
TP-0903 Is Active in Preclinical Models of Acute Myeloid Leukemia with TP53 Mutation/Deletion

 , , ,
, , , 
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. Cell Culture
2.3. Generation of the MV4-11 (R248W) Cells and Mutational Analysis
2.4. Characterization of the MV4-11 TP53 Mutant (R248W) Cells via Gamma Irradiation
2.5. Immunoblotting
2.6. Kinase Profiling
2.7. Cell Viability Assessment
2.8. Cell Cycle Analysis
2.9. Apoptosis Assessment and CD11b Expression Assays
2.10. RNA Isolation and RT-PCR
2.11. Growth Curves
2.12. Murine Xenograft Models
2.13. Targeted Gene Sequencing
2.14. Immunohistochemistry
2.15. Statistics
3. Results
3.1. Isolation and Characterization of a TP53 Mutant MV4-11 Clone
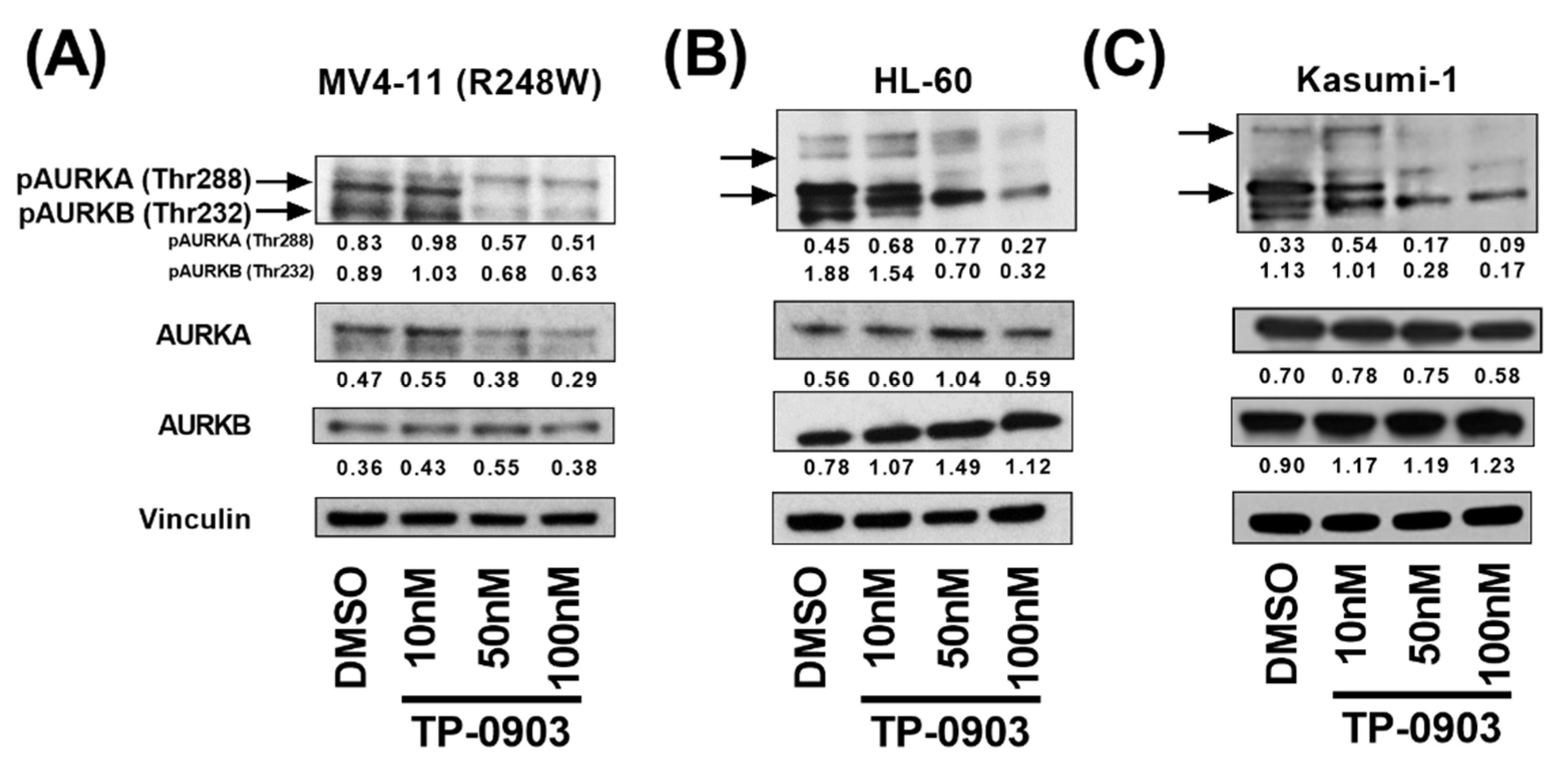
3.2. TP-0903 Inhibits Aurora Kinases
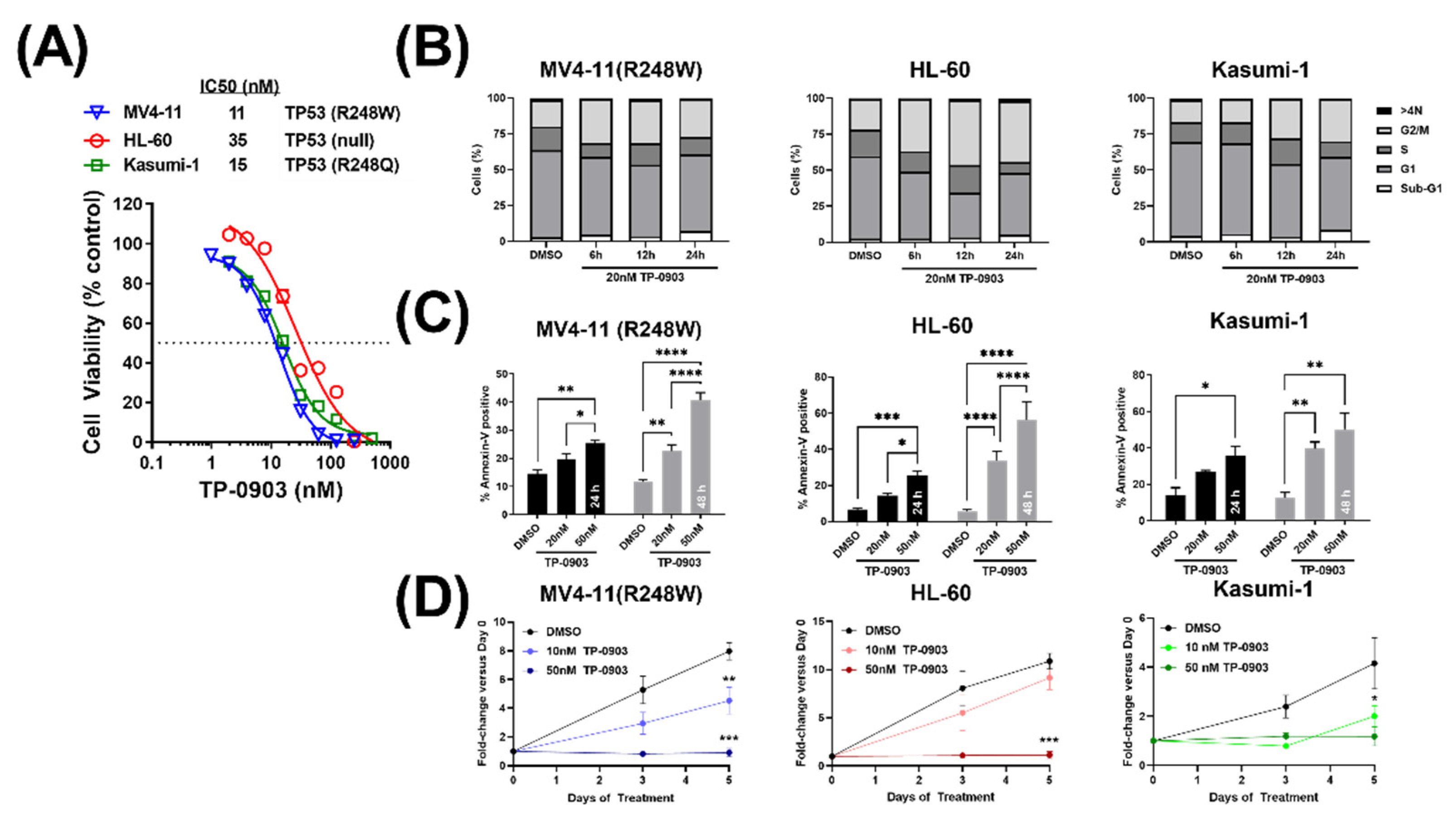
3.3. TP-0903 Has In Vitro Activity in TP53 Mutant AML Cell Lines
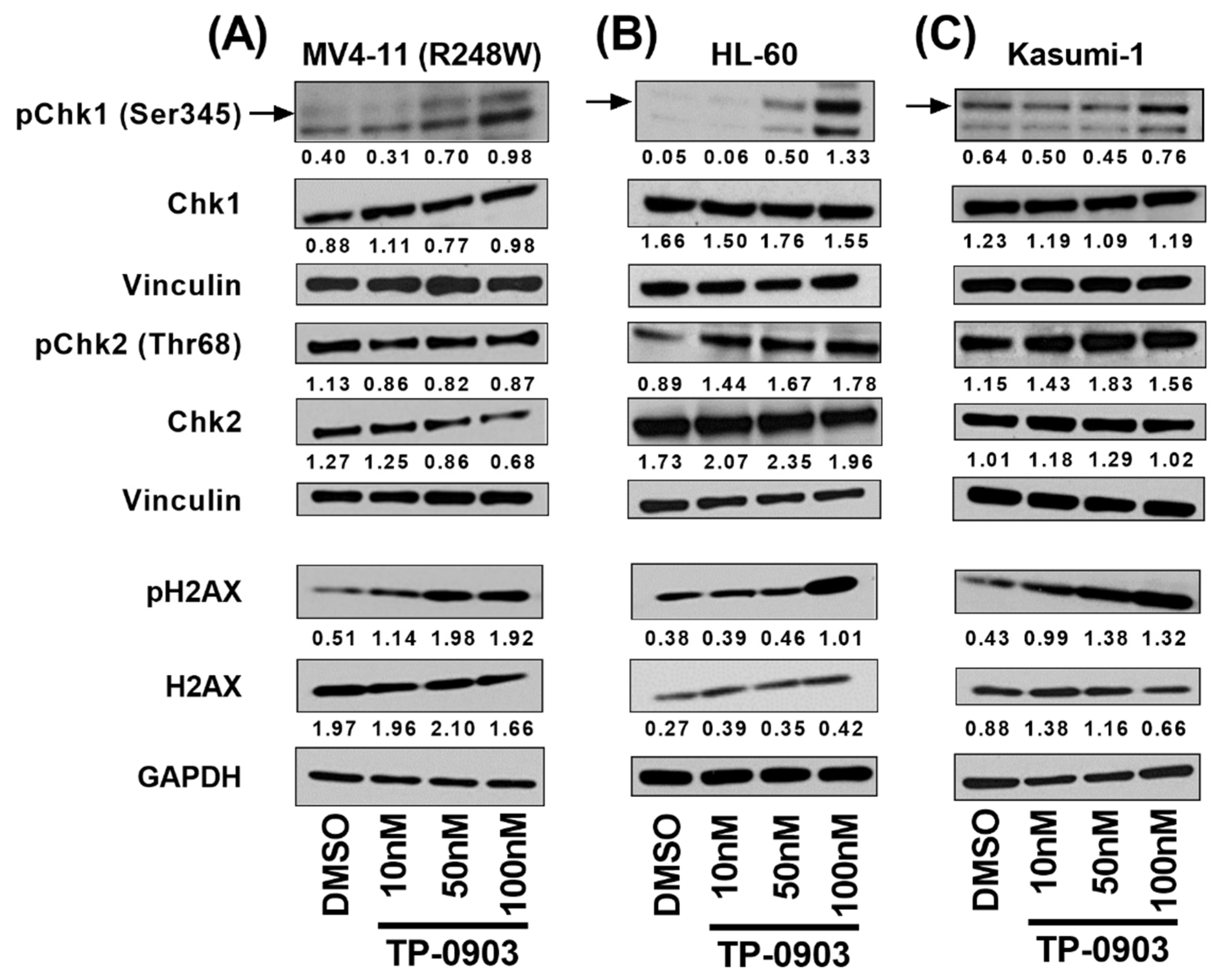
3.4. TP-0903 Inhibits Checkpoint Kinases
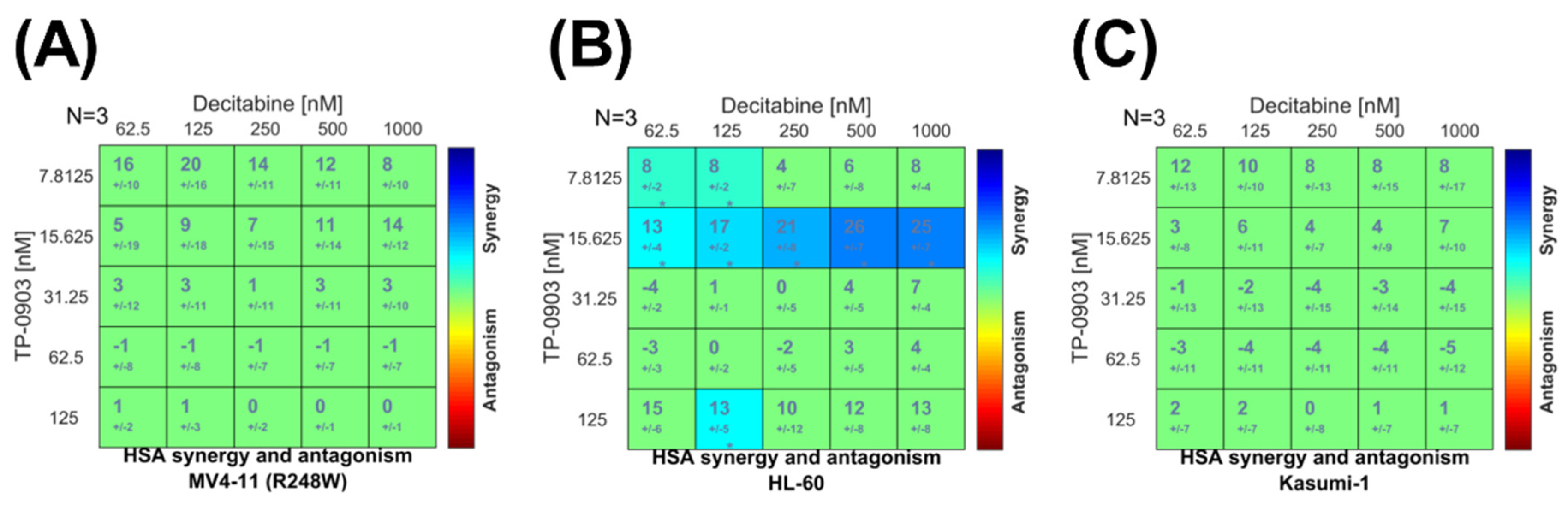
3.5. Combination of TP-0903 and Decitabine Is Active In Vitro
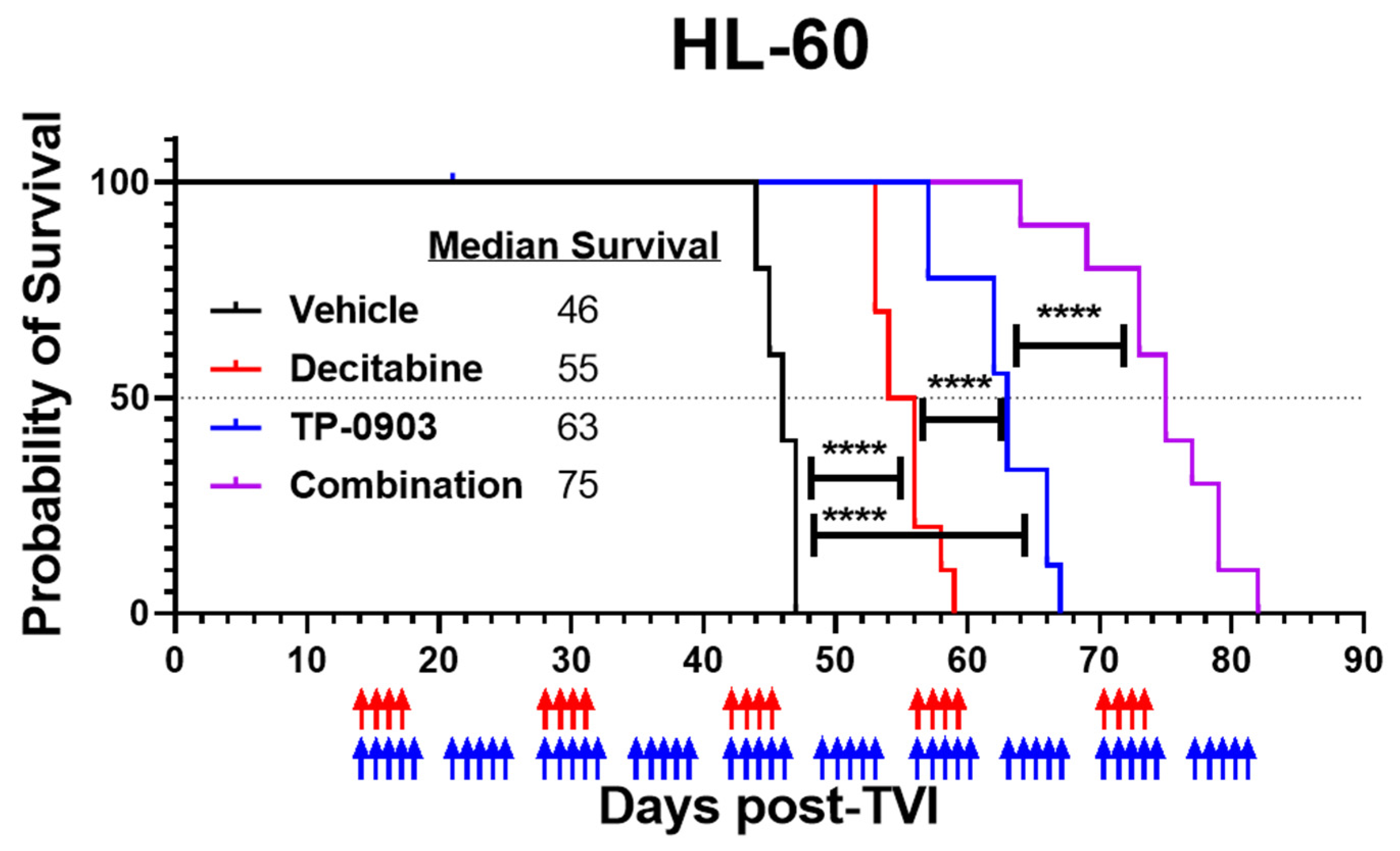
3.6. TP-0903 and Decitabine Improve Survival In Vivo in an HL-60 Xenograft Model
