
Comparison of the Basal Cell Carcinoma (BCC) Tumour Microenvironment to Other Solid Malignancies

 ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition and RNA-Seq Processing
2.2. Cell-Type Enumeration Using RNA Deconvolution
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muzic, J.G.; Schmitt, A.R.; Wright, A.C.; Alniemi, D.T.; Zubair, A.S.; Lourido, J.M.O.; Seda, I.M.S.; Weaver, A.L.; Baum, C.L. Incidence and Trends of Basal Cell Carcinoma and Cutaneous Squamous Cell Carcinoma. Mayo Clin. Proc. 2017, 92, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Chlebicka, I.; Stefaniak, A.; Matusiak, Ł.; Szepietowski, J. Basal cell carcinoma: What new can be learned about the most common human cancer? A cross-sectional prospective study of 180 cases in a single centre. Adv. Dermatol. Allergol. 2021, 38, 1086–1091. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Weinstock, M.A. Nonmelanoma skin cancer in the United States: Incidence. J. Am. Acad. Dermatol. 1994, 30 Pt 1, 774–778. [Google Scholar] [CrossRef]
- Carballo, G.B.; Honorato, J.R.; De Lopes, G.P.F.; de Sampaio e Spohr, T.C.L. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef]
- Marzuka, A.G.; Book, S.E. Basal Cell Carcinoma: Pathogenesis, Epidemiology, Clinical Features, Diagnosis, Histopathology, and Management. Yale J. Boil. Med. 2015, 88, 167–179. [Google Scholar]
- Quinn, A.G.; Epstein, J.E. Patched, Hedgehog, and Skin Cancer; Humana Press: Totowa, NJ, USA, 2003; pp. 85–95. [Google Scholar]
- Rubin, A.I.; Chen, E.H.; Ratner, D. Basal-Cell Carcinoma. N. Engl. J. Med. 2005, 353, 2262–2269. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and Safety of Vismodegib in Advanced Basal-Cell Carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [Green Version]
- Xie, P.; Lefrançois, P. Efficacy, safety, and comparison of sonic hedgehog inhibitors in basal cell carcinomas: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 2018, 79, 1089–1100.e17. [Google Scholar] [CrossRef]
- Mohan, S.V.; Chang, A.L.S. Advanced Basal Cell Carcinoma: Epidemiology and Therapeutic Innovations. Curr. Dermatol. Rep. 2014, 3, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.-P.; Shen, Q.-S.; Yang, C.-P.; Chen, Y.-B. Establishment of basal cell carcinoma animal model in Chinese tree shrew (Tupaia belangeri chinensis). Zool. Res. 2017, 38, 180–190. [Google Scholar] [CrossRef] [Green Version]
- Noubissi, F.K.; Kim, T.; Kawahara, T.N.; Aughenbaugh, W.D.; Berg, E.; Longley, B.; Athar, M.; Spiegelman, V.S. Role of CRD-BP in the Growth of Human Basal Cell Carcinoma Cells. J. Investig. Dermatol. 2014, 134, 1718–1724. [Google Scholar] [CrossRef] [PubMed]
