharya, A.; Joosten, S.E.P.; Da Silva, A.M.; Padrao, N.; et al. MYC promotes immune-suppression in triple-negative breast cancer via inhibition of interferon signaling. Nat. Commun. 2022, 13, 6579. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ma, D.; Zhao, S.; Suo, C.; Shi, J.; Xue, M.Z.; Ruan, M.; Wang, H.; Zhao, J.; Li, Q.; et al. Multi-Omics Profiling Reveals Distinct Microenvironment Characterization and Suggests Immune Escape Mechanisms of Triple-Negative Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5002–5014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, T.; Hiraki, M.; Jin, C.; Rajabi, H.; Tagde, A.; Alam, M.; Bouillez, A.; Hu, X.; Suzuki, Y.; Miyo, M.; et al. MUC1-C Induces PD-L1 and Immune Evasion in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Zhou, W.; Li, X.; Peng, F.; Yan, M.; Zhan, Y.; An, F.; Li, X.; Liu, Y.; Liu, Q.; et al. Nuclear Aurora kinase A triggers programmed death-ligand 1-mediated immune suppression by activating MYC transcription in triple-negative breast cancer. Cancer Commun. 2021, 41, 851–866. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, R.; Lin, Z.; Zhang, S.; Chen, Y.; Tang, J.; Hong, J.; Zhou, X.; Zong, Y.; Xu, Y.; et al. CDK7 inhibitor THZ1 enhances antiPD-1 therapy efficacy via the p38α/MYC/PD-L1 signaling in non-small cell lung cancer. J. Hematol. Oncol. 2020, 13, 99. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Ding, X.; Xu, S.; Zhu, B.; Jia, Q. Gene expression profiling identified TP53(Mut)PIK3CA(Wild) as a potential biomarker for patients with triple-negative breast cancer treated with immune checkpoint inhibitors. Oncol. Lett. 2020, 19, 2817–2824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Ye, Y.; Chan, L.C.; Li, Y.; Liang, K.; Lin, A.; Egranov, S.D.; Zhang, Y.; Xia, W.; Gong, J.; et al. Oncogenic lncRNA downregulates cancer cell antigen presentation and intrinsic tumor suppression. Nat. Immunol. 2019, 20, 835–851. [Google Scholar] [CrossRef]
- Sasaki, R.; Horimoto, Y.; Yanai, Y.; Kurisaki-Arakawa, A.; Arakawa, A.; Nakai, K.; Saito, M.; Saito, T. Molecular Characteristics of Lymphocyte-predominant Triple-negative Breast Cancer. Anticancer Res. 2021, 41, 2133–2140. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Wang, X.; Yang, Q. PPP2R2B downregulation is associated with immune evasion and predicts poor clinical outcomes in triple-negative breast cancer. Cancer Cell Int. 2021, 21, 13. [Google Scholar] [CrossRef]
- Bareche, Y.; Buisseret, L.; Gruosso, T.; Girard, E.; Venet, D.; Dupont, F.; Desmedt, C.; Larsimont, D.; Park, M.; Rothé, F.; et al. Unraveling Triple-Negative Breast Cancer Tumor Microenvironment Heterogeneity: Towards an Optimized Treatment Approach. J. Natl. Cancer Inst. 2020, 112, 708–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldominos, P.; Barbera-Mourelle, A.; Barreiro, O.; Huang, Y.; Wight, A.; Cho, J.W.; Zhao, X.; Estivill, G.; Adam, I.; Sanchez, X.; et al. Quiescent cancer cells resist T cell attack by forming an immunosuppressive niche. Cell 2022, 185, 1694–1708.e1619. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xu, J.; Jiang, C.; Zhang, J.; Sun, T. Predictive and Prognostic Role of Peripheral Blood T-Cell Subsets in Triple-Negative Breast Cancer. Front. Oncol. 2022, 12, 842705. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Middleton, G. The interplay of immunotherapy and chemotherapy: Harnessing potential synergies. Cancer Immunol. Res. 2015, 3, 436–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Adams, S.; Barrios, C.H.; Diéras, V.; Iwata, H.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Winer, E.P.; Patel, S.; et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 983–993. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Chang, J.Y.; Mehran, R.J.; Feng, L.; Verma, V.; Liao, Z.; Welsh, J.W.; Lin, S.H.; O’Reilly, M.S.; Jeter, M.D.; Balter, P.A.; et al. Stereotactic ablative radiotherapy for operable stage I non-small-cell lung cancer (revised STARS): Long-term results of a single-arm, prospective trial with prespecified comparison to surgery. Lancet Oncol. 2021, 22, 1448–1457. [Google Scholar] [CrossRef]
- Yeo, E.L.L.; Li, Y.Q.; Soo, K.C.; Wee, J.T.S.; Chua, M.L.K. Combinatorial strategies of radiotherapy and immunotherapy in nasopharyngeal carcinoma. Chin. Clin. Oncol. 2018, 7, 15. [Google Scholar] [CrossRef]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef]
- Demaria, S.; Kawashima, N.; Yang, A.M.; Devitt, M.L.; Babb, J.S.; Allison, J.P.; Formenti, S.C. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 728–734. [Google Scholar] [CrossRef]
- Song, H.N.; Jin, H.; Kim, J.H.; Ha, I.B.; Kang, K.M.; Choi, H.S.; Jeong, H.J.; Kim, M.Y.; Kim, H.J.; Jeong, B.K. Abscopal Effect of Radiotherapy Enhanced with Immune Checkpoint Inhibitors of Triple Negative Breast Cancer in 4T1 Mammary Carcinoma Model. Int. J. Mol. Sci. 2021, 22, 10476. [Google Scholar] [CrossRef] [PubMed]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.Y.; Barker, C.A.; Arnold, B.B.; Powell, S.N.; Hu, Z.I.; Gucalp, A.; Lebron-Zapata, L.; Wen, H.Y.; Kallman, C.; D’Agnolo, A.; et al. A phase 2 clinical trial assessing the efficacy and safety of pembrolizumab and radiotherapy in patients with metastatic triple-negative breast cancer. Cancer 2020, 126, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Leighl, N.B.; Redman, M.W.; Rizvi, N.; Hirsch, F.R.; Mack, P.C.; Schwartz, L.H.; Wade, J.L.; Irvin, W.J.; Reddy, S.C.; Crawford, J.; et al. Phase II study of durvalumab plus tremelimumab as therapy for patients with previously treated anti-PD-1/PD-L1 resistant stage IV squamous cell lung cancer (Lung-MAP substudy S1400F, NCT03373760). J. Immunother. Cancer 2021, 9, e002973. [Google Scholar] [CrossRef] [PubMed]
- Rupp, T.; Genest, L.; Babin, D.; Legrand, C.; Hunault, M.; Froget, G.; Castagné, V. Anti-CTLA-4 and anti-PD-1 immunotherapies repress tumor progression in preclinical breast and colon model with independent regulatory T cells response. Transl. Oncol. 2022, 20, 101405. [Google Scholar] [CrossRef]
- Joller, N.; Hafler, J.P.; Brynedal, B.; Kassam, N.; Spoerl, S.; Levin, S.D.; Sharpe, A.H.; Kuchroo, V.K. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 2011, 186, 1338–1342. [Google Scholar] [CrossRef] [Green Version]