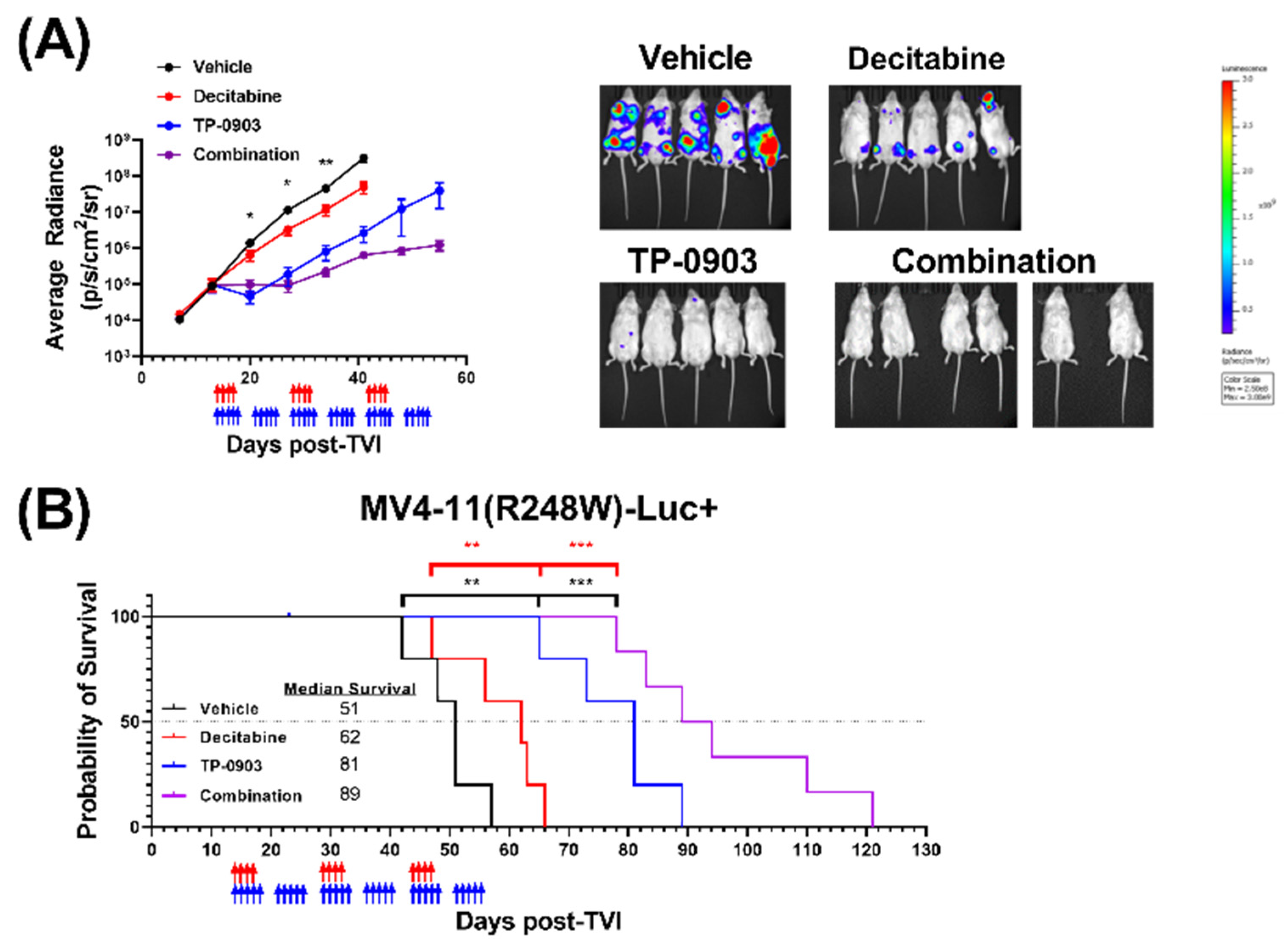
3.7. TP-0903 Is Active in a MV4-11 (R248W)-Luc+ Xenograft Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute Myeloid Leukemia: Current Progress and Future Directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef]
- Hou, H.-A.; Chou, W.-C.; Kuo, Y.-Y.; Liu, C.-Y.; Lin, L.-I.; Tseng, M.-H.; Chiang, Y.-C.; Liu, M.-C.; Liu, C.-W.; Tang, J.-L.; et al. TP53 Mutations in de Novo Acute Myeloid Leukemia Patients: Longitudinal Follow-Ups Show the Mutation Is Stable during Disease Evolution. Blood Cancer J. 2015, 