
Epidemiologic, Genetic, Pathogenic, Metabolic, Epigenetic Aspects Involved in NASH-HCC: Current Therapeutic Strategies

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Epidemiology of NASH/HCC
2.1. Prevalence of NASH
2.2. Prevalence of HCC
2.3. Incidence of HCC in Patients with NASH
3. Risk Factors for NASH and HCC
3.1. Obesity Is Associated with NASH and HCC
3.2. Metabolic Syndrome Is a Risk Factor for NASH and HCC
3.3. Diabetes Increase the Risk of NASH and HCC
3.4. NAFLD Is the Main Risk Factor of NASH and HCC
4. Genetic Factors and Gene Expression Affecting Development of NASH and HCC
4.1. Genetic Risk Factors for NASH
4.2. Genetic Risk Factors for HCC
4.3. Genetic Instability and Altered Gene Expression Affecting NASH and HCC Diseases
4.3.1. Somatic Mutations in NASH and HCC [77,88]
4.3.2. Pathways Affected by Gene Mutations in a Mice NASH-HCC Model and Human NASH-HCCs
4.3.3. Dysregulated Gene Expression in NASH and HCC
4.3.4. Circulating miRNA Signature Associated with NASH and NASH-HCC
4.3.5. LncRNAs Involved in NASH and NASH-HCC
4.3.6. CircRNAs Expressed in NASH
5. Pathogenesis of NASH-Related HCC
5.1. Role of Lipotoxicity and Glucotoxicity in NASH and HCC Development
5.2. Oxidative Stress in NASH-HCC
5.3. Chronic Inflammation in NASH-HCC
5.4. Mitochondrial Dysfunction Plays a Key Role in the Transition from NASH to HCC
5.5. Dysregulation of Autophagy in NASH-Derived HCC
5.6. Immune Dysregulation in NASH-HCC
6. Metabolic Reprogramming in NASH and NASH-Related HCC
6.1. Alteration of Carbohydrate Metabolism in HSC
6.2. Carbohydrate Metabolism in HCC
Gluconeogenesis and Tricarboxylic Acid Cycle Are Reduced in HCC
6.3. Role of Lipid Metabolism in NASH-HCC
6.4. Cholesterol Plays an Important Role in the Transition from NASH to HCC
7. Epigenetic Modifications in NASH and HCC
7.1. DNA Methylation Patterns in NASH
7.2. Aberrant DNA Methylation in HCC
7.3. Histone Modifications Involved in HCC
8. Current Pharmacological Therapies and Emerging Therapies for NASH/HCC
8.1. Therapeutic Strategies for Preventing and Treating of NASH
8.1.1. Lifestyle Modification and Exercise
8.1.2. Vitamin E (Antioxidant)
8.1.3. Pharmacological Treatment Aimed at Reducing Steatosis and Glucose Levels in NASH
Thiazolidinediones
Liraglutide
Metformin
Obeticholic Acid
Statins
Omega-3 Polyunsaturated Fatty Acids (PUFAs)
8.1.4. Pentoxifylline (Anti-Inflammatory)
8.1.5. Antifibrotic Drugs
Cenicriviroc
Pirfenidone
8.2. Current Pharmacological Therapies and Treatment Options for NASH-HCC
8.2.1. First-Line Treatment for HCC
Bevacizumab Plus Atezolizumab
Sorafenib
Lenvatinib
8.2.2. Second-Line Treatment for HCC
Regorafenib
Cabozantinib
Ramucirumab
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gutierrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Pathophysiological Molecular Mechanisms of Obesity: A Link between MAFLD and NASH with Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 11629. [Google Scholar] [CrossRef] [PubMed]
- Starley, B.Q.; Calcagno, C.J.; Harrison, S.A. Nonalcoholic fatty liver disease and hepatocellular carcinoma: A weighty connection. Hepatology 2010, 51, 1820–1832. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, A.; Alt, Y.; Koch, S.; Nelles, C.; Duber, C.; Lang, H.; Otto, G.; Zimmermann, T.; Marquardt, J.U.; Galle, P.R.; et al. Treatment and survival of non-alcoholic steatohepatitis associated hepatocellular carcinoma. BMC Cancer 2015, 15, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62 (Suppl. S1), S47–S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braghini, M.R.; Lo Re, O.; Romito, I.; Fernandez-Barrena, M.G.; Barbaro, B.; Pomella, S.; Rota, R.; Vinciguerra, M.; Avila, M.A.; Alisi, A. Epigenetic remodelling in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 107. [Google Scholar] [CrossRef]
- Du, D.; Liu, C.; Qin, M.; Zhang, X.; Xi, T.; Yuan, S.; Hao, H.; Xiong, J. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm. Sin. B 2022, 12, 558–580. [Google Scholar] [CrossRef]
- Nakagawa, H.; Hayata, Y.; Kawamura, S.; Yamada, T.; Fujiwara, N.; Koike, K. Lipid Metabolic Reprogramming in Hepatocellular Carcinoma. Cancers 2018, 10, 447. [Google Scholar] [CrossRef] [Green Version]
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of NASH. Int. J. Mol. Sci. 2016, 17, 1575. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Kohjima, M.; Tanaka, M.; Goya, T.; Itoh, S.; Yoshizumi, T.; Mori, M.; Tsuda, M.; Takahashi, M.; Kurokawa, M.; et al. Metabolic Alteration in Hepatocellular Carcinoma: Mechanism of Lipid Accumulation in Well-Differentiated Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2021, 2021, 8813410. [Google Scholar] [CrossRef]
- Chan, L.L.; Chan, S.L. Novel Perspectives in Immune Checkpoint Inhibitors and the Management of Non-Alcoholic Steatohepatitis-Related Hepatocellular Carcinoma. Cancers 2022, 14, 1526. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Bohinc, B.N.; Diehl, A.M. Mechanisms of disease progression in NASH: New paradigms. Clin. Liver Dis. 2012, 16, 549–565. [Google Scholar] [CrossRef]
- Lazaridis, N.; Tsochatzis, E. Current and future treatment options in non-alcoholic steatohepatitis (NASH). Expert Rev. Gastroenterol. Hepatol. 2017, 11, 357–369. [Google Scholar] [CrossRef]
- Feng, M.Y.; Chan, L.L.; Chan, S.L. Drug Treatment for Advanced Hepatocellular Carcinoma: First-Line and Beyond. Curr. Oncol. 2022, 29, 5489–5507. [Google Scholar] [CrossRef]
- Falette Puisieux, M.; Pellat, A.; Assaf, A.; Ginestet, C.; Brezault, C.; Dhooge, M.; Soyer, P.; Coriat, R. Therapeutic Management of Advanced Hepatocellular Carcinoma: An Updated Review. Cancers 2022, 14, 2357. [Google Scholar] [CrossRef]
- Caldwell, S.H.; Ikura, Y.; Iezzoni, J.C.; Liu, Z. Has natural selection in human populations produced two types of metabolic syndrome (with and without fatty liver)? J. Gastroenterol. Hepatol. 2007, 22 (Suppl. S1), S11–S19. [Google Scholar] [CrossRef]
- Seyda Seydel, G.; Kucukoglu, O.; Altinbasv, A.; Demir, O.O.; Yilmaz, S.; Akkiz, H.; Otan, E.; Sowa, J.P.; Canbay, A. Economic growth leads to increase of obesity and associated hepatocellular carcinoma in developing countries. Ann. Hepatol. 2016, 15, 662–672. [Google Scholar]
- Younossi, Z.M.; Henry, L. Epidemiology of non-alcoholic fatty liver disease and hepatocellular carcinoma. JHEP Rep. 2021, 3, 100305. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Vuppalanchi, R.; Mladenovic, A.; Samala, N.; Gawrieh, S.; Newsome, P.N.; Chalasani, N. Prevalence of High-risk Nonalcoholic Steatohepatitis (NASH) in the United States: Results From NHANES 2017-2018. Clin. Gastroenterol. Hepatol. 2021, in press. [Google Scholar] [CrossRef]
- Pomenti, S.; Gandle, C.; Abu Sbeih, H.; Phipps, M.; Livanos, A.; Guo, A.; Yeh, J.; Burney, H.; Liu, H.; Dakhoul, L.; et al. Hepatocellular Carcinoma in Hispanic Patients: Trends and Outcomes in a Large United States Cohort. Hepatol. Commun. 2020, 4, 1708–1716. [Google Scholar] [CrossRef]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Wang, L.; Wang, Z.; Chen, S.; Ni, Y.; Jiang, D. Higher non-HDL-cholesterol to HDL-cholesterol ratio linked with increased nonalcoholic steatohepatitis. Lipids Health Dis. 2018, 17, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tampi, R.P.; Wong, V.W.; Wong, G.L.; Shu, S.S.; Chan, H.L.; Fung, J.; Stepanova, M.; Younossi, Z.M. Modelling the economic and clinical burden of non-alcoholic steatohepatitis in East Asia: Data from Hong Kong. Hepatol. Res. 2020, 50, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Chan, H.L.Y.; Chien, R.N.; Chuang, W.L.; Fung, J.; Goh, G.B.; Hu, T.H.; Huang, J.F.; Jang, B.K.; Jun, D.W.; et al. Modelling NAFLD disease burden in four Asian regions-2019–2030. Aliment. Pharmacol. Ther. 2020, 51, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, Y.; Wong, G.; Lee, E.I.; Akhtar, O.; Lopes, R.; Sumida, Y. Epidemiology of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in Japan: A focused literature review. JGH Open 2020, 4, 808–817. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Altekruse, S.F.; McGlynn, K.A.; Reichman, M.E. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J. Clin. Oncol. 2009, 27, 1485–1491. [Google Scholar] [CrossRef] [Green Version]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef]
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 2020, 72, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.H.; Cheng, Y.; Zhang, S.; Fan, J.; Gao, Q. Changing epidemiology of hepatocellular carcinoma in Asia. Liver Int. 2022, 42, 2029–2041. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.; Hallouch, O.; Chernyak, V.; Kamaya, A.; Sirlin, C.B. Epidemiology of hepatocellular carcinoma: Target population for surveillance and diagnosis. Abdom. Radiol. 2018, 43, 13–25. [Google Scholar] [CrossRef]
- Ha, J.; Chaudhri, A.; Avirineni, A.; Pan, J.J. Burden of hepatocellular carcinoma among hispanics in South Texas: A systematic review. Biomark. Res. 2017, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef]
- Kulik, L.; El-Serag, H.B. Epidemiology and Management of Hepatocellular Carcinoma. Gastroenterology 2019, 156, 477–491.e1. [Google Scholar] [CrossRef]
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar]
- Cholankeril, G.; Patel, R.; Khurana, S.; Satapathy, S.K. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Current knowledge and implications for management. World J. Hepatol. 2017, 9, 533–543. [Google Scholar] [CrossRef]
- Stras, W.; Malkowski, P.; Tronina, O. Hepatocellular carcinoma in patients with non-alcoholic steatohepatitis—Epidemiology, risk factors, clinical implications and treatment. Clin. Exp. Hepatol. 2020, 6, 170–175. [Google Scholar] [CrossRef]
- Ioannou, G.N. Epidemiology and risk-stratification of NAFLD-associated HCC. J. Hepatol. 2021, 75, 1476–1484. [Google Scholar] [CrossRef]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, B.; Stal, P.; Wahlin, S.; Bjorkstrom, N.K.; Hagstrom, H. Characteristics and outcome of hepatocellular carcinoma in patients with NAFLD without cirrhosis. Liver Int. 2019, 39, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.G.; Nguyen, P.P.; Dang, H.; Kumari, R.; Garcia, G.; Esquivel, C.O.; Nguyen, M.H. Temporal trends in disease presentation and survival of patients with hepatocellular carcinoma: A real-world experience from 1998 to 2015. Cancer 2018, 124, 2588–2598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asfari, M.M.; Talal Sarmini, M.; Alomari, M.; Lopez, R.; Dasarathy, S.; McCullough, A.J. The association of nonalcoholic steatohepatitis and hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2020, 32, 1566–1570. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 2019, 17, 748–755.e3. [Google Scholar] [CrossRef] [Green Version]
- Larsson, S.C.; Wolk, A. Overweight, obesity and risk of liver cancer: A meta-analysis of cohort studies. Br. J. Cancer. 2007, 97, 1005–1008. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.; Wang, J.; Yan, Z.; Luo, J. Excess body weight and the risk of primary liver cancer: An updated meta-analysis of prospective studies. Eur. J. Cancer. 2012, 48, 2137–2145. [Google Scholar] [CrossRef]