- Sarhan, D.; Cichocki, F.; Zhang, B.; Yingst, A.; Spellman, S.R.; Cooley, S.; Verneris, M.R.; Blazar, B.R.; Miller, J.S. Adaptive NK Cells with Low TIGIT Expression Are Inherently Resistant to Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5696–5706. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, S.; Inozume, T.; Kawazu, M.; Ueno, T.; Nagasaki, J.; Tanji, E.; Honobe, A.; Ohnuma, T.; Kawamura, T.; Umeda, Y.; et al. TIGIT/CD155 axis mediates resistance to immunotherapy in patients with melanoma with the inflamed tumor microenvironment. J. Immunother. Cancer 2021, 9, e003134. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y.; Xun, X.; Wang, S.; Xiang, X.; Hu, S.; Cheng, Q.; Guo, J.; Li, Z.; Zhu, J. TIGIT Can Exert Immunosuppressive Effects on CD8+ T Cells by the CD155/TIGIT Signaling Pathway for Hepatocellular Carcinoma In Vitro. J. Immunother. 2020, 43, 236–243. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Sun, L.; Li, C.F.; Wang, Y.H.; Yao, J.; Li, H.; Yan, M.; Chang, W.C.; Hsu, J.M.; Cha, J.H.; et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 2021, 12, 832. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, J.W.; Lorentzen, C.L.; Martinenaite, E.; Ellebaek, E.; Donia, M.; Holmstroem, R.B.; Klausen, T.W.; Madsen, C.O.; Ahmed, S.M.; Weis-Banke, S.E.; et al. A phase 1/2 trial of an immune-modulatory vaccine against IDO/PD-L1 in combination with nivolumab in metastatic melanoma. Nat. Med. 2021, 27, 2212–2223. [Google Scholar] [CrossRef]
- Hong, D.S.; Schoffski, P.; Calvo, A.; Sarantopoulos, J.; Olza, M.O.D.; Carvajal, R.D.; Prawira, A.; Kyi, C.; Esaki, T.; Akerley, W.L.; et al. Phase I/II study of LAG525 ± spartalizumab (PDR001) in patients (pts) with advanced malignancies. J. Clin. Oncol. 2018, 36, 3012. [Google Scholar] [CrossRef]
- Spahn, J.; Peng, J.; Lorenzana, E.; Kan, D.; Hunsaker, T.; Segal, E.; Mautino, M.; Brincks, E.; Pirzkall, A.; Kelley, S.; et al. Improved anti-tumor immunity and efficacy upon combination of the IDO1 inhibitor GDC-0919 with anti-PD-l1 blockade versus anti-PD-l1 alone in preclinical tumor models. J. Immunother. Cancer 2015, 3, P303. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.H.; LoRusso, P.; Burris, H.; Gordon, M.; Bang, Y.J.; Hellmann, M.D.; Cervantes, A.; Ochoa de Olza, M.; Marabelle, A.; Hodi, F.S.; et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) Administered with PD-L1 Inhibitor (Atezolizumab) in Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3220–3228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Amé, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. BioEssays News Rev. Mol. Cell. Dev. Biol. 2004, 26, 882–893. [Google Scholar] [CrossRef]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef]
- Garufi, G.; Palazzo, A.; Paris, I.; Orlandi, A.; Cassano, A.; Tortora, G.; Scambia, G.; Bria, E.; Carbognin, L. Neoadjuvant therapy for triple-negative breast cancer: Potential predictive biomarkers of activity and efficacy of platinum chemotherapy, PARP- and immune-checkpoint-inhibitors. Expert Opin. Pharmacother. 2020, 21, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Labrie, M.; Li, A.; Creason, A.; Betts, C.; Keck, J.; Johnson, B.; Sivagnanam, S.; Boniface, C.; Ma, H.; Blucher, A.; et al. Multiomics analysis of serial PARP inhibitor treated metastatic TNBC inform on rational combination therapies. npj Precis. Oncol. 2021, 5, 92. [Google Scholar] [CrossRef] [PubMed]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.A.; Park, Y.H.; Delord, J.P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, E.S.; Witkiewicz, A.K. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 2017, 3, 39–55. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.M.; Goss, A.; Garber, J.E.; Torous, V.; Richardson, E.T.; Haviland, M.J.; Hacker, M.R.; Freeman, G.J.; Nalven, T.; Alexander, B.; et al. Retinoblastoma protein expression and its predictors in triple-negative breast cancer. npj Breast Cancer 2020, 6, 19. [Google Scholar] [CrossRef]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Tolba, M.F.; Omar, H.A. Immunotherapy, an evolving approach for the management of triple negative breast cancer: Converting non-responders to responders. Crit. Rev. Oncol. Hematol. 2018, 122, 202–207. [Google Scholar] [CrossRef]