- Lefrançois, P.; Xie, P.; Gunn, S.; Gantchev, J.; Villarreal, A.M.; Sasseville, D.; Litvinov, I.V. In silico analyses of the tumor microenvironment highlight tumoral inflammation, a Th2 cytokine shift and a mesenchymal stem cell-like phenotype in advanced in basal cell carcinomas. J. Cell Commun. Signal. 2020, 14, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, I.V.; Xie, P.; Gunn, S.; Sasseville, D.; Lefrançois, P. The transcriptional landscape analysis of basal cell carcinomas reveals novel signalling pathways and actionable targets. Life Sci. Alliance 2021, 4, e202000651. [Google Scholar] [CrossRef] [PubMed]
- Alkallas, R.; Lajoie, M.; Moldoveanu, D.; Hoang, K.V.; Lefrançois, P.; Lingrand, M.; Ahanfeshar-Adams, M.; Watters, K.; Spatz, A.; Zippin, J.H.; et al. Multi-omic analysis reveals significantly mutated genes and DDX3X as a sex-specific tumor suppressor in cutaneous melanoma. Nat. Cancer 2020, 1, 635–652. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Lefrançois, P.; Xie, P.; Wang, L.; Tetzlaff, M.T.; Moreau, L.; Watters, A.K.; Netchiporouk, E.; Provost, N.; Gilbert, M.; Ni, X.; et al. Gene expression profiling and immune cell-type deconvolution highlight robust disease progression and survival markers in multiple cohorts of CTCL patients. OncoImmunology 2018, 7, e1467856. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Kovaleva, O.V.; Samoilova, D.V.; Shitova, M.S.; Gratchev, A. Tumor Associated Macrophages in Kidney Cancer. Anal. Cell. Pathol. 2016, 2016, 9307549. [Google Scholar] [CrossRef] [Green Version]
- Goldenberg, G.; Karagiannis, T.; Palmer, J.B.; Lotya, J.; O’Neill, C.; Kisa, R.; Herrera, V.; Siegel, D.M. Incidence and prevalence of basal cell carcinoma (BCC) and locally advanced BCC (LABCC) in a large commercially insured population in the United States: A retrospective cohort study. J. Am. Acad. Dermatol. 2016, 75, 957–966.e2. [Google Scholar] [CrossRef]
- Volpe, A.; Novara, G.; Antonelli, A.; Bertini, R.; Billia, M.; Carmignani, G.; Cunico, S.C.; Longo, N.; Martignoni, G.; Minervini, A.; et al. Chromophobe renal cell carcinoma (RCC): Oncological outcomes and prognostic factors in a large multicentre series. BJU Int. 2011, 110, 76–83. [Google Scholar] [CrossRef]
- Piva de Freitas, P.; Senna, C.G.; Tabai, M.; Chone, C.T.; Altemani, A. Metastatic Basal Cell Carcinoma: A Rare Manifestation of a Common Disease. Case Rep. Med. 2017, 2017, 8929745. [Google Scholar] [CrossRef] [PubMed]
- Shea, C.R.; McNutt, N.S.; Volkenandt, M.; Lugo, J.; Prioleau, P.G.; Albino, A.P. Overexpression of p53 protein in basal cell carcinomas of human skin. Am. J. Pathol. 1992, 141, 25–29. [Google Scholar] [PubMed]