- Hassan, M.M.; Abdel-Wahab, R.; Kaseb, A.; Shalaby, A.; Phan, A.T.; El-Serag, H.B.; Hawk, E.; Morris, J.; Singh Raghav, K.P.; Lee, J.S.; et al. Obesity Early in Adulthood Increases Risk but Does Not Affect Outcomes of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Das, A.; Majumder, K.; Arora, N.; Mayo, H.G.; Singh, P.P.; Beg, M.S.; Singh, S. Obesity is Independently Associated With Increased Risk of Hepatocellular Cancer-related Mortality: A Systematic Review and Meta-Analysis. Am. J. Clin. Oncol. 2018, 41, 874–881. [Google Scholar] [CrossRef]
- Jinjuvadia, R.; Patel, S.; Liangpunsakul, S. The association between metabolic syndrome and hepatocellular carcinoma: Systemic review and meta-analysis. J. Clin. Gastroenterol. 2014, 48, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Wang, J.; Gao, Y.; Yang, F.; Huang, W. Metabolic syndrome and liver-related events: A systematic review and meta-analysis. BMC Endocr. Disord. 2019, 19, 40. [Google Scholar] [CrossRef] [Green Version]
- Makarova-Rusher, O.V.; Altekruse, S.F.; McNeel, T.S.; Ulahannan, S.; Duffy, A.G.; Graubard, B.I.; Greten, T.F.; McGlynn, K.A. Population attributable fractions of risk factors for hepatocellular carcinoma in the United States. Cancer 2016, 122, 1757–1765. [Google Scholar] [CrossRef]
- Bian, H.; Zhu, X.; Xia, M.; Yan, H.; Chang, X.; Hu, X.; Pan, B.; Guo, W.; Li, X.; Gao, X. Impact of Type 2 Diabetes on Nonalcoholic Steatohepatitis and Advanced Fibrosis in Patients with Nonalcoholic Fatty Liver Disease. Endocr. Pract. 2020, 26, 444–453. [Google Scholar] [CrossRef]
- Wang, Q.; You, H.; Ou, X.; Zhao, X.; Sun, Y.; Wang, M.; Wang, P.; Wang, Y.; Duan, W.; Wang, X.; et al. Non-obese histologically confirmed NASH patients with abnormal liver biochemistry have more advanced fibrosis. Hepatol. Int. 2019, 13, 766–776. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Hampel, H.; Javadi, F. The association between diabetes and hepatocellular carcinoma: A systematic review of epidemiologic evidence. Clin. Gastroenterol. Hepatol. 2006, 4, 369–380. [Google Scholar] [CrossRef]
- Wang, P.; Kang, D.; Cao, W.; Wang, Y.; Liu, Z. Diabetes mellitus and risk of hepatocellular carcinoma: A systematic review and meta-analysis. Diabetes Metab. Res. Rev. 2012, 28, 109–122. [Google Scholar] [CrossRef]
- Argo, C.K.; Northup, P.G.; Al-Osaimi, A.M.; Caldwell, S.H. Systematic review of risk factors for fibrosis progression in non-alcoholic steatohepatitis. J. Hepatol. 2009, 51, 371–379. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.; Sada, Y.H.; El-Serag, H.B.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Temporal trends of nonalcoholic fatty liver disease-related hepatocellular carcinoma in the veteran affairs population. Clin. Gastroenterol. Hepatol. 2015, 13, 594–601.e1. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Stepanova, M.; Negro, F.; Hallaji, S.; Younossi, Y.; Lam, B.; Srishord, M. Nonalcoholic fatty liver disease in lean individuals in the United States. Medicine 2012, 91, 319–327. [Google Scholar] [CrossRef]
- Pingitore, P.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Indiveri, C.; Pujia, A.; Montalcini, T.; Hedfalk, K.; Romeo, S. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim. Biophys. Acta 2014, 1841, 574–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirazzi, C.; Valenti, L.; Motta, B.M.; Pingitore, P.; Hedfalk, K.; Mancina, R.M.; Burza, M.A.; Indiveri, C.; Ferro, Y.; Montalcini, T.; et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 2014, 23, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenknecht, L.E.; Palmer, N.D.; Bowden, D.W.; Rotter, J.I.; Norris, J.M.; Ziegler, J.; Chen, Y.D.; Haffner, S.; Scherzinger, A.; Langefeld, C.D. Association of PNPLA3 with non-alcoholic fatty liver disease in a minority cohort: The Insulin Resistance Atherosclerosis Family Study. Liver Int. 2011, 31, 412–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef]
- Zain, S.M.; Mohamed, R.; Mahadeva, S.; Cheah, P.L.; Rampal, S.; Basu, R.C.; Mohamed, Z. A multi-ethnic study of a PNPLA3 gene variant and its association with disease severity in non-alcoholic fatty liver disease. Hum. Genet. 2012, 131, 1145–1152. [Google Scholar] [CrossRef] [Green Version]
- Tai, C.M.; Huang, C.K.; Tu, H.P.; Hwang, J.C.; Chang, C.Y.; Yu, M.L. PNPLA3 genotype increases susceptibility of nonalcoholic steatohepatitis among obese patients with nonalcoholic fatty liver disease. Surg. Obes. Relat. Dis. 2015, 11, 888–894. [Google Scholar] [CrossRef]
- Linden, D.; Ahnmark, A.; Pingitore, P.; Ciociola, E.; Ahlstedt, I.; Andreasson, A.C.; Sasidharan, K.; Madeyski-Bengtson, K.; Zurek, M.; Mancina, R.M.; et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol. Metab. 2019, 22, 49–61. [Google Scholar] [CrossRef]
- Pang, J.; Xu, W.; Zhang, X.; Wong, G.L.; Chan, A.W.; Chan, H.Y.; Tse, C.H.; Shu, S.S.; Choi, P.C.; Chan, H.L.; et al. Significant positive association of endotoxemia with histological severity in 237 patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2017, 46, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Tai, C.M.; Huang, C.K.; Tu, H.P.; Hwang, J.C.; Yeh, M.L.; Huang, C.F.; Huang, J.F.; Dai, C.Y.; Chuang, W.L.; Yu, M.L. Interactions of a PPARGC1A Variant and a PNPLA3 Variant Affect Nonalcoholic Steatohepatitis in Severely Obese Taiwanese Patients. Medicine 2016, 95, e3120. [Google Scholar] [CrossRef]
- Zhang, R.N.; Shen, F.; Pan, Q.; Cao, H.X.; Chen, G.Y.; Fan, J.G. PPARGC1A rs8192678 G>A polymorphism affects the severity of hepatic histological features and nonalcoholic steatohepatitis in patients with nonalcoholic fatty liver disease. World J. Gastroenterol. 2021, 27, 3863–3876. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, J.; Sheng, H.; You, N.; Chen, J.; Mi, X.; Yang, W.; Zang, S.; Shi, J.; Chinese NAFLD Clinical Research Network (CNAFLD CRN). Haptoglobin 2-2 Genotype is Associated with More Advanced Disease in Subjects with Non-Alcoholic Steatohepatitis: A Retrospective Study. Adv. Ther. 2019, 36, 880–895. [Google Scholar] [CrossRef]
- Namikawa, C.; Shu-Ping, Z.; Vyselaar, J.R.; Nozaki, Y.; Nemoto, Y.; Ono, M.; Akisawa, N.; Saibara, T.; Hiroi, M.; Enzan, H.; et al. Polymorphisms of microsomal triglyceride transfer protein gene and manganese superoxide dismutase gene in non-alcoholic steatohepatitis. J. Hepatol. 2004, 40, 781–786. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef]
- Pirola, C.J.; Garaycoechea, M.; Flichman, D.; Arrese, M.; San Martino, J.; Gazzi, C.; Castano, G.O.; Sookoian, S. Splice variant rs72613567 prevents worst histologic outcomes in patients with nonalcoholic fatty liver disease. J. Lipid. Res. 2019, 60, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Botello-Manilla, A.E.; Chavez-Tapia, N.C.; Uribe, M.; Nuno-Lambarri, N. Genetics and epigenetics purpose in nonalcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 2020, 14, 733–748. [Google Scholar] [CrossRef]
- Yang, J.; Trepo, E.; Nahon, P.; Cao, Q.; Moreno, C.; Letouze, E.; Imbeaud, S.; Bayard, Q.; Gustot, T.; Deviere, J.; et al. A 17-Beta-Hydroxysteroid Dehydrogenase 13 Variant Protects From Hepatocellular Carcinoma Development in Alcoholic Liver Disease. Hepatology 2019, 70, 231–240. [Google Scholar] [CrossRef]
- Bianco, C.; Jamialahmadi, O.; Pelusi, S.; Baselli, G.; Dongiovanni, P.; Zanoni, I.; Santoro, L.; Maier, S.; Liguori, A.; Meroni, M.; et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J. Hepatol. 2021, 74, 775–782. [Google Scholar] [CrossRef]
- Gellert-Kristensen, H.; Richardson, T.G.; Davey Smith, G.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Stender, S. Combined Effect of PNPLA3, TM6SF2, and HSD17B13 Variants on Risk of Cirrhosis and Hepatocellular Carcinoma in the General Population. Hepatology 2020, 72, 845–856. [Google Scholar] [CrossRef]
- Yang, J.; Trepo, E.; Nahon, P.; Cao, Q.; Moreno, C.; Letouze, E.; Imbeaud, S.; Gustot, T.; Deviere, J.; Debette, S.; et al. PNPLA3 and TM6SF2 variants as risk factors of hepatocellular carcinoma across various etiologies and severity of underlying liver diseases. Int. J. Cancer 2019, 144, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.; Allison, M.E.; Alexander, G.J.; Piguet, A.C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, M.; Stokes, C.S.; Romeo, S.; Lammert, F. HCC and liver disease risks in homozygous PNPLA3 p.I148M carriers approach monogenic inheritance. J. Hepatol. 2015, 62, 980–981. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.G.; Manjunath, H.; Yopp, A.C.; Beg, M.S.; Marrero, J.A.; Gopal, P.; Waljee, A.K. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Q.; Qian, B.X.; Yin, W.L.; Wang, F.M.; Han, T. Association between the HFE C282Y.; H63D Polymorphisms and the Risks of Non-Alcoholic Fatty Liver Disease, Liver Cirrhosis and Hepatocellular Carcinoma: An Updated Systematic Review and Meta-Analysis of 5,758 Cases and 14,741 Controls. PLoS ONE 2016, 11, e0163423. [Google Scholar] [CrossRef] [Green Version]
- Donati, B.; Dongiovanni, P.; Romeo, S.; Meroni, M.; McCain, M.; Miele, L.; Petta, S.; Maier, S.; Rosso, C.; De Luca, L.; et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 2017, 7, 4492. [Google Scholar] [CrossRef] [Green Version]
- Valenti, L.; Motta, B.M.; Alisi, A.; Sartorelli, R.; Buonaiuto, G.; Dongiovanni, P.; Rametta, R.; Pelusi, S.; Fargion, S.; Nobili, V. LPIN1 rs13412852 polymorphism in pediatric nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 588–593. [Google Scholar] [CrossRef]
- Choudhary, N.S.; Duseja, A. Genetic and epigenetic disease modifiers: Non-alcoholic fatty liver disease (NAFLD) and alcoholic liver disease (ALD). Transl. Gastroenterol. Hepatol. 2021, 6, 2. [Google Scholar] [CrossRef]
- Del Campo, J.A.; Gallego-Duran, R.; Gallego, P.; Grande, L. Genetic and Epigenetic Regulation in Nonalcoholic Fatty Liver Disease (NAFLD). Int. J. Mol. Sci. 2018, 19, 911. [Google Scholar] [CrossRef] [Green Version]
- Juanola, O.; Martinez-Lopez, S.; Frances, R.; Gomez-Hurtado, I. Non-Alcoholic Fatty Liver Disease: Metabolic, Genetic, Epigenetic and Environmental Risk Factors. Int. J. Environ. Res. Public Health 2021, 18, 5227. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Shima, T.; Mizuno, M.; Mitsumoto, Y.; Umemura, A.; Kanbara, Y.; Tanaka, S.; Sumida, Y.; Yasui, K.; Takahashi, M.; et al. Risk estimation model for nonalcoholic fatty liver disease in the Japanese using multiple genetic markers. PLoS ONE 2018, 13, e0185490. [Google Scholar] [CrossRef]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjaerg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef]
- Nelson, J.E.; Bhattacharya, R.; Lindor, K.D.; Chalasani, N.; Raaka, S.; Heathcote, E.J.; Miskovsky, E.; Shaffer, E.; Rulyak, S.J.; Kowdley, K.V. HFE C282Y mutations are associated with advanced hepatic fibrosis in Caucasians with nonalcoholic steatohepatitis. Hepatology 2007, 46, 723–729. [Google Scholar] [CrossRef]
- Desterke, C.; Chiappini, F. Lipid Related Genes Altered in NASH Connect Inflammation in Liver Pathogenesis Progression to HCC. A Canonical Pathway. Int. J. Mol. Sci. 2019, 20, 5594. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.F.; Zhao, Q.; Hu, H.; Liao, J.Z.; Lin, J.S.; Xia, C.; Chang, Y.; Liu, J.; Guo, A.Y.; He, X.X. The ASH1-miR-375-YWHAZ Signaling Axis Regulates Tumor Properties in Hepatocellular Carcinoma. Mol. Ther. Nucleic Acids 2018, 11, 538–553. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [Green Version]