5, e331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ok, C.Y.; Patel, K.P.; Garcia-Manero, G.; Routbort, M.J.; Peng, J.; Tang, G.; Goswami, M.; Young, K.H.; Singh, R.; Medeiros, L.J.; et al. TP53 Mutation Characteristics in Therapy-Related Myelodysplastic Syndromes and Acute Myeloid Leukemia Is Similar to de Novo Diseases. J. Hematol. Oncol. 2015, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Burd, A.; Levine, R.L.; Ruppert, A.S.; Mims, A.S.; Borate, U.; Stein, E.M.; Patel, P.; Baer, M.R.; Stock, W.; Deininger, M.; et al. Precision Medicine Treatment in Acute Myeloid Leukemia Using Prospective Genomic Profiling: Feasibility and Preliminary Efficacy of the Beat AML Master Trial. Nat. Med. 2020, 26, 1852–1858. [Google Scholar] [CrossRef]
- Prochazka, K.T.; Pregartner, G.; Rücker, F.G.; Heitzer, E.; Pabst, G.; Wölfler, A.; Zebisch, A.; Berghold, A.; Döhner, K.; Sill, H. Clinical Implications of Subclonal TP53 Mutations in Acute Myeloid Leukemia. Haematologica 2019, 104, 516–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Comprehensive Cancer Network Acute Myeloid Leukemia (Version 1.2022). Available online: https://www.nccn.org/professionals/physician_gls/pdf/aml.pdf (accessed on 1 September 2022).