- Jayaraman, S.S.; Rayhan, D.J.; Hazany, S.; Kolodney, M.S. Mutational Landscape of Basal Cell Carcinomas by Whole-Exome Sequencing. J. Investig. Dermatol. 2014, 134, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griewank, K.G.; Murali, R.; Schilling, B.; Schimming, T.; Möller, I.; Moll, I.; Schwamborn, M.; Sucker, A.; Zimmer, L.; Schadendorf, D.; et al. TERT Promoter Mutations Are Frequent in Cutaneous Basal Cell Carcinoma and Squamous Cell Carcinoma. PLoS ONE 2013, 8, e80354. [Google Scholar] [CrossRef]
- Pópulo, H.; Boaventura, P.; Vinagre, J.; Batista, R.; Mendes, A.; Caldas, R.; Pardal, J.; Azevedo, F.; Honavar, M.; Guimarães, I.; et al. TERT Promoter Mutations in Skin Cancer: The Effects of Sun Exposure and X-Irradiation. J. Investig. Dermatol. 2014, 134, 2251–2257. [Google Scholar] [CrossRef] [Green Version]
- Scott, G.A.; Laughlin, T.S.; Rothberg, P.G. Mutations of the TERT promoter are common in basal cell carcinoma and squamous cell carcinoma. Mod. Pathol. 2014, 27, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Garje, R.; Elhag, D.; A Yasin, H.; Acharya, L.; Vaena, D.; Dahmoush, L. Comprehensive review of chromophobe renal cell carcinoma. Crit. Rev. Oncol./Hematol. 2021, 160, 103287. [Google Scholar] [CrossRef]
- Papanikolaou, D.; Ioannidou, P.; Koukourikis, P.; Moysidis, K.; Meditskou, S.; Koutsoumparis, D.; Hatzimouratidis, K.; Hatzivassiliou, E. Systemic therapy for chromophobe renal cell carcinoma: A systematic review. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 137–149. [Google Scholar] [CrossRef]
- Clemente, O.; Ottaiano, A.; Di Lorenzo, G.; Bracigliano, A.; Lamia, S.; Cannella, L.; Pizzolorusso, A.; Di Marzo, M.; Santorsola, M.; De Chiara, A.; et al. Is immunotherapy in the future of therapeutic management of sarcomas? J. Transl. Med. 2021, 19, 173. [Google Scholar] [CrossRef]
- Dancsok, A.R.; Asleh-Aburaya, K.; Nielsen, T.O. Advances in sarcoma diagnostics and treatment. Oncotarget 2016, 8, 7068–7093. [Google Scholar] [CrossRef] [Green Version]
- Navarrete-Dechent, C.; Mori, S.; Barker, C.A.; Dickson, M.A.; Nehal, K.S. Imatinib Treatment for Locally Advanced or Metastatic Dermatofibrosarcoma Protuberans. JAMA Dermatol. 2019, 155, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Kaporis, H.G.; Guttman-Yassky, E.; A Lowes, M.; Haider, A.S.; Fuentes-Duculan, J.; Darabi, K.; Whynot-Ertelt, J.; Khatcherian, A.; Cardinale, I.; Novitskaya, I.; et al. Human Basal Cell Carcinoma Is Associated with Foxp3+ T cells in a Th2 Dominant Microenvironment. J. Investig. Dermatol. 2007, 127, 2391–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackiewicz-Wysocka, M.; Bowszyc-Dmochowska, M.; Strzelecka-Węklar, D.; Dańczak-Pazdrowska, A.; Adamski, Z. Reviews Basal cell carcinoma—Diagnosis. Contemp. Oncol./Współczesna Onkol. 