- Pinyol, R.; Torrecilla, S.; Wang, H.; Montironi, C.; Pique-Gili, M.; Torres-Martin, M.; Wei-Qiang, L.; Willoughby, C.E.; Ramadori, P.; Andreu-Oller, C.; et al. Corrigendum to ‘Molecular characterisation of hepatocellular carcinoma in patients with non-alcoholic steatohepatitis’. J. Hepatol. 2021, 75, 865–878, Corrigendum to 2021, 75, 1515. [Google Scholar] [CrossRef]
- Yao, S.; Johnson, C.; Hu, Q.; Yan, L.; Liu, B.; Ambrosone, C.B.; Wang, J.; Liu, S. Differences in somatic mutation landscape of hepatocellular carcinoma in Asian American and European American populations. Oncotarget 2016, 7, 40491–40499. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.Q.; Teoh, N.; Xu, L.; Pok, S.; Li, X.; Chu, E.S.H.; Chiu, J.; Dong, L.; Arfianti, E.; Haigh, W.G.; et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat. Commun. 2018, 9, 4490. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.B.; Barlos, D.; Hauser, C.J. Free cholesterol alters lipid raft structure and function regulating neutrophil Ca2+ entry and respiratory burst: Correlations with calcium channel raft trafficking. J. Immunol. 2007, 178, 5253–5261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteith, G.R.; Davis, F.M.; Roberts-Thomson, S.J. Calcium channels and pumps in cancer: Changes and consequences. J. Biol. Chem. 2012, 287, 31666–31673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Yang, W.; Tian, Y.; Zeng, X.; Zhou, J.; Mok, M.T.S.; Tang, W.; Feng, Y.; Xu, L.; Chan, A.W.H.; et al. An inflammatory-CCRK circuitry drives mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat. Commun. 2018, 9, 5214. [Google Scholar] [CrossRef] [PubMed]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity During Development of Steatohepatitis in Mice. Gastroenterology 2015, 149, 468–480.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, M.; Chen, X.; Xu, H.; Wenping; Fang, M.; Xu, Y. Hepatocyte-specific deletion of Brg1 alleviates methionine-and-choline-deficient diet (MCD) induced non-alcoholic steatohepatitis in mice. Biochem. Biophys. Res. Commun. 2018, 503, 344–351. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, H.; Bu, Q.; Wei, S.; Li, L.; Zhou, J.; Zhou, S.; Su, W.; Liu, M.; Liu, Z.; et al. Role of XBP1 in regulating the progression of non-alcoholic steatohepatitis. J. Hepatol. 2022, 77, 312–325. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Clarke, J.D.; Novak, P.; Lake, A.D.; Shipkova, P.; Aranibar, N.; Robertson, D.; Severson, P.L.; Reily, M.D.; Futscher, B.W.; Lehman-McKeeman, L.D.; et al. Characterization of hepatocellular carcinoma related genes and metabolites in human nonalcoholic fatty liver disease. Dig. Dis. Sci. 2014, 59, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Dechassa, M.L.; Tryndyak, V.; de Conti, A.; Xiao, W.; Beland, F.A.; Pogribny, I.P. Identification of chromatin-accessible domains in non-alcoholic steatohepatitis-derived hepatocellular carcinoma. Mol. Carcinog. 2018, 57, 978–987. [Google Scholar] [CrossRef]
- Kang, M.; Kim, J.; An, H.T.; Ko, J. Human leucine zipper protein promotes hepatic steatosis via induction of apolipoprotein A-IV. FASEB J. 2017, 31, 2548–2561. [Google Scholar] [CrossRef] [Green Version]
- Longerich, T.; Haller, M.T.; Mogler, C.; Aulmann, S.; Lohmann, V.; Schirmacher, P.; Brand, K. Annexin A2 as a differential diagnostic marker of hepatocellular tumors. Pathol. Res. Pract. 2011, 207, 8–14. [Google Scholar] [CrossRef]
- Zhang, H.J.; Yao, D.F.; Yao, M.; Huang, H.; Wu, W.; Yan, M.J.; Yan, X.D.; Chen, J. Expression characteristics and diagnostic value of annexin A2 in hepatocellular carcinoma. World J. Gastroenterol. 2012, 18, 5897–5904. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Rustichelli, A.; Dongiovanni, P. Nutrition and Genetics in NAFLD: The Perfect Binomium. Int. J. Mol. Sci. 2020, 21, 2986. [Google Scholar] [CrossRef]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef] [Green Version]
- Sulaiman, S.A.; Muhsin, N.I.A.; Jamal, R. Regulatory Non-coding RNAs Network in Non-alcoholic Fatty Liver Disease. Front. Physiol. 2019, 10, 279. [Google Scholar] [CrossRef]
- Qiang, J.; Tao, Y.F.; Bao, J.W.; Chen, J.; Li, H.X.; He, J.; Xu, P. High Fat Diet-Induced miR-122 Regulates Lipid Metabolism and Fat Deposition in Genetically Improved Farmed Tilapia (GIFT.; Oreochromis niloticus) Liver. Front. Physiol. 2018, 9, 1422. [Google Scholar] [CrossRef] [Green Version]
- Long, J.K.; Dai, W.; Zheng, Y.W.; Zhao, S.P. miR-122 promotes hepatic lipogenesis via inhibiting the LKB1/AMPK pathway by targeting Sirt1 in non-alcoholic fatty liver disease. Mol. Med. 2019, 25, 26. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.H.; Yang, Y.L.; Wang, F.S. The Role of miR-29a in the Regulation, Function, and Signaling of Liver Fibrosis. Int. J. Mol. Sci. 2018, 19, 1889. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.L.; Kuo, H.C.; Wang, F.S.; Huang, Y.H. MicroRNA-29a Disrupts DNMT3b to Ameliorate Diet-Induced Non-Alcoholic Steatohepatitis in Mice. Int. J. Mol. Sci. 2019, 20, 1499. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.X.; Gao, M.; Li, C.Z.; Yu, C.Z.; Yan, H.; Peng, C.; Li, Y.; Li, C.G.; Ma, Z.L.; Zhao, Y.; et al. Dicer1/miR-29/HMGCR axis contributes to hepatic free cholesterol accumulation in mouse non-alcoholic steatohepatitis. Acta. Pharmacol. Sin. 2017, 38, 660–671. [Google Scholar] [CrossRef]
- Loyer, X.; Paradis, V.; Henique, C.; Vion, A.C.; Colnot, N.; Guerin, C.L.; Devue, C.; On, S.; Scetbun, J.; Romain, M.; et al. Liver microRNA-21 is overexpressed in non-alcoholic steatohepatitis and contributes to the disease in experimental models by inhibiting PPARalpha expression. Gut 2016, 65, 1882–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braza-Boils, A.; Mari-Alexandre, J.; Molina, P.; Arnau, M.A.; Barcelo-Molina, M.; Domingo, D.; Girbes, J.; Giner, J.; Martinez-Dolz, L.; Zorio, E. Deregulated hepatic microRNAs underlie the association between non-alcoholic fatty liver disease and coronary artery disease. Liver Int. 2016, 36, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Formichi, C.; Nigi, L.; Grieco, G.E.; Maccora, C.; Fignani, D.; Brusco, N.; Licata, G.; Sebastiani, G.; Dotta, F. Non-Coding RNAs: Novel Players in Insulin Resistance and Related Diseases. Int. J. Mol. Sci. 2021, 22, 7716. [Google Scholar] [CrossRef] [PubMed]
- Markovic, J.; Sharma, A.D.; Balakrishnan, A. MicroRNA-221: A Fine Tuner and Potential Biomarker of Chronic Liver Injury. Cells 2020, 9, 1767. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Starlard-Davenport, A.; Tryndyak, V.P.; Han, T.; Ross, S.A.; Rusyn, I.; Beland, F.A. Difference in expression of hepatic microRNAs miR-29c, miR-34a, miR-155, and miR-200b is associated with strain-specific susceptibility to dietary nonalcoholic steatohepatitis in mice. Lab. Investig. 2010, 90, 1437–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estep, M.; Armistead, D.; Hossain, N.; Elarainy, H.; Goodman, Z.; Baranova, A.; Chandhoke, V.; Younossi, Z.M. Differential expression of miRNAs in the visceral adipose tissue of patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2010, 32, 487–497. [Google Scholar] [CrossRef]
- Pirola, C.J.; Fernandez Gianotti, T.; Castano, G.O.; Mallardi, P.; San Martino, J.; Mora Gonzalez Lopez Ledesma, M.; Flichman, D.; Mirshahi, F.; Sanyal, A.J.; Sookoian, S. Circulating microRNA signature in non-alcoholic fatty liver disease: From serum non-coding RNAs to liver histology and disease pathogenesis. Gut 2015, 64, 800–812. [Google Scholar] [CrossRef] [Green Version]
- Khalifa, O.; Errafii, K.; Al-Akl, N.S.; Arredouani, A. Noncoding RNAs in Nonalcoholic Fatty Liver Disease: Potential Diagnosis and Prognosis Biomarkers. Dis. Markers 2020, 2020, 8822859. [Google Scholar] [CrossRef]
- Lopez-Riera, M.; Conde, I.; Quintas, G.; Pedrola, L.; Zaragoza, A.; Perez-Rojas, J.; Salcedo, M.; Benlloch, S.; Castell, J.V.; Jover, R. Non-invasive prediction of NAFLD severity: A comprehensive, independent validation of previously postulated serum microRNA biomarkers. Sci. Rep. 2018, 8, 10606. [Google Scholar] [CrossRef] [Green Version]
- Vulf, M.; Shunkina, D.; Komar, A.; Bograya, M.; Zatolokin, P.; Kirienkova, E.; Gazatova, N.; Kozlov, I.; Litvinova, L. Analysis of miRNAs Profiles in Serum of Patients With Steatosis and Steatohepatitis. Front. Cell Dev. Biol. 2021, 9, 736677. [Google Scholar] [CrossRef]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334.e7. [Google Scholar] [CrossRef] [Green Version]
- Sui, C.; Zhang, L.; Hu, Y. MicroRNAlet7a inhibition inhibits LPSinduced inflammatory injury of chondrocytes by targeting IL6R. Mol. Med. Rep. 2019, 20, 2633–2640. [Google Scholar]
- Furuta, K.; Guo, Q.; Pavelko, K.D.; Lee, J.H.; Robertson, K.D.; Nakao, Y.; Melek, J.; Shah, V.H.; Hirsova, P.; Ibrahim, S.H. Lipid-induced endothelial vascular cell adhesion molecule 1 promotes nonalcoholic steatohepatitis pathogenesis. J. Clin. Investig. 2021, 131, e143690. [Google Scholar] [CrossRef]
- Wang, B.; Majumder, S.; Nuovo, G.; Kutay, H.; Volinia, S.; Patel, T.; Schmittgen, T.D.; Croce, C.; Ghoshal, K.; Jacob, S.T. Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet in C57BL/6 mice. Hepatology 2009, 50, 1152–1161. [Google Scholar] [CrossRef]
- Gori, M.; Arciello, M.; Balsano, C. MicroRNAs in nonalcoholic fatty liver disease: Novel biomarkers and prognostic tools during the transition from steatosis to hepatocarcinoma. Biomed. Res. Int. 2014, 2014, 741465. [Google Scholar] [CrossRef] [Green Version]
- Tessitore, A.; Cicciarelli, G.; Del Vecchio, F.; Gaggiano, A.; Verzella, D.; Fischietti, M.; Mastroiaco, V.; Vetuschi, A.; Sferra, R.; Barnabei, R.; et al. MicroRNA expression analysis in high fat diet-induced NAFLD-NASH-HCC progression: Study on C57BL/6J mice. BMC Cancer 2016, 16, 3. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.L.; Jia, Y.L.; Chen, L.; Zeng, Q.; Zhou, J.N.; Fu, C.J.; Chen, H.X.; Yuan, H.F.; Li, Z.W.; Shi, L.; et al. Hepatocellular carcinoma-associated mesenchymal stem cells promote hepatocarcinoma progression: Role of the S100A4-miR155-SOCS1-MMP9 axis. Hepatology 2013, 57, 2274–2286. [Google Scholar] [CrossRef]
- Tsai, W.C.; Hsu, S.D.; Hsu, C.S.; Lai, T.C.; Chen, S.J.; Shen, R.; Huang, Y.; Chen, H.C.; Lee, C.H.; Tsai, T.F.; et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Investig. 2012, 122, 2884–2897. [Google Scholar] [CrossRef] [Green Version]
- Li, C.W.; Chiu, Y.K.; Chen, B.S. Investigating Pathogenic and Hepatocarcinogenic Mechanisms from Normal Liver to HCC by Constructing Genetic and Epigenetic Networks via Big Genetic and Epigenetic Data Mining and Genome-Wide NGS Data Identification. Dis. Markers 2018, 2018, 8635329. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Mani, R.S.; Ateeq, B.; Dhanasekaran, S.M.; Asangani, I.A.; Prensner, J.R.; Kim, J.H.; Brenner, J.C.; Jing, X.; Cao, X.; et al. Coordinated regulation of polycomb group complexes through microRNAs in cancer. Cancer Cell 2011, 20, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasovska, B.; Rensen, S.S.; Marsman, G.; Shiri-Sverdlov, R.; Withoff, S.; Kuipers, F.; Wijmenga, C.; van de Sluis, B.; Fu, J. Long Non-Coding RNAs Involved in Progression of Non-Alcoholic Fatty Liver Disease to Steatohepatitis. Cells 2021, 10, 1883. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, J.; Zhang, K.; Feng, B.; Wang, R.; Chen, L. Progress and Prospects of Long Noncoding RNAs (lncRNAs) in Hepatocellular Carcinoma. Cell Physiol. Biochem. 2015, 36, 423–434. [Google Scholar] [CrossRef]