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef]
- Kim, K.; Maiti, A.; Loghavi, S.; Pourebrahim, R.; Kadia, T.M.; Rausch, C.R.; Furudate, K.; Daver, N.G.; Alvarado, Y.; Ohanian, M.; et al. Outcomes of TP53 -mutant Acute Myeloid Leukemia with Decitabine and Venetoclax. Cancer 2021, 127, 3772–3781. [Google Scholar] [CrossRef]
- Abou Zahr, A.; Borthakur, G. Emerging Cell Cycle Inhibitors for Acute Myeloid Leukemia. Expert Opin. Emerg. Drugs 2017, 22, 137–148. [Google Scholar] [CrossRef]
- Dixon, H.; Norbury, C.J. Therapeutic Exploitation of Checkpoint Defects in Cancer Cells Lacking P53 Function. Cell Cycle 2002, 1, 362–368. [Google Scholar] [CrossRef]
- Neizer-Ashun, F.; Bhattacharya, R. Reality CHEK: Understanding the Biology and Clinical Potential of CHK1. Cancer Lett. 2021, 497, 202–211. [Google Scholar] [CrossRef]
- Dar, A.A.; Belkhiri, A.; Ecsedy, J.; Zaika, A.; El-Rifai, W. Aurora Kinase A Inhibition Leads to P73-Dependent Apoptosis in P53-Deficient Cancer Cells. Cancer Res. 2008, 68, 8998–9004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marxer, M.; Ma, H.T.; Man, W.Y.; Poon, R.Y.C. P53 Deficiency Enhances Mitotic Arrest and Slippage Induced by Pharmacological Inhibition of Aurora Kinases. Oncogene 2014, 33, 3550–3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borah, N.A.; Reddy, M.M. Aurora Kinase B Inhibition: A Potential Therapeutic Strategy for Cancer. Molecules 2021, 26, 1981. [Google Scholar] [CrossRef]
- Gali-Muhtasib, H.; Kuester, D.; Mawrin, C.; Bajbouj, K.; Diestel, A.; Ocker, M.; Habold, C.; Foltzer-Jourdainne, C.; Schoenfeld, P.; Peters, B.; et al. Thymoquinone Triggers Inactivation of the Stress Response Pathway Sensor CHEK1 and Contributes to Apoptosis in Colorectal Cancer Cells. Cancer Res. 2008, 68, 5609–5618. [Google Scholar] [CrossRef] [Green Version]
- Jammal, N.; Rausch, C.R.; Kadia, T.M.; Pemmaraju, N. Cell Cycle Inhibitors for the Treatment of Acute Myeloid Leukemia: A Review of Phase 2 & 3 Clinical Trials. Expert Opin. Emerg. Drugs 2020, 25, 491–499. [Google Scholar] [CrossRef]
- Alcaraz-Sanabria, A.; Nieto-Jiménez, C.; Corrales-Sánchez, V.; Serrano-Oviedo, L.; Andrés-Pretel, F.; Montero, J.C.; Burgos, M.; Llopis, J.; Galán-Moya, E.M.; Pandiella, A.; et al. Synthetic Lethality Interaction between Aurora Kinases and CHEK1 Inhibitors in Ovarian Cancer. Mol. Cancer Ther. 2017, 16, 2552–2562. [Google Scholar] [CrossRef] [Green Version]
- Mollard, A.; Warner, S.L.; Call, L.T.; Wade, M.L.; Bearss, J.J.; Verma, A.; Sharma, S.; Vankayalapati, H.; Bearss, D.J. Design, Synthesis, and Biological Evaluation of a Series of Novel AXL Kinase Inhibitors. ACS Med. Chem. Lett. 2011, 2, 907–912. [Google Scholar] [CrossRef]
- Jeon, J.Y.; Buelow, D.R.; Garrison, D.A.; Niu, M.; Eisenmann, E.D.; Huang, K.M.; Zavorka Thomas, M.E.; Weber, R.H.; Whatcott, C.J.; Warner, S.L.; et al. TP-0903 Is Active in Models of Drug-Resistant Acute Myeloid Leukemia. JCI Insight 2020, 5, e140169. [Google Scholar] [CrossRef]
- Kumagai, Y.; Oishi, J.; Nakamura, M.; Foulks, J.M.; Whatcott, C.J.; Warner, S.L.; Bearss, D.J.; Goto, M. Abstract 3969: TP-0903, a Potent AXL Receptor Tyrosine Kinase Inhibitor, Enhances the Activity of Anti-PD-1 Therapy in a Metastatic Preclinical Syngeneic Model of Breast Cancer. Cancer Res. 2019, 79, 3969. [Google Scholar] [CrossRef]
- Cheng, Y.; Funakoshi, Y.; Wang, X.; Warner, S.; Bearss, D.; Ueno, N. Abstract P2-06-05: TP-0903, an AXL Kinase Inhibitor, Reduces Inflammatory Breast Cancer Aggressiveness and Macrophage Polarization through Additional Mechanisms That May Include JAK2 and Aurora B. Cancer Res. 2019, 79, P2-06. [Google Scholar] [CrossRef]
- Zhang, Y.; Arner, E.N.; Rizvi, A.; Toombs, J.E.; Huang, H.; Warner, S.L.; Foulks, J.M.; Brekken, R.A. AXL Inhibitor TP-0903 Reduces Metastasis and Therapy Resistance in Pancreatic Cancer. Mol. Cancer Ther. 2022, 21, 38–47. [Google Scholar] [CrossRef]