2013, 4, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Nishio, J.; Iwasaki, H.; Nabeshima, K.; Naito, M. Cytogenetics and Molecular Genetics of Myxoid Soft-Tissue Sarcomas. Genet. Res. Int. 2011, 2011, 497148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkiewicz, A.K.; Chung, S.; Brough, R.; Vail, P.; Franco, J.; Lord, C.; Knudsen, E.S. Targeting the Vulnerability of RB Tumor Suppressor Loss in Triple-Negative Breast Cancer. Cell Rep. 2018, 22, 1185–1199. [Google Scholar] [CrossRef] [Green Version]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [Green Version]
- I Daud, A.; Daud; Soon, C.W. Treatment of cutaneous melanoma: Current approaches and future prospects. Cancer Manag. Res. 2010, 2, 197–211. [Google Scholar] [CrossRef]
- Libra, M.; Malaponte, G.; Bevelacqua, V.; Siciliano, R.; Castrogiovanni, P.; Fulvi, A.; Micali, G.; Ligresti, G.; Mazzarino, M.C.; Stivala, F.; et al. Absence of BRAF Gene Mutation in Non-Melanoma Skin Tumors. Cell Cycle 2006, 5, 968–970. [Google Scholar] [CrossRef] [Green Version]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic Activation of KIT in Distinct Subtypes of Melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef]
- Terada, T. Expression of NCAM (CD56), chromogranin A, synaptophysin, c-KIT (CD117) and PDGFRA in normal non-neoplastic skin and basal cell carcinoma: An immunohistochemical study of 66 consecutive cases. Med. Oncol. 2013, 30, 444. [Google Scholar] [CrossRef]
- Leon, A.; Ceauşu, Z.; Ceauşu, M.; Ardeleanu, C.; Mehedinţi, R. Mast cells and dendritic cells in basal cell carcinoma. Rom. J. Morphol. Embryol. 2009, 50, 85–90. [Google Scholar] [PubMed]
- Castillo, J.-M.; Knol, A.-C.; Nguyen, J.-M.; Khammari, A.; Saint-Jean, M.; Dreno, B. Immunohistochemical markers of advanced basal cell carcinoma: CD56 is associated with a lack of response to vismodegib. Eur. J. Dermatol. 2016, 26, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Dessauvagie, B.F.; Wood, B.A. CD117 and CD43 are useful adjuncts in the distinction of adenoid cystic carcinoma from adenoid basal cell carcinoma. Pathology 2015, 47, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.T.; Holden, J.A.; Florell, S.R. CD117, CK20, TTF-1, and DNA topoisomerase II-α antigen expression in small cell tumors. J. Cutan. Pathol. 2004, 31, 254–261. [Google Scholar] [CrossRef]
- Verma, D.; Kantarjian, H.; Strom, S.S.; Rios, M.B.; Jabbour, E.; Quintas-Cardama, A.; Verstovsek, S.; Ravandi, F.; O’Brien, S.; Cortes, J. Malignancies occurring during therapy with tyrosine kinase inhibitors (TKIs) for chronic myeloid leukemia (CML) and other hematologic malignancies. Blood 2011, 118, 4353–4358. [Google Scholar] [CrossRef]