- Leti, F.; Legendre, C.; Still, C.D.; Chu, X.; Petrick, A.; Gerhard, G.S.; DiStefano, J.K. Altered expression of MALAT1 lncRNA in nonalcoholic steatohepatitis fibrosis regulates CXCL5 in hepatic stellate cells. Transl. Res. 2017, 190, 25–39.e21. [Google Scholar] [CrossRef]
- DiStefano, J.K.; Gerhard, G.S. Long Noncoding RNAs and Human Liver Disease. Annu. Rev. Pathol. 2022, 17, 1–21. [Google Scholar] [CrossRef]
- Atanasovska, B.; Rensen, S.S.; van der Sijde, M.R.; Marsman, G.; Kumar, V.; Jonkers, I.; Withoff, S.; Shiri-Sverdlov, R.; Greve, J.W.M.; Faber, K.N.; et al. A liver-specific long noncoding RNA with a role in cell viability is elevated in human nonalcoholic steatohepatitis. Hepatology 2017, 66, 794–808. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yang, Z.; Trottier, J.; Barbier, O.; Wang, L. Long noncoding RNA MEG3 induces cholestatic liver injury by interaction with PTBP1 to facilitate shp mRNA decay. Hepatology 2017, 65, 604–615. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Feng, C.Y.; Xiang, Z.; Chen, Y.P.; Li, Y.M. CircRNA expression pattern and circRNA-miRNA-mRNA network in the pathogenesis of nonalcoholic steatohepatitis. Oncotarget 2016, 7, 66455–66467. [Google Scholar] [CrossRef] [Green Version]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Ramai, D.; Facciorusso, A.; Vigandt, E.; Schaf, B.; Saadedeen, W.; Chauhan, A.; di Nunzio, S.; Shah, A.; Giacomelli, L.; Sacco, R. Progressive Liver Fibrosis in Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 3401. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Toulis, K.A.; Goulis, D.G.; Zavos, C.; Kountouras, J. Serum total adiponectin in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Metabolism 2011, 60, 313–326. [Google Scholar] [CrossRef]
- Longo, M.; Paolini, E.; Meroni, M.; Dongiovanni, P. Remodeling of Mitochondrial Plasticity. The Key Switch from NAFLD/NASH to HCC. Int. J. Mol. Sci. 2021, 22, 4173. [Google Scholar] [CrossRef]
- Leclercq, I.A.; Farrell, G.C.; Schriemer, R.; Robertson, G.R. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J. Hepatol. 2002, 37, 206–213. [Google Scholar] [CrossRef]
- Stefanou, N.; Papanikolaou, V.; Furukawa, Y.; Nakamura, Y.; Tsezou, A. Leptin as a critical regulator of hepatocellular carcinoma development through modulation of human telomerase reverse transcriptase. BMC Cancer 2010, 10, 442. [Google Scholar] [CrossRef] [Green Version]
- Kitade, M.; Yoshiji, H.; Kojima, H.; Ikenaka, Y.; Noguchi, R.; Kaji, K.; Yoshii, J.; Yanase, K.; Namisaki, T.; Asada, K.; et al. Leptin-mediated neovascularization is a prerequisite for progression of nonalcoholic steatohepatitis in rats. Hepatology 2006, 44, 983–991. [Google Scholar] [CrossRef]
- Bachelder, R.E.; Wendt, M.A.; Mercurio, A.M. Vascular endothelial growth factor promotes breast carcinoma invasion in an autocrine manner by regulating the chemokine receptor CXCR4. Cancer Res. 2002, 62, 7203–7206. [Google Scholar]
- Nardo, A.D.; Grun, N.G.; Zeyda, M.; Dumanic, M.; Oberhuber, G.; Rivelles, E.; Helbich, T.H.; Markgraf, D.F.; Roden, M.; Claudel, T.; et al. Impact of osteopontin on the development of non-alcoholic liver disease and related hepatocellular carcinoma. Liver Int. 2020, 40, 1620–1633. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajesh, Y.; Sarkar, D. Molecular Mechanisms Regulating Obesity-Associated Hepatocellular Carcinoma. Cancers 2020, 12, 1290. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhang, Z.; Zhang, P.; Yu, M.; Yang, T. Role of canonical Hedgehog signaling pathway in liver. Int. J. Biol. Sci. 2018, 14, 1636–1644. [Google Scholar] [CrossRef]
- Ye, J.; Li, T.S.; Xu, G.; Zhao, Y.M.; Zhang, N.P.; Fan, J.; Wu, J. JCAD Promotes Progression of Nonalcoholic Steatohepatitis to Liver Cancer by Inhibiting LATS2 Kinase Activity. Cancer Res. 2017, 77, 5287–5300. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Martin, R.C.; Shi, X.; Pandit, H.; Yu, Y.; Liu, X.; Guo, W.; Tan, M.; Bai, O.; Meng, X.; et al. Lack of FGF21 promotes NASH-HCC transition via hepatocyte-TLR4-IL-17A signaling. Theranostics 2020, 10, 9923–9936. [Google Scholar] [CrossRef]
- Takakura, K.; Oikawa, T.; Nakano, M.; Saeki, C.; Torisu, Y.; Kajihara, M.; Saruta, M. Recent Insights into the Multiple Pathways Driving Non-alcoholic Steatohepatitis-Derived Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 762. [Google Scholar] [CrossRef] [Green Version]
- Uchida, D.; Takaki, A.; Adachi, T.; Okada, H. Beneficial and Paradoxical Roles of Anti-Oxidative Nutritional Support for Non-Alcoholic Fatty Liver Disease. Nutrients 2018, 10, 977. [Google Scholar] [CrossRef] [Green Version]
- Page, J.M.; Harrison, S.A. NASH and HCC. Clin. Liver Dis. 2009, 13, 631–647. [Google Scholar] [CrossRef]
- Aubert, J.; Begriche, K.; Knockaert, L.; Robin, M.A.; Fromenty, B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: Mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 630–637. [Google Scholar] [CrossRef]
- Sodum, N.; Kumar, G.; Bojja, S.L.; Kumar, N.; Rao, C.M. Epigenetics in NAFLD/NASH: Targets and therapy. Pharmacol. Res. 2021, 167, 105484. [Google Scholar] [CrossRef]
- Campisano, S.; La Colla, A.; Echarte, S.M.; Chisari, A.N. Interplay between early-life malnutrition, epigenetic modulation of the immune function and liver diseases. Nutr. Res. Rev. 2019, 32, 128–145. [Google Scholar] [CrossRef]
- Dewdney, B.; Roberts, A.; Qiao, L.; George, J.; Hebbard, L. A Sweet Connection? Fructose’s Role in Hepatocellular Carcinoma. Biomolecules 2020, 10, 496. [Google Scholar] [CrossRef] [Green Version]
- Dongiovanni, P.; Meroni, M.; Longo, M.; Fargion, S.; Fracanzani, A.L. Genetics, Immunity and Nutrition Boost the Switching from NASH to HCC. Biomedicines 2021, 9, 1524. [Google Scholar] [CrossRef]
- Wegermann, K.; Hyun, J.; Diehl, A.M. Molecular Mechanisms Linking Nonalcoholic Steatohepatitis to Cancer. Clin. Liver Dis. 2021, 17, 6–10. [Google Scholar] [CrossRef]
- Lequoy, M.; Gigante, E.; Couty, J.P.; Desbois-Mouthon, C. Hepatocellular carcinoma in the context of non-alcoholic steatohepatitis (NASH): Recent advances in the pathogenic mechanisms. Horm. Mol. Biol. Clin. Investig. 2020, 41, 20190044. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Inaba, Y.; Watanabe, H.; Matsukawa, T.; Matsumoto, M.; Inoue, H. Nicotinic alpha-7 acetylcholine receptor deficiency exacerbates hepatic inflammation and fibrosis in a mouse model of non-alcoholic steatohepatitis. J. Diabetes Investig. 2019, 10, 659–666. [Google Scholar] [CrossRef] [Green Version]
- van der Windt, D.J.; Sud, V.; Zhang, H.; Varley, P.R.; Goswami, J.; Yazdani, H.O.; Tohme, S.; Loughran, P.; O’Doherty, R.M.; Minervini, M.I.; et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 2018, 68, 1347–1360. [Google Scholar] [CrossRef] [Green Version]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate Immunity and Inflammation in NAFLD/NASH. Dig. Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, L.; Mittenbuhler, M.J.; Vesting, A.J.; Ostermann, A.L.; Wunderlich, C.M.; Wunderlich, F.T. Obesity-Induced TNFalpha and IL-6 Signaling: The Missing Link between Obesity and Inflammation-Driven Liver and Colorectal Cancers. Cancers 2018, 11, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsova, P.; Weng, P.; Salim, W.; Bronk, S.F.; Griffith, T.S.; Ibrahim, S.H.; Gores, G.J. TRAIL Deletion Prevents Liver, but Not Adipose Tissue, Inflammation during Murine Diet-Induced Obesity. Hepatol. Commun. 2017, 1, 648–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular Pathogenesis of Nonalcoholic Steatohepatitis- (NASH-) Related Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 8543763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Targher, G.; Bertolini, L.; Rodella, S.; Lippi, G.; Franchini, M.; Zoppini, G.; Muggeo, M.; Day, C.P. NASH predicts plasma inflammatory biomarkers independently of visceral fat in men. Obesity 2008, 16, 1394–1399. [Google Scholar] [CrossRef]
- Hirsova, P.; Bamidele, A.O.; Wang, H.; Povero, D.; Revelo, X.S. Emerging Roles of T Cells in the Pathogenesis of Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. Front. Endocrinol. 2021, 12, 760860. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e1. [Google Scholar] [CrossRef] [Green Version]
- Kanda, T.; Goto, T.; Hirotsu, Y.; Masuzaki, R.; Moriyama, M.; Omata, M. Molecular Mechanisms: Connections between Nonalcoholic Fatty Liver Disease, Steatohepatitis and Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 1525. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- Sinton, M.C.; Hay, D.C.; Drake, A.J. Metabolic control of gene transcription in non-alcoholic fatty liver disease: The role of the epigenome. Clin. Epigenet. 2019, 11, 104. [Google Scholar] [CrossRef] [Green Version]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef]
- Krishnasamy, Y.; Gooz, M.; Li, L.; Lemasters, J.J.; Zhong, Z. Role of mitochondrial depolarization and disrupted mitochondrial homeostasis in non-alcoholic steatohepatitis and fibrosis in mice. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 190–204. [Google Scholar]
- Rautou, P.E.; Cazals-Hatem, D.; Feldmann, G.; Mansouri, A.; Grodet, A.; Barge, S.; Martinot-Peignoux, M.; Duces, A.; Bieche, I.; Lebrec, D.; et al. Changes in autophagic response in patients with chronic hepatitis C virus infection. Am. J. Pathol. 2011, 178, 2708–2715. [Google Scholar] [CrossRef] [Green Version]
- Piccinin, E.; Villani, G.; Moschetta, A. Metabolic aspects in NAFLD.; NASH and hepatocellular carcinoma: The role of PGC1 coactivators. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 160–174. [Google Scholar] [CrossRef]
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Ou, L.; Twaddel, W.; Fang, H.B.; Vafai, S.B.; Vazquez, F.; Puigserver, P.; Boros, L.; et al. PGC1alpha promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 2011, 71, 6888–6898. [Google Scholar] [CrossRef]
- Peng, K.Y.; Watt, M.J.; Rensen, S.; Greve, J.W.; Huynh, K.; Jayawardana, K.S.; Meikle, P.J.; Meex, R.C.R. Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J. Lipid. Res. 2018, 59, 1977–1986. [Google Scholar] [CrossRef] [Green Version]
- Apostolopoulou, M.; Gordillo, R.; Koliaki, C.; Gancheva, S.; Jelenik, T.; De Filippo, E.; Herder, C.; Markgraf, D.; Jankowiak, F.; Esposito, I.; et al. Specific Hepatic Sphingolipids Relate to Insulin Resistance, Oxidative Stress, and Inflammation in Nonalcoholic Steatohepatitis. Diabetes Care 2018, 41, 1235–1243. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Arima, T.; Hada, H.; Fukushima, M.; Watanabe, J.; Nagashima, H. Human hepatocellular carcinoma is associated with quantitative and qualitative changes in glycolipids. Liver 1985, 5, 226–235. [Google Scholar] [CrossRef]
- Le, T.H.; Caldwell, S.H.; Redick, J.A.; Sheppard, B.L.; Davis, C.A.; Arseneau, K.O.; Iezzoni, J.C.; Hespenheide, E.E.; Al-Osaimi, A.; Peterson, T.C. The zonal distribution of megamitochondria with crystalline inclusions in nonalcoholic steatohepatitis. Hepatology 2004, 39, 1423–1429. [Google Scholar] [CrossRef]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Bockowska, S.B.; Lebensztejn, D.M. Pediatric non-alcoholic steatohepatitis: The first report on the ultrastructure of hepatocyte mitochondria. World J. Gastroenterol. 2014, 20, 4335–4340. [Google Scholar] [CrossRef] [PubMed]