- Maennling, A.E.; Tur, M.K.; Niebert, M.; Klockenbring, T.; Zeppernick, F.; Gattenlöhner, S.; Meinhold-Heerlein, I.; Hussain, A.F. Molecular Targeting Therapy against EGFR Family in Breast Cancer: Progress and Future Potentials. Cancers 2019, 11, 1826. [Google Scholar] [CrossRef]
- Changavi, A.A.; Shashikala, A.; Ramji, A.S. Epidermal Growth Factor Receptor Expression in Triple Negative and Nontriple Negative Breast Carcinomas. J. Lab. Physicians 2015, 7, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Hung, M.C.; Yamaguchi, H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar] [PubMed]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.W.; Kim, I.A.; et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 2014, 27, 1212–1222. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Conchas, G.A.; Rodriguez-Romo, L.; Hernandez-Barajas, D.; Gonzalez-Guerrero, J.F.; Rodriguez-Fernandez, I.A.; Verdines-Perez, A.; Templeton, A.J.; Ocana, A.; Seruga, B.; Tannock, I.F.; et al. Epidermal growth factor receptor overexpression and outcomes in early breast cancer: A systematic review and a meta-analysis. Cancer Treat. Rev. 2018, 62, 1–8. [Google Scholar] [CrossRef]
- Liang, H.; Liu, X.; Wang, M. Immunotherapy combined with epidermal growth factor receptor-tyrosine kinase inhibitors in non-small-cell lung cancer treatment. OncoTargets Ther. 2018, 11, 6189–6196. [Google Scholar] [CrossRef] [Green Version]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.; Zheng, Z.; Liu, J.Y.; Chen, Y.J.; Ding, J.; Hu, G.S.; Hu, Y.H.; Liu, S.; Luo, W.X.; Xia, N.S.; et al. Targeting Triple-Negative Breast Cancer with Combination Therapy of EGFR CAR T Cells and CDK7 Inhibition. Cancer Immunol. Res. 2021, 9, 707–722. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Y.; Huang, K.H.; Li, Y.; Fang, X.; An, L.; Wang, F.; Chen, Q.; Zhang, Y.; Shi, A.; et al. EGFR-specific CAR-T cells trigger cell lysis in EGFR-positive TNBC. Aging 2019, 11, 11054–11072. [Google Scholar] [CrossRef]
- Kowanetz, M.; Ferrara, N. Vascular endothelial growth factor signaling pathways: Therapeutic perspective. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 5018–5022. [Google Scholar] [CrossRef] [Green Version]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Yasuda, S.; Sho, M.; Yamato, I.; Yoshiji, H.; Wakatsuki, K.; Nishiwada, S.; Yagita, H.; Nakajima, Y. Simultaneous blockade of programmed death 1 and vascular endothelial growth factor receptor 2 (VEGFR2) induces synergistic anti-tumour effect in vivo. Clin. Exp. Immunol. 2013, 172, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhang, H.; Liang, J.; Li, K.; Zhu, W.; Fu, L.; Wang, F.; Zheng, X.; Shi, H.; Wu, S.; et al. Identification and characterization of alphavirus M1 as a selective oncolytic virus targeting ZAP-defective human cancers. Proc. Natl. Acad. Sci. USA 2014, 111, E4504–E4512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgeois-Daigneault, M.C.; Roy, D.G.; Aitken, A.S.; El Sayes, N.; Martin, N.T.; Varette, O.; Falls, T.; St-Germain, L.E.; Pelin, A.; Lichty, B.D.; et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018, 10, eaao1641. [Google Scholar] [CrossRef] [Green Version]