- Jin, Y.-J.; Park, I.; Hong, I.-K.; Byun, H.-J.; Choi, J.; Kim, Y.-M.; Lee, H. Fibronectin and vitronectin induce AP-1-mediated matrix metalloproteinase-9 expression through integrin α5β1/αvβ3-dependent Akt, ERK and JNK signaling pathways in human umbilical vein endothelial cells. Cell. Signal. 2011, 23, 125–134. [Google Scholar] [CrossRef]
- Qin, Y.; Milton, D.R.; Oba, J.; Ding, Z.; Lizée, G.; Ekmekcioglu, S.; Grimm, E.A. Inflammatory IL-1β-driven JNK activation in stage III melanoma. Pigment Cell Melanoma Res. 2015, 28, 236–239. [Google Scholar] [CrossRef]
- Santarlasci, V.; Cosmi, L.; Maggi, L.; Liotta, F.; Annunziato, F. IL-1 and T Helper Immune Responses. Front. Immunol. 2013, 4, 182. [Google Scholar] [CrossRef] [Green Version]
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 2020, 9, 857. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly Recurrent TERT Promoter Mutations in Human Melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [Green Version]
- Negrini, S.; De Palma, R.; Filaci, G. Anti-cancer Immunotherapies Targeting Telomerase. Cancers 2020, 12, 2260. [Google Scholar] [CrossRef] [PubMed]
- Scatena, C.; Murtas, D.; Tomei, S. Cutaneous Melanoma Classification: The Importance of High-Throughput Genomic Technologies. Front. Oncol. 2021, 11, 635488. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kudchadkar, R.; Trefzer, U.; Gogov, S.; et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): A multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015, 16, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, A.; Galdo, G.; Aieta, M.; Fabrizio, T.; Villonio, A.; Conca, R. A Vismodegib Experience in Elderly Patients with Basal Cell Carcinoma: Case Reports and Review of the Literature. Int. J. Mol. Sci. 2020, 21, 8596. [Google Scholar] [CrossRef] [PubMed]
- Biehs, B.; Dijkgraaf, G.J.P.; Piskol, R.; Alicke, B.; Boumahdi, S.; Peale, F.; Gould, S.E.; de Sauvage, F.J. A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 2018, 562, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Stratigos, A.J.; Sekulic, A.; Peris, K.; Bechter, O.; Prey, S.; Kaatz, M.; Lewis, K.D.; Basset-Seguin, N.; Chang, A.L.S.; Dalle, S.; et al. Cemiplimab in locally advanced basal cell carcinoma after hedgehog inhibitor therapy: An open-label, multi-centre, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 848–857. [Google Scholar] [CrossRef]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.S.; et al. Smoothened Variants Explain the Majority of Drug Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic Analysis of Smoothened Inhibitor Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
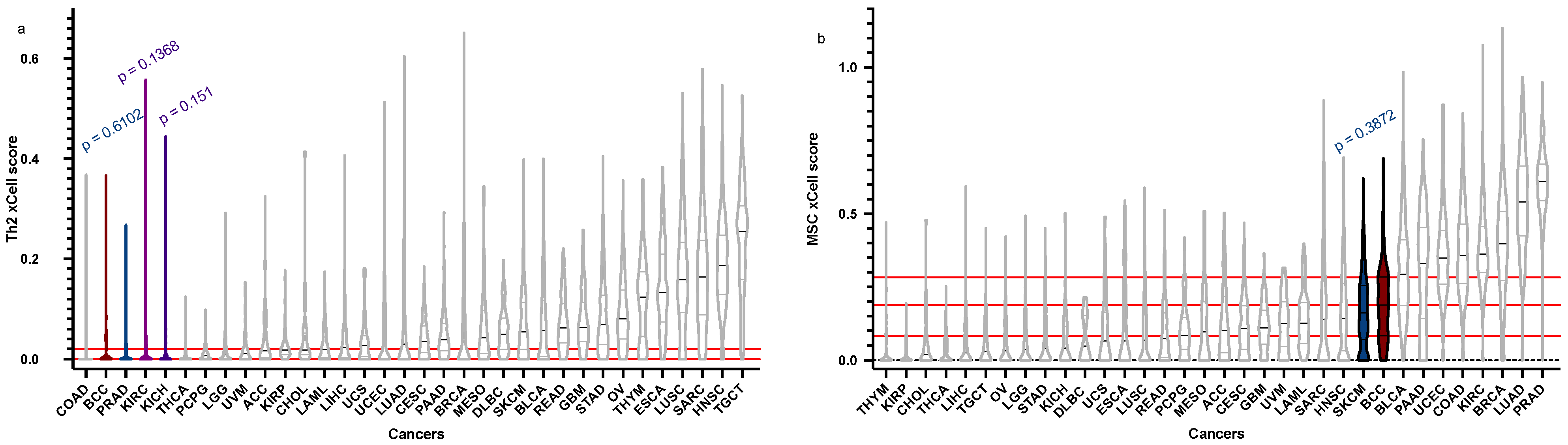
| Cell Type | BCC Score (Median), Q1, Q3 | BCC “Relatives” Score (Median) | Q1 | Q3 | p-Value |
|---|---|---|---|---|---|