- Onal, G.; Kutlu, O.; Gozuacik, D.; Dokmeci Emre, S. Lipid Droplets in Health and Disease. Lipids Health Dis. 2017, 16, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yang, P.; Zhao, L.; Chen, Y.; Zhang, X.; Zeng, S.; Wei, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; et al. CD36 plays a negative role in the regulation of lipophagy in hepatocytes through an AMPK-dependent pathway. J. Lipid Res. 2019, 60, 844–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udoh, U.S.; Rajan, P.K.; Nakafuku, Y.; Finley, R.; Sanabria, J.R. Cell Autophagy in NASH and NASH-Related Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 7734. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [Green Version]
- Inokuchi-Shimizu, S.; Park, E.J.; Roh, Y.S.; Yang, L.; Zhang, B.; Song, J.; Liang, S.; Pimienta, M.; Taniguchi, K.; Wu, X.; et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J. Clin. Investig. 2014, 124, 3566–3578. [Google Scholar] [CrossRef]
- Kazankov, K.; Jorgensen, S.M.D.; Thomsen, K.L.; Moller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Gronbaek, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Kitade, H.; Chen, G.; Ni, Y.; Ota, T. Nonalcoholic Fatty Liver Disease and Insulin Resistance: New Insights and Potential New Treatments. Nutrients 2017, 9, 387. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Wu, Z.; Xu, Y.; Liu, Y.; Liu, W.; Wang, T.; Li, C.; Zhang, C.; Yi, F.; Gao, L.; et al. Increased Tim-3 expression alleviates liver injury by regulating macrophage activation in MCD-induced NASH mice. Cell. Mol. Immunol. 2019, 16, 878–886. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat. Med. 2019, 25, 641–655. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, K.; Abe, M.; Tada, F.; Tokumoto, Y.; Chen, S.; Miyake, T.; Furukawa, S.; Matsuura, B.; Hiasa, Y.; Onji, M. Blockade of B-cell-activating factor signaling enhances hepatic steatosis induced by a high-fat diet and improves insulin sensitivity. Lab. Investig. 2013, 93, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M. Impaired Response to Immunotherapy in Non-Alcoholic Steatohepatitis-Related Hepatocellular Carcinoma? Liver Cancer 2021, 10, 289–295. [Google Scholar] [CrossRef]
- Soderberg, C.; Marmur, J.; Eckes, K.; Glaumann, H.; Sallberg, M.; Frelin, L.; Rosenberg, P.; Stal, P.; Hultcrantz, R. Microvesicular fat, inter cellular adhesion molecule-1 and regulatory T-lymphocytes are of importance for the inflammatory process in livers with non-alcoholic steatohepatitis. APMIS 2011, 119, 412–420. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, H.; Yao, Y.; Zhang, X.; Guan, Y.; Zheng, F. CD4(+) T cell activation and inflammation in NASH-related fibrosis. Front. Immunol. 2022, 13, 967410. [Google Scholar] [CrossRef]
- Luo, X.Y.; Takahara, T.; Kawai, K.; Fujino, M.; Sugiyama, T.; Tsuneyama, K.; Tsukada, K.; Nakae, S.; Zhong, L.; Li, X.K. IFN-gamma deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G891–G899. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, H.; Wang, Y.; Brown, Z.J.; Xia, Y.; Huang, Z.; Shen, C.; Hu, Z.; Beane, J.; Ansa-Addo, E.A.; et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 1271–1283. [Google Scholar] [CrossRef]
- Syn, W.K.; Oo, Y.H.; Pereira, T.A.; Karaca, G.F.; Jung, Y.; Omenetti, A.; Witek, R.P.; Choi, S.S.; Guy, C.D.; Fearing, C.M.; et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 2010, 51, 1998–2007. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, J.; Kumar, J.M.; Arindkar, S.; Das, B.; Pramod, U.; Juyal, R.C.; Majumdar, S.S.; Nagarajan, P. Role of immunodeficient animal models in the development of fructose induced NAFLD. J. Nutr. Biochem. 2014, 25, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Satriano, L.; Lewinska, M.; Rodrigues, P.M.; Banales, J.M.; Andersen, J.B. Metabolic rearrangements in primary liver cancers: Cause and consequences. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 748–766. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Mendez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial Adaptation in Nonalcoholic Fatty Liver Disease. Novel Mechanisms and Treatment Strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.E.; Cardenas, B.I.; Farran, N.; Fernandez, M. Metabolic Reprogramming of Liver Fibrosis. Cells 2021, 10, 3604. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Wang, S.; Friedman, S.L. The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells. Cell Metab. 2021, 33, 242–257. [Google Scholar] [CrossRef]
- Chen, Y.; Choi, S.S.; Michelotti, G.A.; Chan, I.S.; Swiderska-Syn, M.; Karaca, G.F.; Xie, G.; Moylan, C.A.; Garibaldi, F.; Premont, R.; et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 2012, 143, 1319–1329.e11. [Google Scholar] [CrossRef] [Green Version]
- Geschwind, J.F.; Georgiades, C.S.; Ko, Y.H.; Pedersen, P.L. Recently elucidated energy catabolism pathways provide opportunities for novel treatments in hepatocellular carcinoma. Expert Rev. Anticancer Ther. 2004, 4, 449–457. [Google Scholar] [CrossRef]
- Wilson, J.E. Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J. Exp. Biol. 2003, 206, 2049–2057. [Google Scholar] [CrossRef] [Green Version]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Puszyk, W.M.; Trinh, T.L.; Chapple, S.J.; Liu, C. Linking metabolism and epigenetic regulation in development of hepatocellular carcinoma. Lab. Investig. 2013, 93, 983–990. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yang, S.; Chen, J.; Su, Z. Unraveling the Regulation of Hepatic Gluconeogenesis. Front. Endocrinol. 2018, 9, 802. [Google Scholar] [CrossRef] [Green Version]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Li, J.; Guo, Z.; Sun, L.; Juan, C.; Zhou, Y.; Gu, H.; Yu, Y.; Hu, Q.; Kan, Q.; et al. Overexpression of Pyruvate Dehydrogenase E1alpha Subunit Inhibits Warburg Effect and Induces Cell Apoptosis Through Mitochondria-Mediated Pathway in Hepatocellular Carcinoma. Oncol. Res. 2019, 27, 407–414. [Google Scholar] [CrossRef]
- Tian, G.Y.; Zang, S.F.; Wang, L.; Luo, Y.; Shi, J.P.; Lou, G.Q. Isocitrate Dehydrogenase 2 Suppresses the Invasion of Hepatocellular Carcinoma Cells via Matrix Metalloproteinase 9. Cell. Physiol. Biochem. 2015, 37, 2405–2414. [Google Scholar] [CrossRef]
- Eisenberg, S.; Levy, R.I. Lipoprotein metabolism. Adv. Lipid Res. 1975, 13, 1–89. [Google Scholar]
- Castillo-Leon, E.; Connelly, M.A.; Konomi, J.V.; Caltharp, S.; Cleeton, R.; Vos, M.B. Increased atherogenic lipoprotein profile in children with non-alcoholic steatohepatitis. Pediatr. Obes. 2020, 15, e12648. [Google Scholar] [CrossRef]
- Uehara, K.; Sostre-Colon, J.; Gavin, M.; Santoleri, D.; Leonard, K.A.; Jacobs, R.L.; Titchenell, P.M. Activation of Liver mTORC1 Protects Against NASH via Dual Regulation of VLDL-TAG Secretion and De Novo Lipogenesis. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1625–1647. [Google Scholar] [CrossRef]
- Agosti, P.; Sabba, C.; Mazzocca, A. Emerging metabolic risk factors in hepatocellular carcinoma and their influence on the liver microenvironment. Biochim. Biophys. Acta Mol. Basis. Dis. 2018, 1864, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial Oxidative Stress and Antioxidants Balance in Fatty Liver Disease. Hepatol. Commun. 2018, 2, 1425–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, N.; Nakagawa, H.; Enooku, K.; Kudo, Y.; Hayata, Y.; Nakatsuka, T.; Tanaka, Y.; Tateishi, R.; Hikiba, Y.; Misumi, K.; et al. CPT2 downregulation adapts HCC to lipid-rich environment and promotes carcinogenesis via acylcarnitine accumulation in obesity. Gut 2018, 67, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Bellanti, F.; Tamborra, R.; Rollo, T.; Capitanio, N.; Romano, A.D.; Sastre, J.; Vendemiale, G.; Altomare, E. Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut 2008, 57, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef]
- Liu, X.; Liang, Y.; Song, R.; Yang, G.; Han, J.; Lan, Y.; Pan, S.; Zhu, M.; Liu, Y.; Wang, Y.; et al. Long non-coding RNA NEAT1-modulated abnormal lipolysis via ATGL drives hepatocellular carcinoma proliferation. Mol. Cancer 2018, 17, 90. [Google Scholar] [CrossRef]
- Zhu, W.; Zhao, Y.; Zhou, J.; Wang, X.; Pan, Q.; Zhang, N.; Wang, L.; Wang, M.; Zhan, D.; Liu, Z.; et al. Monoacylglycerol lipase promotes progression of hepatocellular carcinoma via NF-kappaB-mediated epithelial-mesenchymal transition. J. Hematol. Oncol. 2016, 9, 127. [Google Scholar] [CrossRef]
- Nath, A.; Li, I.; Roberts, L.R.; Chan, C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci. Rep. 2015, 5, 14752. [Google Scholar] [CrossRef] [Green Version]
- Horn, P.; Newsome, P.N. Emerging therapeutic targets for NASH: Key innovations at the preclinical level. Expert Opin. Ther. Targets 2020, 24, 175–186. [Google Scholar] [CrossRef]
- Lally, J.S.V.; Ghoshal, S.; DePeralta, D.K.; Moaven, O.; Wei, L.; Masia, R.; Erstad, D.J.; Fujiwara, N.; Leong, V.; Houde, V.P.; et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019, 29, 174–182.e5. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.K.F.; Lau, E.Y.T.; Leung, D.H.W.; Lo, J.; Ho, N.P.Y.; Cheng, L.K.W.; Ma, S.; Lin, C.H.; Copland, J.A.; Ding, J.; et al. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J. Hepatol. 2017, 67, 979–990. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.B.; Jang, K.; Jun, D.W.; Lee, B.H.; Shin, K.J. Expression of liver X receptor correlates with intrahepatic inflammation and fibrosis in patients with nonalcoholic fatty liver disease. Dig. Dis. Sci. 2014, 59, 2975–2982. [Google Scholar] [CrossRef]
- Wolfe, A.; Thomas, A.; Edwards, G.; Jaseja, R.; Guo, G.L.; Apte, U. Increased activation of the Wnt/beta-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J. Pharmacol. Exp. Ther. 2011, 338, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Giudetti, A.M.; De Domenico, S.; Ragusa, A.; Lunetti, P.; Gaballo, A.; Franck, J.; Simeone, P.; Nicolardi, G.; De Nuccio, F.; Santino, A.; et al. A specific lipid metabolic profile is associated with the epithelial mesenchymal transition program. Biochim. Biophys. Acta Mol. Cell Biol Lipids 2019, 1864, 344–357. [Google Scholar] [CrossRef]
- Buechler, C.; Aslanidis, C. Role of lipids in pathophysiology, diagnosis and therapy of hepatocellular carcinoma. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158658. [Google Scholar] [CrossRef]
- Mannisto, V.T.; Simonen, M.; Hyysalo, J.; Soininen, P.; Kangas, A.J.; Kaminska, D.; Matte, A.K.; Venesmaa, S.; Kakela, P.; Karja, V.; et al. Ketone body production is differentially altered in steatosis and non-alcoholic steatohepatitis in obese humans. Liver Int. 2015, 35, 1853–1861. [Google Scholar] [CrossRef]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef]
- Ichimura, M.; Kawase, M.; Masuzumi, M.; Sakaki, M.; Nagata, Y.; Tanaka, K.; Suruga, K.; Tamaru, S.; Kato, S.; Tsuneyama, K.; et al. High-fat and high-cholesterol diet rapidly induces non-alcoholic steatohepatitis with advanced fibrosis in Sprague-Dawley rats. Hepatol. Res. 2015, 45, 458–469. [Google Scholar] [CrossRef]