- Martin, N.T.; Roy, D.G.; Workenhe, S.T.; van den Wollenberg, D.J.M.; Hoeben, R.C.; Mossman, K.L.; Bell, J.C.; Bourgeois-Daigneault, M.C. Pre-surgical neoadjuvant oncolytic virotherapy confers protection against rechallenge in a murine model of breast cancer. Sci. Rep. 2019, 9, 1865. [Google Scholar] [CrossRef] [Green Version]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal Transduct Target Ther. 2022, 20, 62. [Google Scholar] [CrossRef]
- Al-Awadhi, A.; Lee Murray, J.; Ibrahim, N.K. Developing anti-HER2 vaccines: Breast cancer experience. Int. J. Cancer 2018, 143, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Cannon, B.A.; Castro-Sanchez, A.; Barragan-Carrillo, R.; de la Rosa Pacheco, S.; Platas, A.; Fonseca, A.; Vega, Y.; Bojorquez-Velazquez, K.; Bargallo-Rocha, J.E.; Mohar, A.; et al. Adherence to Adjuvant Tamoxifen in Mexican Young Women with Breast Cancer. Patient Prefer. Adherence 2021, 15, 1039–1049. [Google Scholar] [CrossRef]
- Barve, M.; Aaron, P.; Manning, L.; Bognar, E.; Wallraven, G.; Horvath, S.; Stanbery, L.; Nemunaitis, J. Pilot Study of Combination Gemogenovatucel-T (Vigil) and Durvalumab in Women With Relapsed BRCA-wt Triple-Negative Breast or Ovarian Cancer. Clin. Med. Insights Oncol. 2022, 16, 11795549221110501. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Tian, H.; Zhang, T.; Qin, S.; Huang, Z.; Zhou, L.; Shi, J.; Nice, E.C.; Xie, N.; Huang, C.; Shen, Z. Enhancing the therapeutic efficacy of nanoparticles for cancer treatment using versatile targeted strategies. J. Hematol. Oncol. 2022, 15, 132. [Google Scholar] [CrossRef]
- LaMotte-Mohs, R.; Shah, K.; Smith, D.; Gorlatov, S.; Ciccarone, V.; Tamura, J.; Li, H.; Rillema, J.; Licea, M.; He, L.; et al. Abstract 3217: MGD013, a bispecific PD-1 x LAG-3 Dual-Affinity Re-Targeting (DART®) protein with T-cell immunomodulatory activity for cancer treatment. Cancer Res. 2016, 76, 3217. [Google Scholar] [CrossRef] [Green Version]
- Catenacci, D.V.; Rosales, M.; Chung, H.C.; Yoon, H.H.; Shen, L.; Moehler, M.; Kang, Y.K. MAHOGANY: Margetuximab combination in HER2+ unresectable/metastatic gastric/gastroesophageal junction adenocarcinoma. Future Oncol. 2021, 17, 1155–1164. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Feng, L.; Huang, Y.; Wu, Y.; Xie, N. Mechanisms and Strategies to Overcome PD-1/PD-L1 Blockade Resistance in Triple-Negative Breast Cancer. Cancers 2023, 15, 104. https://doi.org/10.3390/cancers15010104
Chen X, Feng L, Huang Y, Wu Y, Xie N. Mechanisms and Strategies to Overcome PD-1/PD-L1 Blockade Resistance in Triple-Negative Breast Cancer. Cancers. 2023; 15(1):104. https://doi.org/10.3390/cancers15010104
Chicago/Turabian StyleChen, Xingyu, Lixiang Feng, Yujing Huang, Yi Wu, and Na Xie. 2023. "Mechanisms and Strategies to Overcome PD-1/PD-L1 Blockade Resistance in Triple-Negative Breast Cancer" Cancers 15, no. 1: 104. https://doi.org/10.3390/cancers15010104
APA StyleChen, X., Feng, L., Huang, Y., Wu, Y., & Xie, N. (2023). Mechanisms and Strategies to Overcome PD-1/PD-L1 Blockade Resistance in Triple-Negative Breast Cancer. Cancers, 15(1), 104. https://doi.org/10.3390/cancers15010104