- Yan, B.; Chen, Q.; Xu, J.; Li, W.; Xu, B.; Qiu, Y. Low-Frequency TP53 Hotspot Mutation Contributes to Chemoresistance through Clonal Expansion in Acute Myeloid Leukemia. Leukemia 2020, 34, 1816–1827. [Google Scholar] [CrossRef]
- Eisfeld, A.-K.; Kohlschmidt, J.; Mrózek, K.; Blachly, J.S.; Nicolet, D.; Kroll, K.; Orwick, S.; Carroll, A.J.; Stone, R.M.; de la Chapelle, A. Adult Acute Myeloid Leukemia with Trisomy 11 as the Sole Abnormality Is Characterized by the Presence of Five Distinct Gene Mutations: MLL-PTD, DNMT3A, U2AF1, FLT3-ITD and IDH2. Leukemia 2016, 30, 2254. [Google Scholar] [CrossRef] [Green Version]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An Interactive Platform for the Analysis and Visualization of Drug Combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive Detection of Somatic Point Mutations in Impure and Heterogeneous Cancer Samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Ding, Q.; Zhang, Z.; Liu, J.-J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.-J.; Bartkovitz, D.; Podlaski, F.; Janson, C. Discovery of RG7388, a Potent and Selective P53–MDM2 Inhibitor in Clinical Development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef]
- Karthigeyan, D.; Prasad, S.B.B.; Shandilya, J.; Agrawal, S.; Kundu, T.K. Biology of Aurora A Kinase: Implications in Cancer Manifestation and Therapy: BIOLOGY OF AURORA A KINASE. Med. Res. Rev. 2011, 31, 757–793. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Simon, R. Identification of Potential Synthetic Lethal Genes to P53 Using a Computational Biology Approach. BMC Med. Genom. 2013, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsels, L.A.; Qian, Y.; Tanska, D.M.; Gross, M.; Zhao, L.; Hassan, M.C.; Arumugarajah, S.; Parsels, J.D.; Hylander-Gans, L.; Simeone, D.M.; et al. Assessment of Chk1 Phosphorylation as a Pharmacodynamic Biomarker of Chk1 Inhibition. Clin. Cancer Res. 2011, 17, 3706–3715. [Google Scholar] [CrossRef] [Green Version]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the P53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Mirza, S.A.; Xu, S.; Schulz-Heddergott, R.; Marchenko, N.D.; Moll, U.M. P53 Loss-of-Heterozygosity Is a Necessary Prerequisite for Mutant P53 Stabilization and Gain-of-Function in Vivo. Cell Death Dis. 2017, 8, e2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, T.; Rothenberg-Thurley, M.; Grunwald, V.V.; Janke, H.; Goerlich, D.; Sauerland, M.C.; Konstandin, N.P.; Dufour, A.; Schneider, S.; Neusser, M.; et al. Validation and Refinement of the Revised 2017 European LeukemiaNet Genetic Risk Stratification of Acute Myeloid Leukemia. Leukemia 2020, 34, 3161–3172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prokocimer, M.; Molchadsky, A.; Rotter, V. Dysfunctional Diversity of P53 Proteins in Adult Acute Myeloid Leukemia: Projections on Diagnostic Workup and Therapy. Blood 2017, 130, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Pregartner, G.; Rücker, F.G.; Heitzer, E.; Zebisch, A.; Bullinger, L.; Berghold, A.; Döhner, K.; Sill, H. Functional Classification of TP53 Mutations in Acute Myeloid Leukemia. Cancers 2020, 12, 637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grieselhuber, N.R.; Mims, A.S. Novel Targeted Therapeutics in Acute Myeloid Leukemia: An Embarrassment of Riches. Curr. Hematol. Malig. Rep. 2021, 16, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Ali, D.; Jönsson-Videsäter, K.; Deneberg, S.; Bengtzén, S.; Nahi, H.; Paul, C.; Lehmann, S. APR-246 Exhibits Anti-Leukemic Activity and Synergism with Conventional Chemotherapeutic Drugs in Acute Myeloid Leukemia Cells: APR-246 in Acute Myeloid Leukemia. Eur. J. Haematol. 2011, 86, 206–215. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53 -Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone Des Myélodysplasies (GFM). JCO 2021, 39, 1575–1583. [Google Scholar] [CrossRef]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.-H.; Bally, C.; et al. Synergistic Effects of PRIMA-1 Met (APR-246) and 5-Azacitidine in TP53 -Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia. Haematologica 2020, 105, 1539–1551. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.Y.; Sung Kang, H.; Kim, G.-Y.; Kim, W.-J.; Yoo, Y.H.; Choi, Y.H. Decitabine, a DNA Methyltransferases Inhibitor, Induces Cell Cycle Arrest at G2/M Phase through P53-Independent Pathway in Human Cancer Cells. Biomed. Pharmacother. 2013, 67, 305–311. [Google Scholar] [CrossRef]
- Hollenbach, P.W.; Nguyen, A.N.; Brady, H.; Williams, M.; Ning, Y.; Richard, N.; Krushel, L.; Aukerman, S.L.; Heise, C.; MacBeth, K.J. A Comparison of Azacitidine and Decitabine Activities in Acute Myeloid Leukemia Cell Lines. PLoS ONE 2010, 5, e9001. [Google Scholar] [CrossRef]
- Gore, L.; Triche, T.J.; Farrar, J.E.; Wai, D.; Legendre, C.; Gooden, G.C.; Liang, W.S.; Carpten, J.; Lee, D.; Alvaro, F.; et al. A Multicenter, Randomized Study of Decitabine as Epigenetic Priming with Induction Chemotherapy in Children with AML. Clin. Epigenetics 2017, 9, 108. [Google Scholar] [CrossRef] [Green Version]
- Rudek, M.A.; Zhao, M.; He, P.; Hartke, C.; Gilbert, J.; Gore, S.D.; Carducci, M.A.; Baker, S.D. Pharmacokinetics of 5-Azacitidine Administered with Phenylbutyrate in Patients With Refractory Solid Tumors or Hematologic Malignancies. JCO 2005, 23, 3906–3911. [Google Scholar] [CrossRef]
- Oki, Y.; Kantarjian, H.M.; Gharibyan, V.; Jones, D.; O’brien, S.; Verstovsek, S.; Cortes, J.; Morris, G.M.; Garcia-Manero, G.; Issa, J.-P.J. Phase II Study of Low-Dose Decitabine in Combination with Imatinib Mesylate in Patients with Accelerated or Myeloid Blastic Phase of Chronic Myelogenous Leukemia. Cancer 2007, 109, 899–906. [Google Scholar] [CrossRef]
- Muppidi, M.R.; Portwood, S.; Griffiths, E.A.; Thompson, J.E.; Ford, L.A.; Freyer, C.W.; Wetzler, M.; Wang, E.S. Decitabine and Sorafenib Therapy in FLT-3 ITD-Mutant Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2015, 15, S73–S79. [Google Scholar] [CrossRef]
- Sarantopoulos, J.; Fotopoulos, G.; Tsai, F.Y.-C.; Beg, M.S.; Adjei, A.A.; Lou, Y.; Seetharam, M.; Villalona-Calero, M.A.; Melear, J.; Janat-Amsbury, M.; et al. A Phase Ia/b First-in-Human, Open-Label, Dose-Escalation, Safety, PK and PD Study of TP-0903 in Solid Tumours. Ann. Oncol. 2019, 30, v172. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eisenmann, E.D.; Stromatt, J.C.; Fobare, S.; Huang, K.M.; Buelow, D.R.; Orwick, S.; Jeon, J.Y.; Weber, R.H.; Larsen, B.; Mims, A.S.; et al. TP-0903 Is Active in Preclinical Models of Acute Myeloid Leukemia with TP53 Mutation/Deletion. Cancers 2023, 15, 29. https://doi.org/10.3390/cancers15010029
Eisenmann ED, Stromatt JC, Fobare S, Huang KM, Buelow DR, Orwick S, Jeon JY, Weber RH, Larsen B, Mims AS, et al. TP-0903 Is Active in Preclinical Models of Acute Myeloid Leukemia with TP53 Mutation/Deletion. Cancers. 2023; 15(1):29. https://doi.org/10.3390/cancers15010029
Chicago/Turabian StyleEisenmann, Eric D., Jack C. Stromatt, Sydney Fobare, Kevin M. Huang, Daelynn R. Buelow, Shelley Orwick, Jae Yoon Jeon, Robert H. Weber, Bill Larsen, Alice S. Mims, and et al. 2023. "TP-0903 Is Active in Preclinical Models of Acute Myeloid Leukemia with TP53 Mutation/Deletion" Cancers 15, no. 1: 29. https://doi.org/10.3390/cancers15010029
APA StyleEisenmann, E. D., Stromatt, J. C., Fobare, S., Huang, K. M., Buelow, D. R., Orwick, S., Jeon, J. Y., Weber, R. H., Larsen, B., Mims, A. S., Hertlein, E., Byrd, J. C., & Baker, S. D. (2023). TP-0903 Is Active in Preclinical Models of Acute Myeloid Leukemia with TP53 Mutation/Deletion. Cancers, 15(1), 29. https://doi.org/10.3390/cancers15010029