- Long, J.; Zhang, C.J.; Zhu, N.; Du, K.; Yin, Y.F.; Tan, X.; Liao, D.F.; Qin, L. Lipid metabolism and carcinogenesis, cancer development. Am. J. Cancer Res. 2018, 8, 778–791. [Google Scholar]
- Michiel, D.F.; Oppenheim, J.J. Cytokines as positive and negative regulators of tumor promotion and progression. Semin. Cancer Biol. 1992, 3, 3–15. [Google Scholar] [PubMed]
- Eggens, I.; Ekstrom, T.J.; Aberg, F. Studies on the biosynthesis of polyisoprenols, cholesterol and ubiquinone in highly differentiated human hepatomas. J. Exp. Pathol. 1990, 71, 219–232. [Google Scholar]
- Pirola, C.J.; Sookoian, S. Epigenetics factors in nonalcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 2022, 16, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Iuculano, F.; Pallini, G.; Fargion, S.; Fracanzani, A.L. Nutrients, Genetic Factors, and Their Interaction in Non-Alcoholic Fatty Liver Disease and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 8761. [Google Scholar] [CrossRef] [PubMed]
- Kovacheva, V.P.; Mellott, T.J.; Davison, J.M.; Wagner, N.; Lopez-Coviella, I.; Schnitzler, A.C.; Blusztajn, J.K. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J. Biol. Chem. 2007, 282, 31777–31788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Wong, V.W.; Chan, H.L.; Cheng, A.S. Epigenetic regulation of hepatocellular carcinoma in non-alcoholic fatty liver disease. Semin. Cancer Biol. 2013, 23, 471–482. [Google Scholar] [CrossRef]
- Pirola, C.J.; Gianotti, T.F.; Burgueno, A.L.; Rey-Funes, M.; Loidl, C.F.; Mallardi, P.; Martino, J.S.; Castano, G.O.; Sookoian, S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2013, 62, 1356–1363. [Google Scholar] [CrossRef]
- Raza, S.; Rajak, S.; Anjum, B.; Sinha, R.A. Molecular links between non-alcoholic fatty liver disease and hepatocellular carcinoma. Hepatoma Res. 2019, 5, 42. [Google Scholar] [CrossRef] [Green Version]
- Asif, S.; Morrow, N.M.; Mulvihill, E.E.; Kim, K.H. Understanding Dietary Intervention-Mediated Epigenetic Modifications in Metabolic Diseases. Front. Genet. 2020, 11, 590369. [Google Scholar] [CrossRef]
- Kirchner, H.; Sinha, I.; Gao, H.; Ruby, M.A.; Schonke, M.; Lindvall, J.M.; Barres, R.; Krook, A.; Naslund, E.; Dahlman-Wright, K.; et al. Altered DNA methylation of glycolytic and lipogenic genes in liver from obese and type 2 diabetic patients. Mol. Metab. 2016, 5, 171–183. [Google Scholar] [CrossRef]
- Hardy, T.; Mann, D.A. Epigenetics in liver disease: From biology to therapeutics. Gut 2016, 65, 1895–1905. [Google Scholar] [CrossRef] [Green Version]
- Hardy, T.; Zeybel, M.; Day, C.P.; Dipper, C.; Masson, S.; McPherson, S.; Henderson, E.; Tiniakos, D.; White, S.; French, J.; et al. Plasma DNA methylation: A potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut 2017, 66, 1321–1328. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.L.; Mann, D.A.; Borthwick, L.A. Epigenetic reprogramming in liver fibrosis and cancer. Adv. Drug Deliv. Rev. 2017, 121, 124–132. [Google Scholar] [CrossRef]
- Mann, J.; Chu, D.C.; Maxwell, A.; Oakley, F.; Zhu, N.L.; Tsukamoto, H.; Mann, D.A. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 2010, 138, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Kim, J.; Kwon, J.S.; Sandhu, J.; Tontonoz, P.; Lee, S.K.; Lee, S.; Lee, J.W. Critical Roles of the Histone Methyltransferase MLL4/KMT2D in Murine Hepatic Steatosis Directed by ABL1 and PPARgamma2. Cell Rep. 2016, 17, 1671–1682. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhang, R.; Shen, F.; Yang, R.; Zhou, D.; Cao, H.; Chen, G.; Pan, Q.; Fan, J. Altered DNA Methylation Sites in Peripheral Blood Leukocytes from Patients with Simple Steatosis and Nonalcoholic Steatohepatitis (NASH). Med. Sci. Monit. 2018, 24, 6946–6967. [Google Scholar] [CrossRef]
- Borowa-Mazgaj, B.; de Conti, A.; Tryndyak, V.; Steward, C.R.; Jimenez, L.; Melnyk, S.; Seneshaw, M.; Mirshahi, F.; Rusyn, I.; Beland, F.A.; et al. Gene Expression and DNA Methylation Alterations in the Glycine N-Methyltransferase Gene in Diet-Induced Nonalcoholic Fatty Liver Disease-Associated Carcinogenesis. Toxicol. Sci. 2019, 170, 273–282. [Google Scholar] [CrossRef]
- Zheng, Y.; Tang, L.; Chen, G.; Liu, Z. Comprehensive Bioinformatics Analysis of Key Methyltransferases and Demethylases for Histone Lysines in Hepatocellular Carcinoma. Technol. Cancer Res. Treat. 2020, 19, 1533033820983284. [Google Scholar] [CrossRef]
- Qian, Y.; Li, Y.; Zheng, C.; Lu, T.; Sun, R.; Mao, Y.; Yu, S.; Fan, H.; Zhang, Z. High methylation levels of histone H3 lysine 9 associated with activation of hypoxia-inducible factor 1alpha (HIF-1alpha) predict patients’ worse prognosis in human hepatocellular carcinomas. Cancer Genet. 2020, 245, 17–26. [Google Scholar] [CrossRef]
- Fan, Z.; Li, L.; Li, M.; Zhang, X.; Hao, C.; Yu, L.; Zeng, S.; Xu, H.; Fang, M.; Shen, A.; et al. The histone methyltransferase Suv39h2 contributes to nonalcoholic steatohepatitis in mice. Hepatology 2017, 65, 1904–1919. [Google Scholar] [CrossRef] [Green Version]
- de Conti, A.; Dreval, K.; Tryndyak, V.; Orisakwe, O.E.; Ross, S.A.; Beland, F.A.; Pogribny, I.P. Inhibition of the Cell Death Pathway in Nonalcoholic Steatohepatitis (NASH)-Related Hepatocarcinogenesis Is Associated with Histone H4 lysine 16 Deacetylation. Mol. Cancer Res. 2017, 15, 1163–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Niu, J.; Wang, X.; Zhang, Z.S.; Yang, R.H.; Yao, X.; Liu, F.Y.; Li, W.Q.; Pei, S.H.; Sun, H.; et al. P300-dependent acetylation of histone H3 is required for epidermal growth factor receptor-mediated high-mobility group protein A2 transcription in hepatocellular carcinoma. Cancer Sci. 2021, 112, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Tian, S.; Li, P. Histone Acetyltransferase 1 Promotes Cell Proliferation and Induces Cisplatin Resistance in Hepatocellular Carcinoma. Oncol. Res. 2017, 25, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, H.; Pan, H.; Yang, Y.; Huang, G.; Yang, Y.; Zhou, W.P.; Pan, Z.Y. The histone acetyltransferase hMOF suppresses hepatocellular carcinoma growth. Biochem. Biophys. Res. Commun. 2014, 452, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Pote, N.; Cros, J.; Laouirem, S.; Raffenne, J.; Negrao, M.; Albuquerque, M.; Bedossa, P.; Godinho Ferreira, M.; Ait Si Ali, S.; Fior, R.; et al. The histone acetyltransferase hMOF promotes vascular invasion in hepatocellular carcinoma. Liver Int. 2020, 40, 956–967. [Google Scholar] [CrossRef]
- Shen, Z.T.; Chen, Y.; Huang, G.C.; Zhu, X.X.; Wang, R.; Chen, L.B. Aurora-a confers radioresistance in human hepatocellular carcinoma by activating NF-kappaB signaling pathway. BMC Cancer 2019, 19, 1075. [Google Scholar] [CrossRef]
- Tian, Y.; Wong, V.W.; Wong, G.L.; Yang, W.; Sun, H.; Shen, J.; Tong, J.H.; Go, M.Y.; Cheung, Y.S.; Lai, P.B.; et al. Histone Deacetylase HDAC8 Promotes Insulin Resistance and beta-Catenin Activation in NAFLD-Associated Hepatocellular Carcinoma. Cancer Res. 2015, 75, 4803–4816. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.S.; Lau, S.S.; Chen, Y.; Kondo, Y.; Li, M.S.; Feng, H.; Ching, A.K.; Cheung, K.F.; Wong, H.K.; Tong, J.H.; et al. EZH2-mediated concordant repression of Wnt antagonists promotes beta-catenin-dependent hepatocarcinogenesis. Cancer Res. 2011, 71, 4028–4039. [Google Scholar] [CrossRef] [Green Version]
- Glenisson, W.; Castronovo, V.; Waltregny, D. Histone deacetylase 4 is required for TGFbeta1-induced myofibroblastic differentiation. Biochim. Biophys. Acta 2007, 1773, 1572–1582. [Google Scholar] [CrossRef]
- Huang, B.; Huang, M.; Li, Q. MiR-137 suppresses migration and invasion by targeting EZH2-STAT3 signaling in human hepatocellular carcinoma. Pathol. Res. Pract. 2018, 214, 1980–1986. [Google Scholar] [CrossRef]
- Lin, H.P.; Cheng, Z.L.; He, R.Y.; Song, L.; Tian, M.X.; Zhou, L.S.; Groh, B.S.; Liu, W.R.; Ji, M.B.; Ding, C.; et al. Destabilization of Fatty Acid Synthase by Acetylation Inhibits De Novo Lipogenesis and Tumor Cell Growth. Cancer Res. 2016, 76, 6924–6936. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Tang, F.; Li, B.; Yuan, S.; Xu, Q.; Tomlinson, S.; Jin, J.; Hu, W.; He, S. High USP22 expression indicates poor prognosis in hepatocellular carcinoma. Oncotarget 2015, 6, 12654–12667. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Zhong, X.; Wang, S.; Zeng, K.; Sun, H.; Sun, G.; Zou, R.; Liu, W.; Liu, W.; Lin, L.; et al. ATXN7L3 positively regulates SMAD7 transcription in hepatocellular carcinoma with growth inhibitory function. EBioMedicine 2020, 62, 103108. [Google Scholar] [CrossRef]
- Xu, H.; Wang, H.; Zhao, W.; Fu, S.; Li, Y.; Ni, W.; Xin, Y.; Li, W.; Yang, C.; Bai, Y.; et al. SUMO1 modification of methyltransferase-like 3 promotes tumor progression via regulating Snail mRNA homeostasis in hepatocellular carcinoma. Theranostics 2020, 10, 5671–5686. [Google Scholar] [CrossRef]
- Zubiete-Franco, I.; Garcia-Rodriguez, J.L.; Lopitz-Otsoa, F.; Serrano-Macia, M.; Simon, J.; Fernandez-Tussy, P.; Barbier-Torres, L.; Fernandez-Ramos, D.; Gutierrez-de-Juan, V.; Lopez de Davalillo, S.; et al. SUMOylation regulates LKB1 localization and its oncogenic activity in liver cancer. EBioMedicine 2019, 40, 406–421. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Fu, Z.; Yang, G.; Gao, D.; Wang, T.; Liu, Z.; Li, G.; Wang, Y. Exportin-5 SUMOylation promotes hepatocellular carcinoma progression. Exp. Cell Res. 2020, 395, 112219. [Google Scholar] [CrossRef]
- Borghesan, M.; Fusilli, C.; Rappa, F.; Panebianco, C.; Rizzo, G.; Oben, J.A.; Mazzoccoli, G.; Faulkes, C.; Pata, I.; Agodi, A.; et al. DNA Hypomethylation and Histone Variant macroH2A1 Synergistically Attenuate Chemotherapy-Induced Senescence to Promote Hepatocellular Carcinoma Progression. Cancer Res. 2016, 76, 594–606. [Google Scholar] [CrossRef] [Green Version]
- Lo Re, O.; Fusilli, C.; Rappa, F.; Van Haele, M.; Douet, J.; Pindjakova, J.; Rocha, S.W.; Pata, I.; Valcikova, B.; Uldrijan, S.; et al. Induction of cancer cell stemness by depletion of macrohistone H2A1 in hepatocellular carcinoma. Hepatology 2018, 67, 636–650. [Google Scholar] [CrossRef] [Green Version]
- Rivas Serna, I.M.; Romito, I.; Maugeri, A.; Lo Re, O.; Giallongo, S.; Mazzoccoli, G.; Oben, J.A.; Li Volti, G.; Mazza, T.; Alisi, A.; et al. A Lipidomic Signature Complements Stemness Features Acquisition in Liver Cancer Cells. Int. J. Mol. Sci. 2020, 21, 8452. [Google Scholar] [CrossRef]
- Povsic, M.; Oliver, L.; Jiandani, N.R.; Perry, R.; Bottomley, J. A structured literature review of interventions used in the management of nonalcoholic steatohepatitis (NASH). Pharmacol. Res. Perspect. 2019, 7, e00485. [Google Scholar] [CrossRef]