| Th2 | 4.188 × 10−18, 0, 0.01980129 | PRAD = 1.07 × 10−17 | 0 | 0.01706565 | 0.6102 |
| KICH = 0.001056705 | 0 | 0.007381045 | 0.151 | ||
| KIRC = 2.23 × 10−17 | 0 | 0.03051409 | 0.1368 | ||
| MSC | 0.188433346, 0.08336783, 0.28205374 | SKCM = 0.16096867 | 0.07202425 | 0.25446426 | 0.3872 |
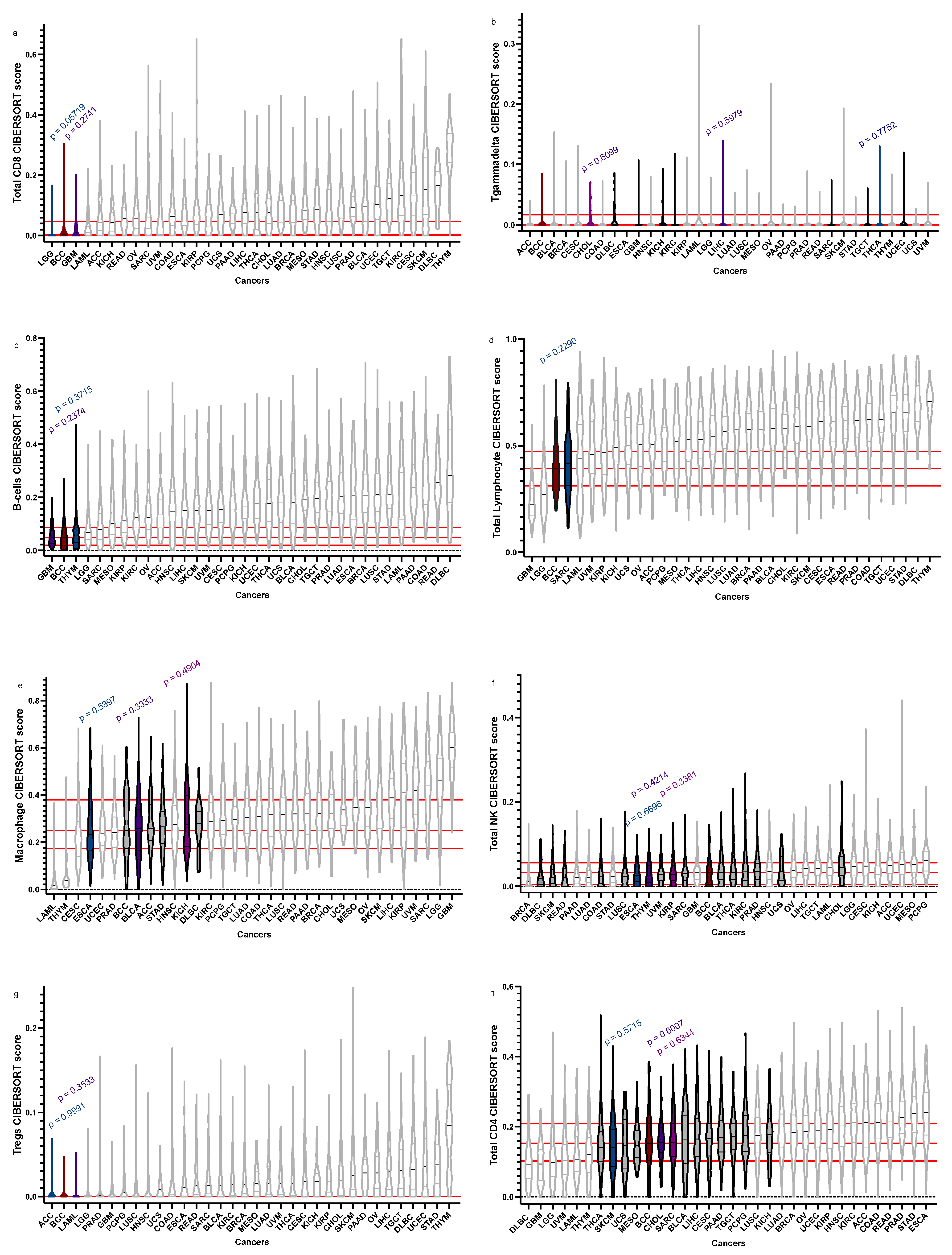
| Total CD8+ | 0.00445073, 0, 0.043739228 | GBM = 0.01048155 | 0 | 0.03021879 | 0.2741 |
| LGG = 0 | 0 | 0.01996548 | 0.05719 | ||
| Tgd | 0, 0, 0.008231755 | THCA = 0 | 0 | 0.008063912 | 0.7752 |
| CHOL = 0 | 0 | 0.01282003 | 0.6099 | ||
| LIHC = 0 | 0 | 0.006720286 | 0.5979 | ||
| B-cells | 0.04857795, 0.020757612, 0.086950544 | THYM = 0.055183104 | 0.03108108 | 0.08455359 | 0.3715 |
| GBM = 0.04815724 | 0.028318876 | 0.075344991 | 0.2374 | ||
| Total Lymphocytes | 0.39140159, 0.3132104, 0.4688854 | SARC = 0.416998654 | 0.3124302 | 0.5153758 | 0.2290 |
| Macrophages | 0.25033106, 0.1763854, 0.3784326 | ESCA = 0.2331247 | 0.1675734 | 0.3352133 | 0.5397 |
| KICH = 0.276729 | 0.1843325 | 0.3949821 | 0.4904 | ||
| BLCA = 0.2590653 | 0.16693727 | 0.33762817 | 0.3333 | ||
| Total NK | 0.0325587, 0.005046464, 0.053041073 | THYM = 0.02682497 | 0.009408674 | 0.049124771 | 0.6696 |
| ESCA = 0.02523973 | 0.01152386 | 0.04604726 | 0.4214 | ||
| KIRP = 0.02878749 | 0.01382830 | 0.04874396 | 0.3381 | ||
| Tregs | 0, 0, 0 | ACC = 0 | 0 | 0 | 0.9991 |
| LAML = 0 | 0 | 0.00315034 | 0.3533 | ||
| Total CD4+ | 0.15296126, 0.1031139, 0.2082608 | CHOL = 0.156189 | 0.1321129 | 0.1952696 | 0.6959 |
| SARC = 0.1562586 | 0.1154291 | 0.2083701 | 0.6334 | ||
| SKCM = 1412604 | 0.08728384 | 0.19168763 | 0.5715 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, E.-R.; Ghezelbash, S.; Xie, P.; Fotovati, M.; Litvinov, I.V.; Lefrançois, P. Comparison of the Basal Cell Carcinoma (BCC) Tumour Microenvironment to Other Solid Malignancies. Cancers 2023, 15, 305. https://doi.org/10.3390/cancers15010305
Zhang E-R, Ghezelbash S, Xie P, Fotovati M, Litvinov IV, Lefrançois P. Comparison of the Basal Cell Carcinoma (BCC) Tumour Microenvironment to Other Solid Malignancies. Cancers. 2023; 15(1):305. https://doi.org/10.3390/cancers15010305
Chicago/Turabian StyleZhang, Eliana-Ruobing, Sarah Ghezelbash, Pingxing Xie, Misha Fotovati, Ivan V. Litvinov, and Philippe Lefrançois. 2023. "Comparison of the Basal Cell Carcinoma (BCC) Tumour Microenvironment to Other Solid Malignancies" Cancers 15, no. 1: 305. https://doi.org/10.3390/cancers15010305
APA StyleZhang, E. -R., Ghezelbash, S., Xie, P., Fotovati, M., Litvinov, I. V., & Lefrançois, P. (2023). Comparison of the Basal Cell Carcinoma (BCC) Tumour Microenvironment to Other Solid Malignancies. Cancers, 15(1), 305. https://doi.org/10.3390/cancers15010305