- Ristic-Medic, D.; Kovacic, M.; Takic, M.; Arsic, A.; Petrovic, S.; Paunovic, M.; Jovicic, M.; Vucic, V. Calorie-Restricted Mediterranean and Low-Fat Diets Affect Fatty Acid Status in Individuals with Nonalcoholic Fatty Liver Disease. Nutrients 2020, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.P.; Wai, J.P.; Tsai, M.K.; Yang, Y.C.; Cheng, T.Y.; Lee, M.C.; Chan, H.T.; Tsao, C.K.; Tsai, S.P.; Wu, X. Minimum amount of physical activity for reduced mortality and extended life expectancy: A prospective cohort study. Lancet 2011, 378, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Miura, I.; Komine, S.; Okada, K.; Wada, S.; Warabi, E.; Uchida, F.; Oh, S.; Suzuki, H.; Mizokami, Y.; Shoda, J. Prevention of non-alcoholic steatohepatitis by long-term exercise via the induction of phenotypic changes in Kupffer cells of hyperphagic obese mice. Physiol. Rep. 2021, 9, e14859. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.M.; Shiroma, E.J.; Lobelo, F.; Puska, P.; Blair, S.N.; Katzmarzyk, P.T.; Lancet Physical Activity Series Working, G. Effect of physical inactivity on major non-communicable diseases worldwide: An analysis of burden of disease and life expectancy. Lancet 2012, 380, 219–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, P.; Breuer, S.; Hoffmann, T.; Vohlen, C.; Janoschek, R.; Schmitz, L.; Appel, S.; Fink, G.; Hunseler, C.; Quaas, A.; et al. Maternal Exercise Mediates Hepatic Metabolic Programming via Activation of AMPK-PGC1alpha Axis in the Offspring of Obese Mothers. Cells 2021, 10, 1247. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Delany, J.P.; Otto, A.D.; Kuller, L.; Vockley, J.; South-Paul, J.E.; Thomas, S.B.; Brown, J.; McTigue, K.; Hames, K.C.; et al. Effects of diet and physical activity interventions on weight loss and cardiometabolic risk factors in severely obese adults: A randomized trial. JAMA 2010, 304, 1795–1802. [Google Scholar] [CrossRef] [Green Version]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef] [Green Version]
- Sumida, Y.; Naito, Y.; Tanaka, S.; Sakai, K.; Inada, Y.; Taketani, H.; Kanemasa, K.; Yasui, K.; Itoh, Y.; Okanoue, T.; et al. Long-term (>=2 yr) efficacy of vitamin E for non-alcoholic steatohepatitis. Hepatogastroenterology 2013, 60, 1445–1450. [Google Scholar]
- Chiu, M.; McBeth, L.; Sindhwani, P.; Hinds, T.D. Deciphering the Roles of Thiazolidinediones and PPARgamma in Bladder Cancer. PPAR Res. 2017, 2017, 4810672. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E.; or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: A Meta-analysis. JAMA Intern. Med. 2017, 177, 633–640. [Google Scholar] [CrossRef]
- Bode, B. An overview of the pharmacokinetics, efficacy and safety of liraglutide. Diabetes Res. Clin. Pract. 2012, 97, 27–42. [Google Scholar] [CrossRef]
- Mells, J.E.; Fu, P.P.; Sharma, S.; Olson, D.; Cheng, L.; Handy, J.A.; Saxena, N.K.; Sorescu, D.; Anania, F.A. Glp-1 analog, liraglutide, ameliorates hepatic steatosis and cardiac hypertrophy in C57BL/6J mice fed a Western diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G225–G235. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; LEAN Trial Team. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Raza, S.; Rajak, S.; Upadhyay, A.; Tewari, A.; Anthony Sinha, R. Current treatment paradigms and emerging therapies for NAFLD/NASH. Front. Biosci. 2021, 26, 206–237. [Google Scholar] [CrossRef]
- Bugianesi, E.; Gentilcore, E.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; David, E.; Rizzetto, M.; Marchesini, G. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am. J. Gastroenterol. 2005, 100, 1082–1090. [Google Scholar] [CrossRef]
- Chen, H.P.; Shieh, J.J.; Chang, C.C.; Chen, T.T.; Lin, J.T.; Wu, M.S.; Lin, J.H.; Wu, C.Y. Metformin decreases hepatocellular carcinoma risk in a dose-dependent manner: Population-based and in vitro studies. Gut 2013, 62, 606–615. [Google Scholar] [CrossRef]
- Pockros, P.J.; Fuchs, M.; Freilich, B.; Schiff, E.; Kohli, A.; Lawitz, E.J.; Hellstern, P.A.; Owens-Grillo, J.; Van Biene, C.; Shringarpure, R.; et al. CONTROL: A randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. 2019, 39, 2082–2093. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]
- Satiya, J.; Snyder, H.S.; Singh, S.P.; Satapathy, S.K. Narrative review of current and emerging pharmacological therapies for nonalcoholic steatohepatitis. Transl. Gastroenterol. Hepatol. 2021, 6, 60. [Google Scholar] [CrossRef]
- Hyogo, H.; Tazuma, S.; Arihiro, K.; Iwamoto, K.; Nabeshima, Y.; Inoue, M.; Ishitobi, T.; Nonaka, M.; Chayama, K. Efficacy of atorvastatin for the treatment of nonalcoholic steatohepatitis with dyslipidemia. Metabolism 2008, 57, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.; Torres, D.M.; Morgan, A.E.; Fincke, C.; Harrison, S.A. A pilot study using simvastatin in the treatment of nonalcoholic steatohepatitis: A randomized placebo-controlled trial. J. Clin. Gastroenterol. 2009, 43, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Hyogo, H.; Ikegami, T.; Tokushige, K.; Hashimoto, E.; Inui, K.; Matsuzaki, Y.; Tokumo, H.; Hino, F.; Tazuma, S. Efficacy of pitavastatin for the treatment of non-alcoholic steatohepatitis with dyslipidemia: An open-label, pilot study. Hepatol. Res. 2011, 41, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, F.; Aron-Wisnewsky, J.; Pais, R.; Tordjman, J.; Poitou, C.; Charlotte, F.; Bedossa, P.; Poynard, T.; Clement, K.; Ratziu, V.; et al. Statins, antidiabetic medications and liver histology in patients with diabetes with non-alcoholic fatty liver disease. BMJ Open Gastroenterol. 2016, 3, e000075. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Petta, S.; Mannisto, V.; Mancina, R.M.; Pipitone, R.; Karja, V.; Maggioni, M.; Kakela, P.; Wiklund, O.; Mozzi, E.; et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J. Hepatol. 2015, 63, 705–712. [Google Scholar] [CrossRef]
- Singh, S.; Singh, P.P.; Singh, A.G.; Murad, M.H.; Sanchez, W. Statins are associated with a reduced risk of hepatocellular cancer: A systematic review and meta-analysis. Gastroenterology 2013, 144, 323–332. [Google Scholar] [CrossRef] [Green Version]
- El-Serag, H.B.; Johnson, M.L.; Hachem, C.; Morgana, R.O. Statins are associated with a reduced risk of hepatocellular carcinoma in a large cohort of patients with diabetes. Gastroenterology 2009, 136, 1601–1608. [Google Scholar] [CrossRef] [Green Version]
- Pinyopornpanish, K.; Al-Yaman, W.; Butler, R.S.; Carey, W.; McCullough, A.; Romero-Marrero, C. Chemopreventive Effect of Statin on Hepatocellular Carcinoma in Patients With Nonalcoholic Steatohepatitis Cirrhosis. Am. J. Gastroenterol. 2021, 116, 2258–2269. [Google Scholar] [CrossRef]
- Shahidi, F.; Ambigaipalan, P. Omega-3 Polyunsaturated Fatty Acids and Their Health Benefits. Annu. Rev. Food Sci. Technol. 2018, 9, 345–381. [Google Scholar] [CrossRef]
- Capanni, M.; Calella, F.; Biagini, M.R.; Genise, S.; Raimondi, L.; Bedogni, G.; Svegliati-Baroni, G.; Sofi, F.; Milani, S.; Abbate, R.; et al. Prolonged n-3 polyunsaturated fatty acid supplementation ameliorates hepatic steatosis in patients with non-alcoholic fatty liver disease: A pilot study. Aliment. Pharmacol. Ther. 2006, 23, 1143–1151. [Google Scholar] [CrossRef]
- Spadaro, L.; Magliocco, O.; Spampinato, D.; Piro, S.; Oliveri, C.; Alagona, C.; Papa, G.; Rabuazzo, A.M.; Purrello, F. Effects of n-3 polyunsaturated fatty acids in subjects with nonalcoholic fatty liver disease. Dig. Liver Dis. 2008, 40, 194–199. [Google Scholar] [CrossRef]
- Scorletti, E.; Bhatia, L.; McCormick, K.G.; Clough, G.F.; Nash, K.; Hodson, L.; Moyses, H.E.; Calder, P.C.; Byrne, C.D.; Study, W. Effects of purified eicosapentaenoic and docosahexaenoic acids in nonalcoholic fatty liver disease: Results from the Welcome* study. Hepatology 2014, 60, 1211–1221. [Google Scholar] [CrossRef]
- Skulas-Ray, A.C.; Wilson, P.W.F.; Harris, W.S.; Brinton, E.A.; Kris-Etherton, P.M.; Richter, C.K.; Jacobson, T.A.; Engler, M.B.; Miller, M.; Robinson, J.G.; et al. Omega-3 Fatty Acids for the Management of Hypertriglyceridemia: A Science Advisory From the American Heart Association. Circulation 2019, 140, e673–e691. [Google Scholar] [CrossRef] [Green Version]
- Zein, C.O.; Yerian, L.M.; Gogate, P.; Lopez, R.; Kirwan, J.P.; Feldstein, A.E.; McCullough, A.J. Pentoxifylline improves nonalcoholic steatohepatitis: A randomized placebo-controlled trial. Hepatology 2011, 54, 1610–1619. [Google Scholar] [CrossRef] [Green Version]
- Van Wagner, L.B.; Koppe, S.W.; Brunt, E.M.; Gottstein, J.; Gardikiotes, K.; Green, R.M.; Rinella, M.E. Pentoxifylline for the treatment of non-alcoholic steatohepatitis: A randomized controlled trial. Ann. Hepatol. 2011, 10, 277–286. [Google Scholar] [CrossRef]
- Du, J.; Ma, Y.Y.; Yu, C.H.; Li, Y.M. Effects of pentoxifylline on nonalcoholic fatty liver disease: A meta-analysis. World J. Gastroenterol. 2014, 20, 569–577. [Google Scholar] [CrossRef]
- Marra, F.; Tacke, F. Roles for chemokines in liver disease. Gastroenterology 2014, 147, 577–594.e1. [Google Scholar] [CrossRef]
- Kruger, A.J.; Fuchs, B.C.; Masia, R.; Holmes, J.A.; Salloum, S.; Sojoodi, M.; Ferreira, D.S.; Rutledge, S.M.; Caravan, P.; Alatrakchi, N.; et al. Prolonged cenicriviroc therapy reduces hepatic fibrosis despite steatohepatitis in a diet-induced mouse model of nonalcoholic steatohepatitis. Hepatol. Commun. 2018, 2, 529–545. [Google Scholar] [CrossRef]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Sanyal, A.; Harrison, S.A.; Wong, V.W.; Francque, S.; Goodman, Z.; Aithal, G.P.; Kowdley, K.V.; Seyedkazemi, S.; Fischer, L.; et al. Cenicriviroc Treatment for Adults With Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology 2020, 72, 892–905. [Google Scholar] [CrossRef]
- Lopez-de la Mora, D.A.; Sanchez-Roque, C.; Montoya-Buelna, M.; Sanchez-Enriquez, S.; Lucano-Landeros, S.; Macias-Barragan, J.; Armendariz-Borunda, J. Role and New Insights of Pirfenidone in Fibrotic Diseases. Int. J. Med. Sci. 2015, 12, 840–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruwanpura, S.M.; Thomas, B.J.; Bardin, P.G. Pirfenidone: Molecular Mechanisms and Potential Clinical Applications in Lung Disease. Am. J. Respir. Cell Mol. Biol. 2020, 62, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Komiya, C.; Tanaka, M.; Tsuchiya, K.; Shimazu, N.; Mori, K.; Furuke, S.; Miyachi, Y.; Shiba, K.; Yamaguchi, S.; Ikeda, K.; et al. Antifibrotic effect of pirfenidone in a mouse model of human nonalcoholic steatohepatitis. Sci. Rep. 2017, 7, 44754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Ni, Y.; Nagata, N.; Xu, L.; Zhuge, F.; Nagashimada, M.; Kaneko, S.; Ota, T. Pirfenidone prevents and reverses hepatic insulin resistance and steatohepatitis by polarizing M2 macrophages. Lab. Investig. 2019, 99, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Rodriguez, A.; Monroy-Ramirez, H.C.; Meza-Rios, A.; Garcia-Banuelos, J.; Vera-Cruz, J.; Gutierrez-Cuevas, J.; Silva-Gomez, J.; Staels, B.; Dominguez-Rosales, J.; Galicia-Moreno, M.; et al. Pirfenidone Is an Agonistic Ligand for PPARalpha and Improves NASH by Activation of SIRT1/LKB1/pAMPK. Hepatol. Commun. 2020, 4, 434–449. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Cuevas, J.; Sandoval-Rodriguez, A.; Monroy-Ramirez, H.C.; Vazquez-Del Mercado, M.; Santos-Garcia, A.; Armendariz-Borunda, J. Prolonged-release pirfenidone prevents obesity-induced cardiac steatosis and fibrosis in a mouse NASH model. Cardiovasc. Drugs Ther. 2021, 35, 927–938. [Google Scholar] [CrossRef]
- Poo, J.L.; Torre, A.; Aguilar-Ramirez, J.R.; Cruz, M.; Mejia-Cuan, L.; Cerda, E.; Velazquez, A.; Patino, A.; Ramirez-Castillo, C.; Cisneros, L.; et al. Benefits of prolonged-release pirfenidone plus standard of care treatment in patients with advanced liver fibrosis: PROMETEO study. Hepatol. Int. 2020, 14, 817–827. [Google Scholar] [CrossRef]
- Jindal, A.; Thadi, A.; Shailubhai, K. Hepatocellular Carcinoma: Etiology and Current and Future Drugs. J. Clin. Exp. Hepatol. 2019, 9, 221–232. [Google Scholar] [CrossRef]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Akce, M.; El-Rayes, B.F.; Bekaii-Saab, T.S. Frontline therapy for advanced hepatocellular carcinoma: An update. Therap. Adv. Gastroenterol. 2022, 15, 17562848221086126. [Google Scholar] [CrossRef]
- Lee, M.S.; Ryoo, B.Y.; Hsu, C.H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.P.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): An open-label, multicentre, phase 1b study. Lancet Oncol. 2020, 21, 808–820. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Cheng, A.L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef]
- Himmelsbach, V.; Pinter, M.; Scheiner, B.; Venerito, M.; Sinner, F.; Zimpel, C.; Marquardt, J.U.; Trojan, J.; Waidmann, O.; Finkelmeier, F. Efficacy and Safety of Atezolizumab and Bevacizumab in the Real-World Treatment of Advanced Hepatocellular Carcinoma: Experience from Four Tertiary Centers. Cancers 2022, 14, 1722. [Google Scholar] [CrossRef]
- Vithayathil, M.; D’Alessio, A.; Fulgenzi, C.A.M.; Nishida, N.; Schonlein, M.; von Felden, J.; Schulze, K.; Wege, H.; Saeed, A.; Wietharn, B.; et al. Impact of older age in patients receiving atezolizumab and bevacizumab for hepatocellular carcinoma. Liver Int. 2022, 42, 2538–2547. [Google Scholar] [CrossRef]
- Rimini, M.; Rimassa, L.; Ueshima, K.; Burgio, V.; Shigeo, S.; Tada, T.; Suda, G.; Yoo, C.; Cheon, J.; Pinato, D.J.; et al. Atezolizumab plus bevacizumab versus lenvatinib or sorafenib in non-viral unresectable hepatocellular carcinoma: An international propensity score matching analysis. ESMO Open 2022, 7, 100591. [Google Scholar] [CrossRef]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [Green Version]
- Mendez-Blanco, C.; Fondevila, F.; Garcia-Palomo, A.; Gonzalez-Gallego, J.; Mauriz, J.L. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Shimose, S.; Hiraoka, A.; Nakano, M.; Iwamoto, H.; Tanaka, M.; Tanaka, T.; Noguchi, K.; Aino, H.; Ogata, K.; Kajiwara, M.; et al. First-line sorafenib sequential therapy and liver disease etiology for unresectable hepatocellular carcinoma using inverse probability weighting: A multicenter retrospective study. Cancer Med. 2021, 10, 8530–8541. [Google Scholar] [CrossRef]
- Jian, C.; Fu, J.; Cheng, X.; Shen, L.J.; Ji, Y.X.; Wang, X.; Pan, S.; Tian, H.; Tian, S.; Liao, R.; et al. Low-Dose Sorafenib Acts as a Mitochondrial Uncoupler and Ameliorates Nonalcoholic Steatohepatitis. Cell Metab. 2020, 31, 1206. [Google Scholar] [CrossRef] [PubMed]
- Romualdo, G.R.; Da Silva, T.C.; de Albuquerque Landi, M.F.; Morais, J.A.; Barbisan, L.F.; Vinken, M.; Oliveira, C.P.; Cogliati, B. Sorafenib reduces steatosis-induced fibrogenesis in a human 3D co-culture model of non-alcoholic fatty liver disease. Environ. Toxicol. 2021, 36, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Kudo, M.; Kawazoe, S.; Osaki, Y.; Ikeda, M.; Okusaka, T.; Tamai, T.; Suzuki, T.; Hisai, T.; Hayato, S.; et al. Phase 2 study of lenvatinib in patients with advanced hepatocellular carcinoma. J. Gastroenterol. 2017, 52, 512–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomonari, T.; Sato, Y.; Tanaka, H.; Mitsuhashi, T.; Hirao, A.; Tanaka, T.; Taniguchi, T.; Okamoto, K.; Sogabe, M.; Miyamoto, H.; et al. Therapeutic efficacy of lenvatinib in nonviral unresectable hepatocellular carcinoma. JGH Open 2021, 5, 1275–1283. [Google Scholar] [CrossRef]
- Rimini, M.; Kudo, M.; Tada, T.; Shigeo, S.; Kang, W.; Suda, G.; Jefremow, A.; Burgio, V.; Iavarone, M.; Tortora, R.; et al. Nonalcoholic steatohepatitis in hepatocarcinoma: New insights about its prognostic role in patients treated with lenvatinib. ESMO Open 2021, 6, 100330. [Google Scholar] [CrossRef]
- Hiraoka, A.; Kumada, T.; Tada, T.; Tani, J.; Kariyama, K.; Fukunishi, S.; Atsukawa, M.; Hirooka, M.; Tsuji, K.; Ishikawa, T.; et al. Efficacy of lenvatinib for unresectable hepatocellular carcinoma based on background liver disease etiology: Multi-center retrospective study. Sci. Rep. 2021, 11, 16663. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Gerolami, R.; Caparello, C.; et al. Outcomes of sequential treatment with sorafenib followed by regorafenib for HCC: Additional analyses from the phase III RESORCE trial. J. Hepatol. 2018, 69, 353–358. [Google Scholar] [CrossRef]
- Schwartz, G.; Darling, J.O.; Mindo, M.; Damicis, L. Management of Adverse Events Associated with Cabozantinib Treatment in Patients with Advanced Hepatocellular Carcinoma. Target Oncol. 2020, 15, 549–565. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Finkelmeier, F.; Scheiner, B.; Leyh, C.; Best, J.; Frundt, T.W.; Czauderna, C.; Beutel, A.; Bettinger, D.; Weiss, J.; Meischl, T.; et al. Cabozantinib in Advanced Hepatocellular Carcinoma: Efficacy and Safety Data from an International Multicenter Real-Life Cohort. Liver Cancer 2021, 10, 360–369. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased alpha-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Zhu, A.X.; Park, J.O.; Ryoo, B.Y.; Yen, C.J.; Poon, R.; Pastorelli, D.; Blanc, J.F.; Chung, H.C.; Baron, A.D.; Pfiffer, T.E.; et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 859–870. [Google Scholar] [CrossRef]






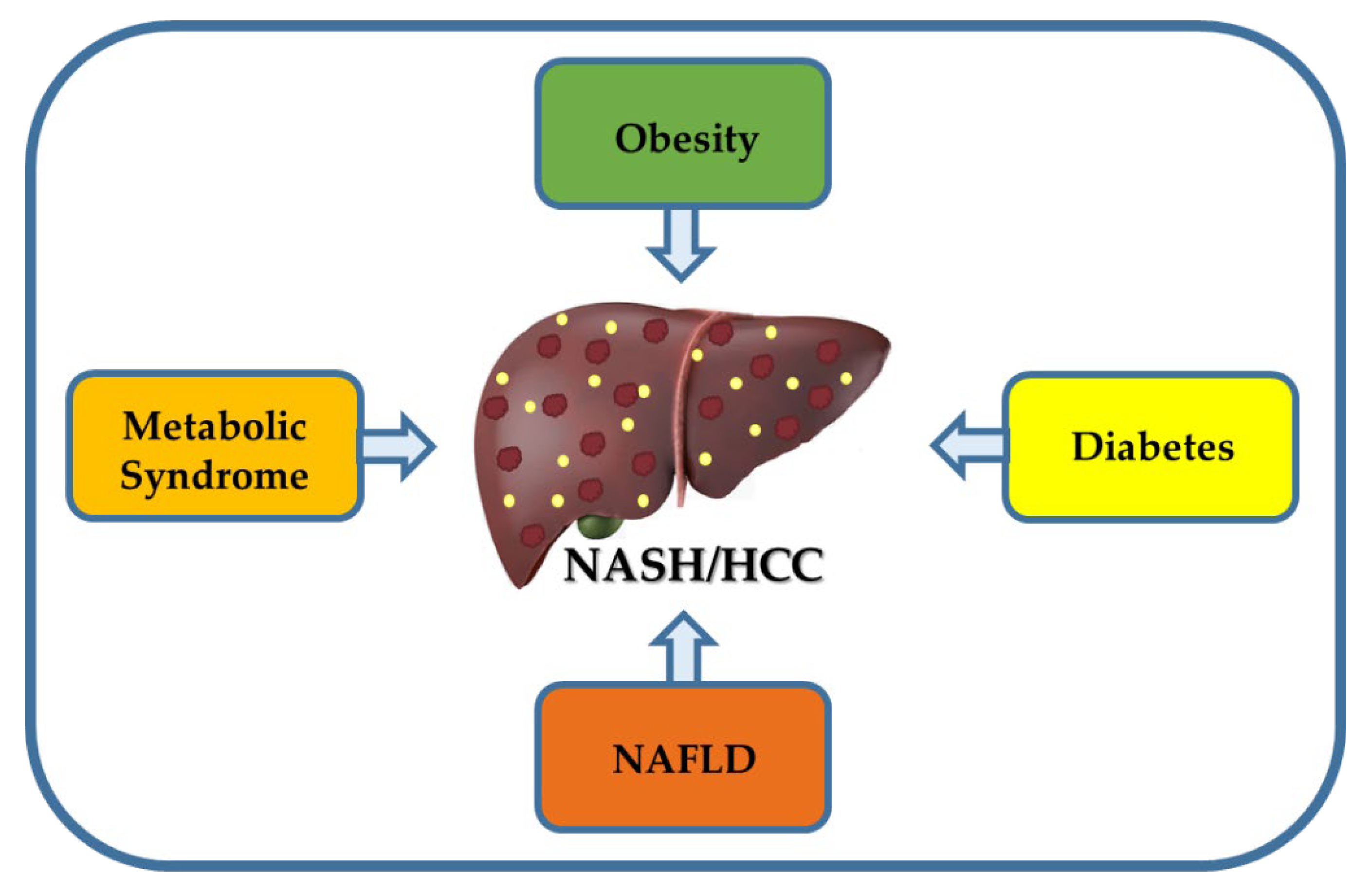{kind=link}
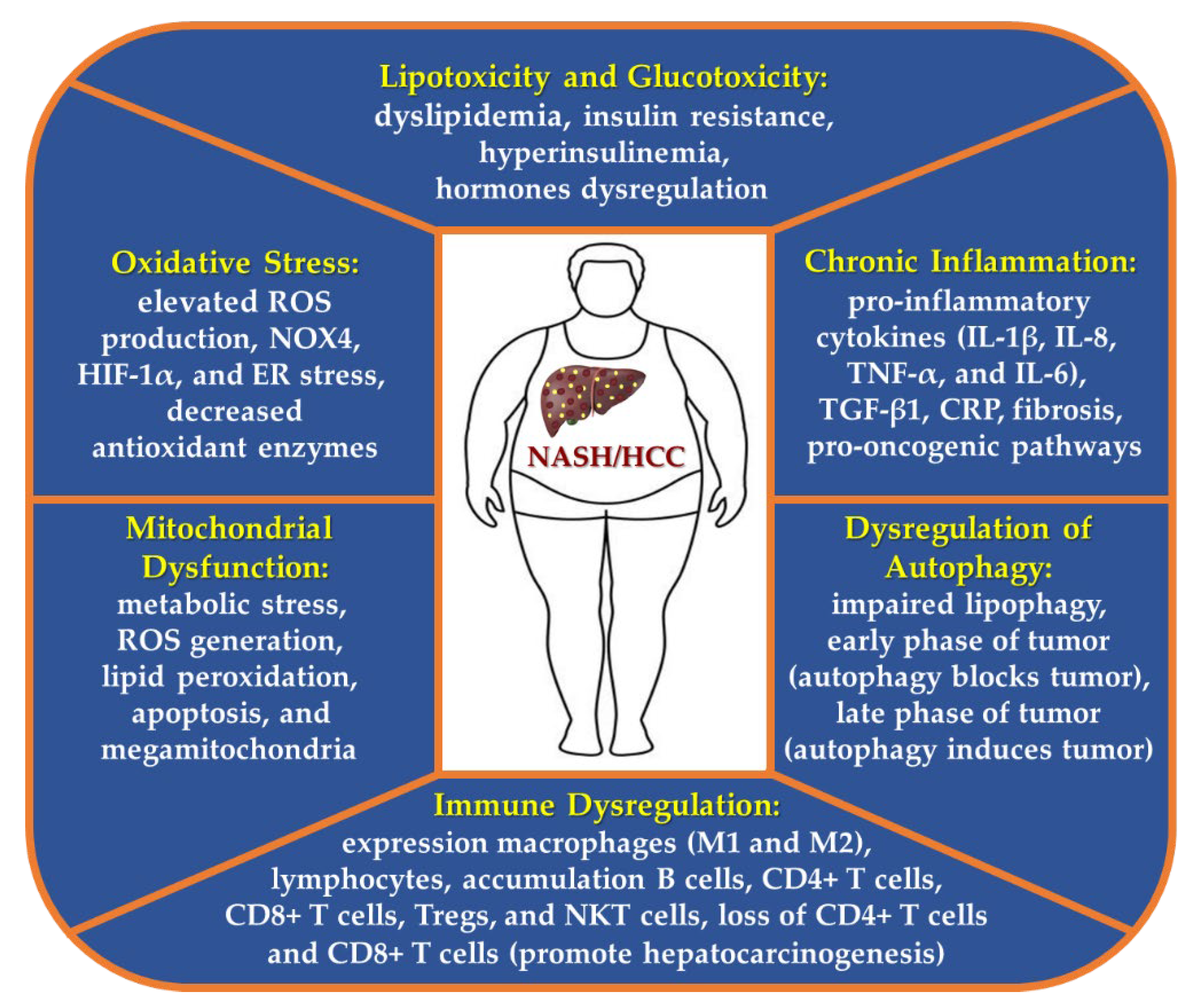{kind=link}
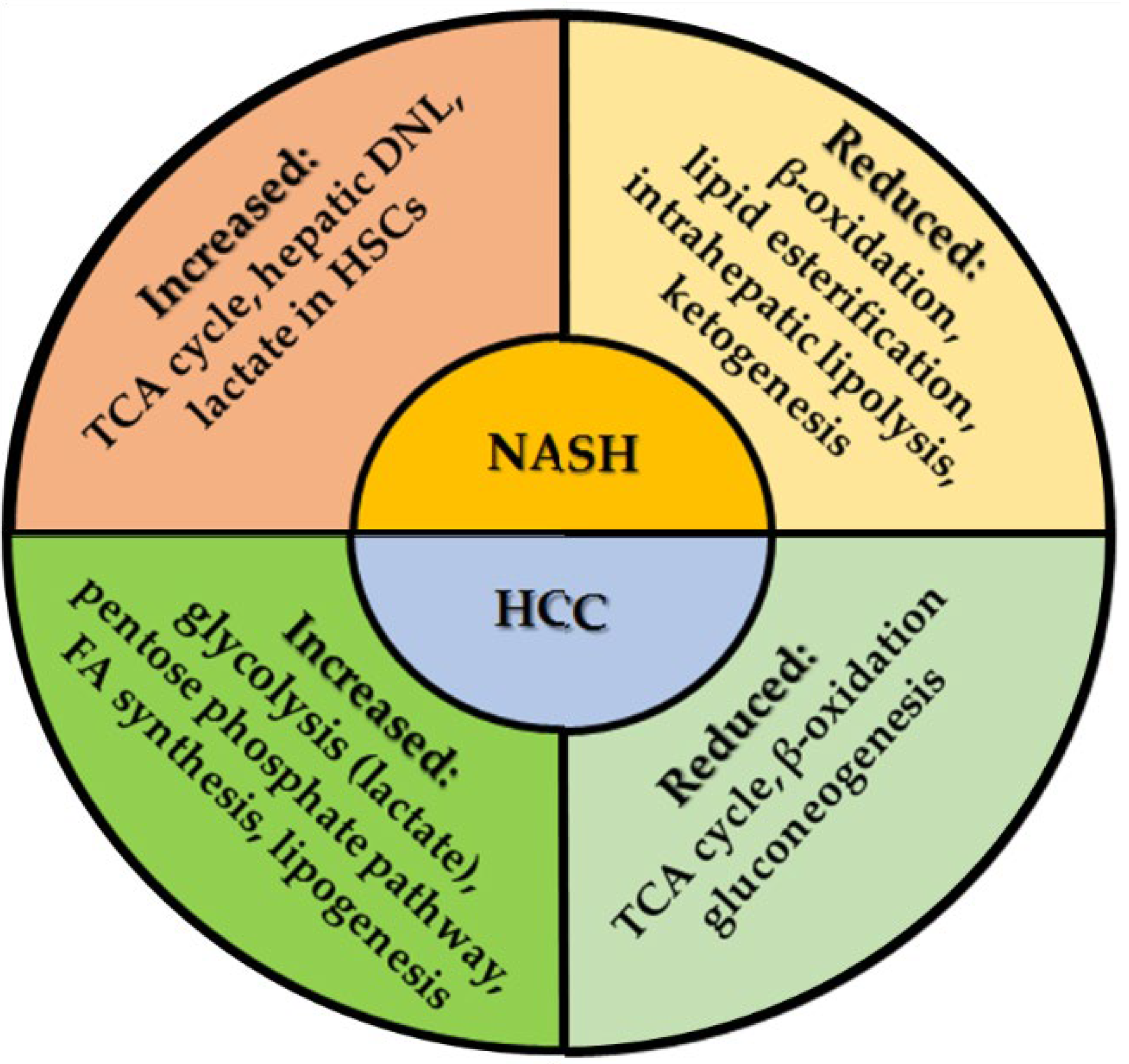{kind=link}
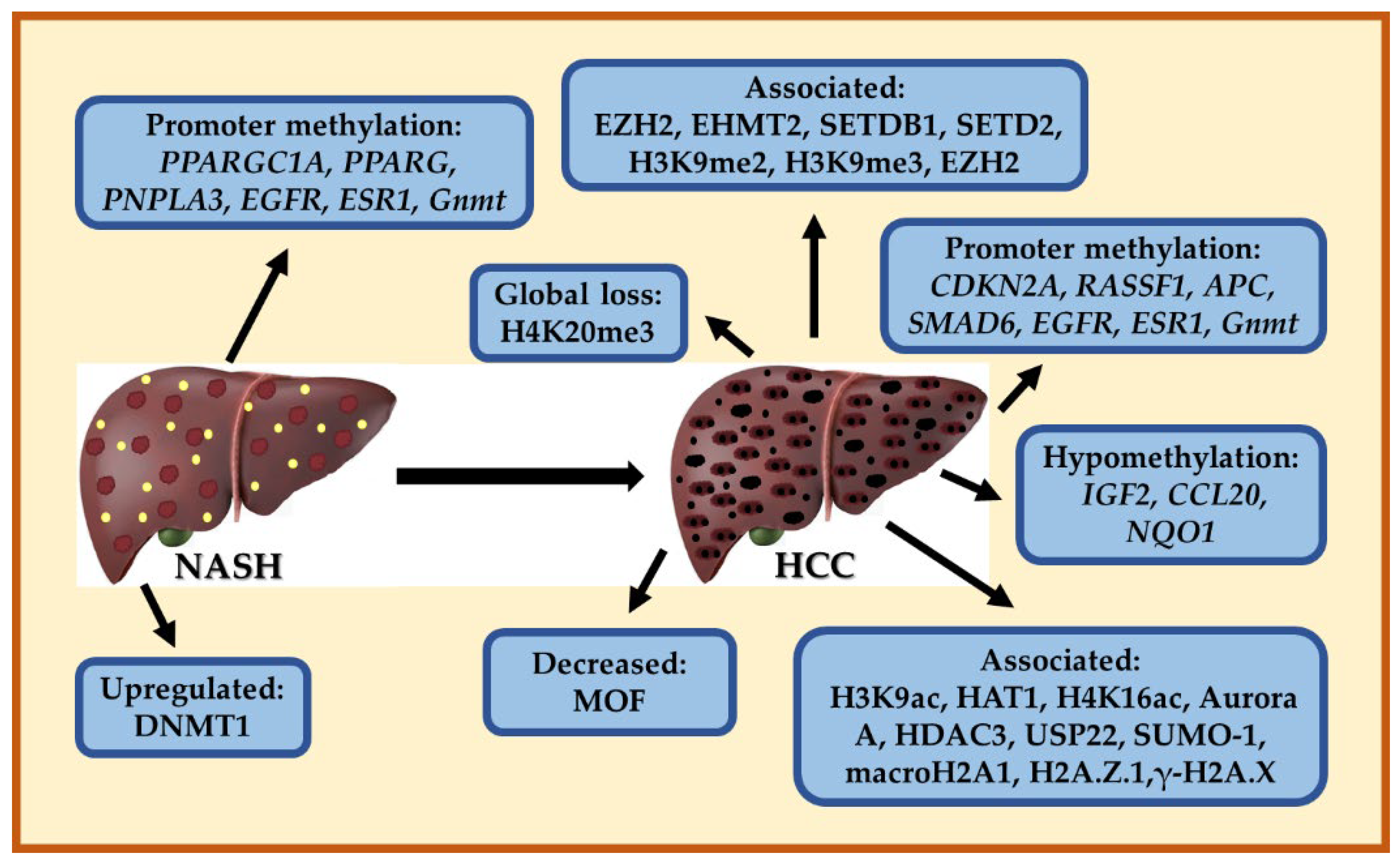{kind=link}
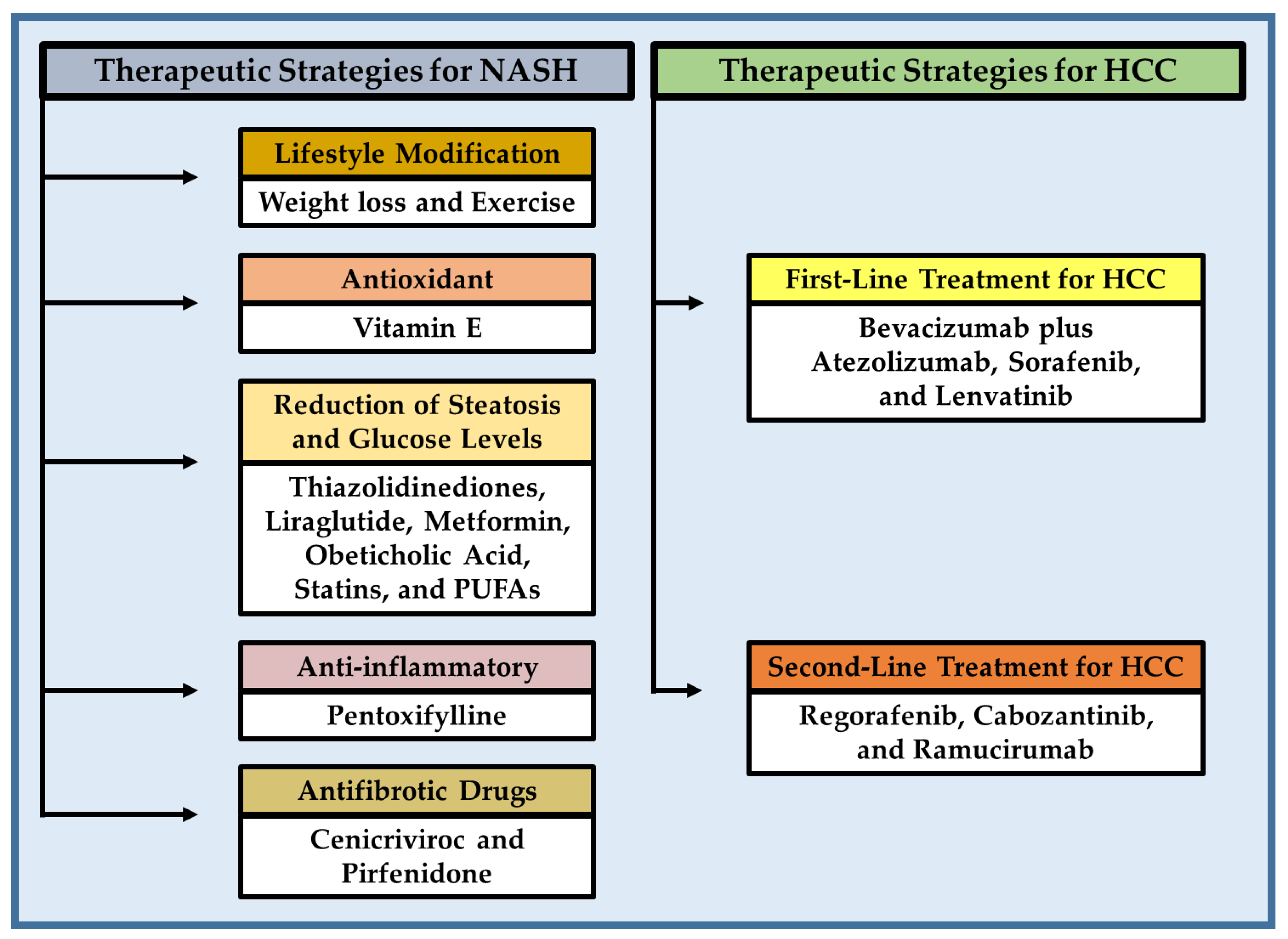{kind=link}
| Pathway | Gen | Polymorphism | Effect |
|---|---|---|---|
| Lipid metabolism | PNPLA3 | rs738409 | Increase severe of NAFLD, NASH, fibrosis, and HCC [64,66,67,83,84] |
| TM6SF2 | rs10401969 (C), rs58542926 (C/T) E167K | Associated with steatosis, NASH, fibrosis/cirrhosis [69,70,82] | |
| Metabolism of phospholipids and triacylglycerols | LPIN1 | rs13412852 C/T | Associated with lipid levels, NASH severity, and hepatic fibrosis in children with NAFLD [87] |
| Lipogenesis | HSD17B13 | rs72613567 | Reduced risk of NASH and progressive liver damage [75,76] |
| Energy metabolism | PPARGC1A | rs8192678 GA/AA | Increased risk of NASH [69,70,71] |
| Insulin resistance | ENPP1 | ENPP1 121Glin | Associated with fibrosis [88] |
| IRS-1 | IRS-1 972Arg | Associated with fibrosis [88] | |
| GCKR | rs780094 and rs1260326, encoding Pro446Leu | Increased serum triglycerides and associated with fibrosis [88] | |
| Transfer of neutral lipids to nascent ApoB | MTP | −493 G/T | Susceptibility for NASH [74] |
| VLDL secretion | APOB | Several | Associated with NAFLD, NASH, fibrosis, and HCC [89] |
| TM6SF2 | rs58542926 C/T | Increase in NAFLD, NASH, and fibrosis [89] | |
| De novo lipogenesis regulation | GCKR | rs780094 A/G | Associated with NAFLD, NASH, and fibrosis [89] |
| KLF6 | rs3750861 G/A | Decrease fibrosis [89] | |
| Fibrosis | AGTR1 | rs3772622 | Associated with steatohepatitis and fibrosis [88] |
| Oxidative stress | GCLC | −129 C/T | Associated with NASH [88] |
| UCP2 | −866 G/A, rs695366 | Reduce the risk of NASH [77,88] | |
| Immune response | TNF | G238A (rs361525), G308A (rs1800629) | Susceptibility for insulin resistance, NAFLD, and NASH [88] |
| IL28B | rs12979860 C/T | Decrease fibrosis [89] | |
| Mitochondrial antioxidant | SOD2 | rs4880 C/T | Increase fibrosis [89] |
| Phosphatidylinositol remodeling | MBOAT7 | rs641738 | Associated with NAFLD, NASH, fibrosis, and HCC [89,90] |
| HSD17B13 | rs72613567 A/T, rs143404524 | Decrease NAFLD, NASH, fibrosis, and HCC [90] | |
| Antioxidant activity | HP | Hp 2-2 genotype | Contribute to NASH development [73] |
| Repairon of the sarcolemmal membrane in skeletal muscle | DYSF | rs17007417 | Associated with NASH-HCC [91] |
| Micro RNAs | Stage Disease | Expression in Hepatic Tissue | Biological Function |
|---|---|---|---|
| miR-122 | NAFLD, NASH, early stages of HCC | Upregulated or Downregulated | Lipid metabolism [113,114,115,127,134,138,139] |
| miR-29a | NASH | Downregulated | Inflammation and fibrosis [118] |
| miR-33 | NASH | Upregulated | Lipid metabolism and transport [115] |
| miR-21 | NASH | Upregulated | Inflammation and fibrosis [115,121] |
| miR-34a-5p | NASH | Upregulated | Lipid metabolism [122] |
| miR-122-5p | NASH | Downregulated | Lipid metabolism [122] |
| miR-21, miR-1290, miR-27b-3p, and miR-192-5p | NASH | Upregulated | Inflammation [115,121,130] |
| miR-221/222 | NASH | Upregulated | Fibrosis [124] |
| miR99b | NASH | Upregulated | Pericellular fibrosis [126] |
| miR-155-5p, miR-423-5p, miR-15b-5p, miR143-3p, and miR-26a-5p | NASH | Upregulated | ATP production [130] |
| miR-let-7c-5p | NASH | Upregulated | Fibrogenic responses [130] |
| miR-423-5p, miR-let-7a-5p, and miR-let-7c-5p | NASH | Upregulated | IL-6R is decreased [130,131,133] |
| miR-155, miR-221/222, and miR-21, | NASH-HCC | Upregulated | Early stages of hepatocarcinogenesis [130,134,137] |
| miR-122 | NASH-HCC | Downregulated | Early stages of hepatocarcinogenesis [134,139] |
| miR-16 | NASH-HCC | Upregulated | Hepatocarcinogenesis [135] |
| miR-155, miR-193b, miR-27a, miR-31, miR-99b, miR-484, miR-574-3p, miR125a-5p, and miR-182 | NAFLD-NASH-HCC | Upregulated | Hepatocarcinogenesis [134,136,137] |
| miR-20a, miR-200c, miR-93, miR-340-5p, and miR-720 | NAFLD-NASH-HCC | Downregulated | Hepatocarcinogenesis [136] |
| miR-21 and miR-155 | HCC | Upregulated | Early stages of HCC [134] |
| miR-192 | NAFLD and NASH | Downregulated | HSC activation [128] |
| miR-34a | NAFLD and NASH | Upregulated | Inflammation [114,123,128,129] |
| miR-375 | NAFLD and NASH | Upregulated | Glucose homeostasis, intestinal permeability modulation, and inflammation [128,129] |
| miR-125b | NAFLD and NASH | Upregulated | Lipid and glucose homeostasis, adipocyte differentiation, and fibrosis [128,129] |
| miR-33a/b | NASH | Upregulated | Lipid and cholesterol homeostasis, glucose homeostasis, and inflammation [128] |
| miR-451 | NAFLD and NASH | Downregulated | Inflammation [128] |
| miR-155 | NASH | Upregulated | Lipid metabolism, intestinal permeability modulation, and inflammation [125,130] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutiérrez-Cuevas, J.; Lucano-Landeros, S.; López-Cifuentes, D.; Santos, A.; Armendariz-Borunda, J. Epidemiologic, Genetic, Pathogenic, Metabolic, Epigenetic Aspects Involved in NASH-HCC: Current Therapeutic Strategies. Cancers 2023, 15, 23. https://doi.org/10.3390/cancers15010023
Gutiérrez-Cuevas J, Lucano-Landeros S, López-Cifuentes D, Santos A, Armendariz-Borunda J. Epidemiologic, Genetic, Pathogenic, Metabolic, Epigenetic Aspects Involved in NASH-HCC: Current Therapeutic Strategies. Cancers. 2023; 15(1):23. https://doi.org/10.3390/cancers15010023
Chicago/Turabian StyleGutiérrez-Cuevas, Jorge, Silvia Lucano-Landeros, Daniel López-Cifuentes, Arturo Santos, and Juan Armendariz-Borunda. 2023. "Epidemiologic, Genetic, Pathogenic, Metabolic, Epigenetic Aspects Involved in NASH-HCC: Current Therapeutic Strategies" Cancers 15, no. 1: 23. https://doi.org/10.3390/cancers15010023
APA StyleGutiérrez-Cuevas, J., Lucano-Landeros, S., López-Cifuentes, D., Santos, A., & Armendariz-Borunda, J. (2023). Epidemiologic, Genetic, Pathogenic, Metabolic, Epigenetic Aspects Involved in NASH-HCC: Current Therapeutic Strategies. Cancers, 15(1), 23. https://doi.org/10.3390/cancers15010023