
Potent Anticancer Activity of CXCR4-Targeted Nanostructured Toxins in Aggressive Endometrial Cancer Models

 , ,
, ,  ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.1.1. Cytotoxic Effect
2.1.2. Apoptosis Assessment by Flow Cytometry
2.1.3. Paraffin-Embedded Cell Blocks
2.1.4. DAPI Fluorescent Staining
2.1.5. Immunocytochemistry
2.2. Animals
2.2.1. Antitumor Effect of Nanotoxins in a Subcutaneous CXCR4+ EC Model
2.2.2. Antimetastatic Effect of T22-DITOX-H6 Using a Highly Metastatic Orthotopic CXCR4+ EC Model
2.2.3. Histological Examination
2.2.4. Immunohistochemistry
2.3. Statistical Analysis
3. Results
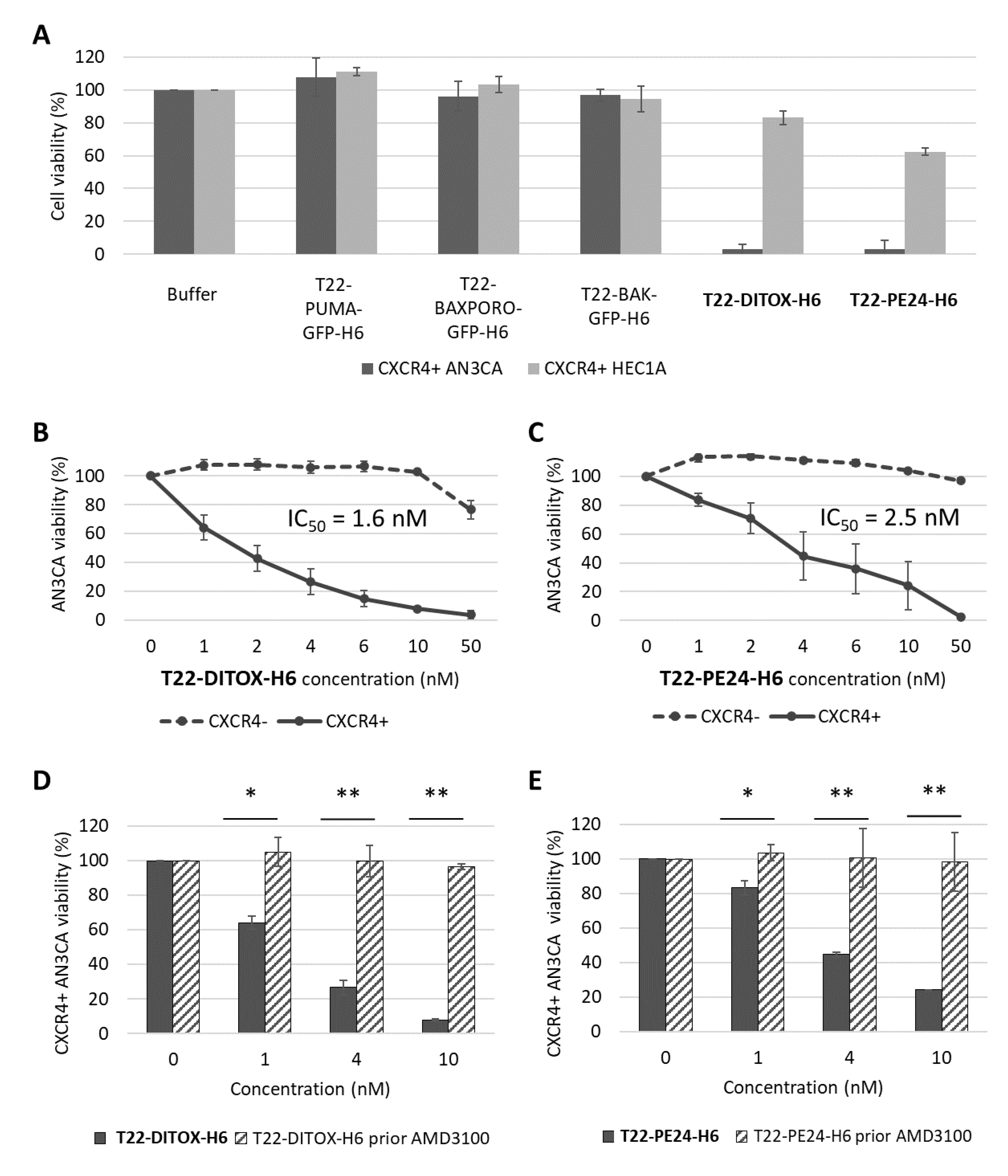
3.1. Potent CXCR4-Dependent Cytotoxic Effect Induced by T22-DITOX-H6 and T22-PE24-H6 in CE Cells In Vitro
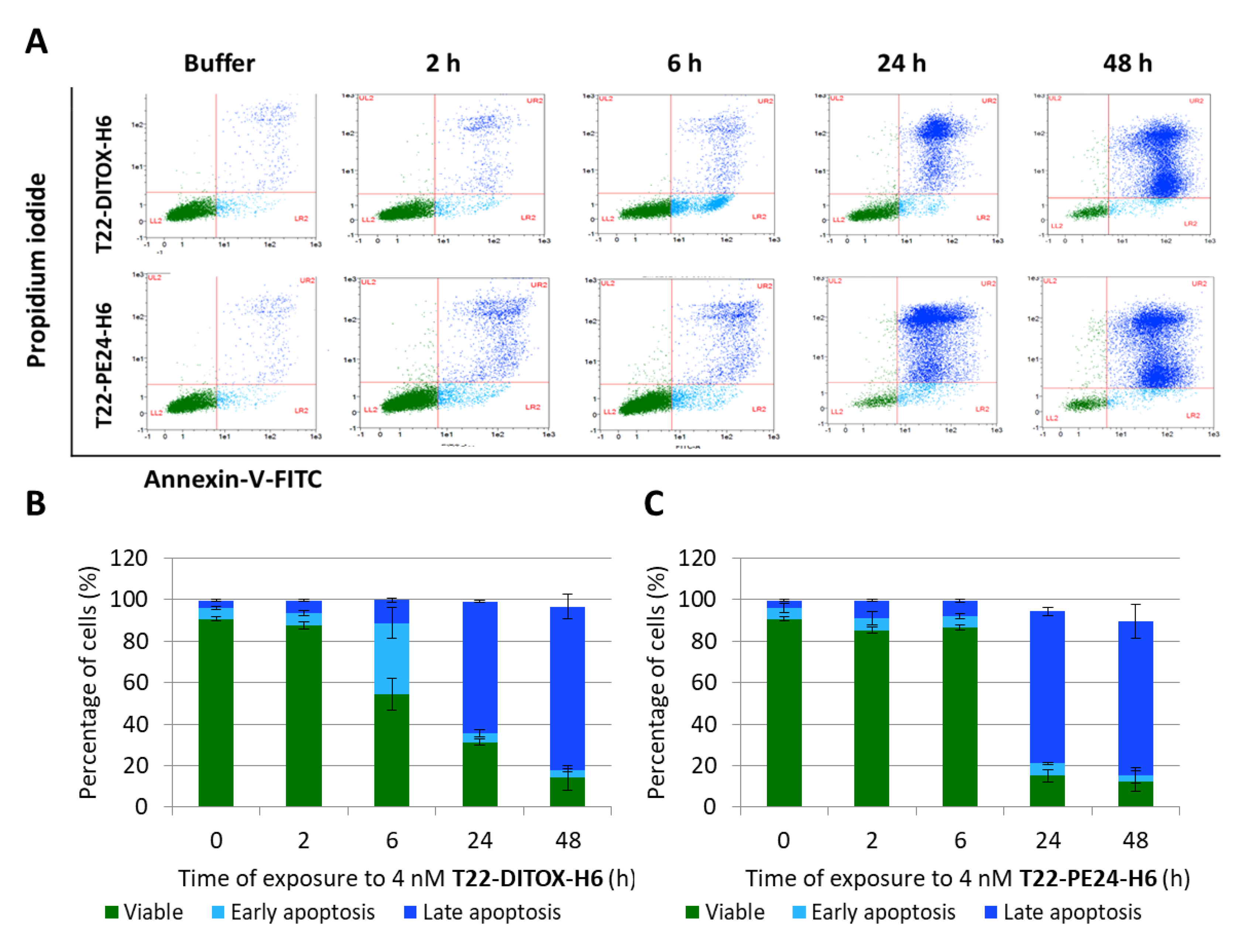
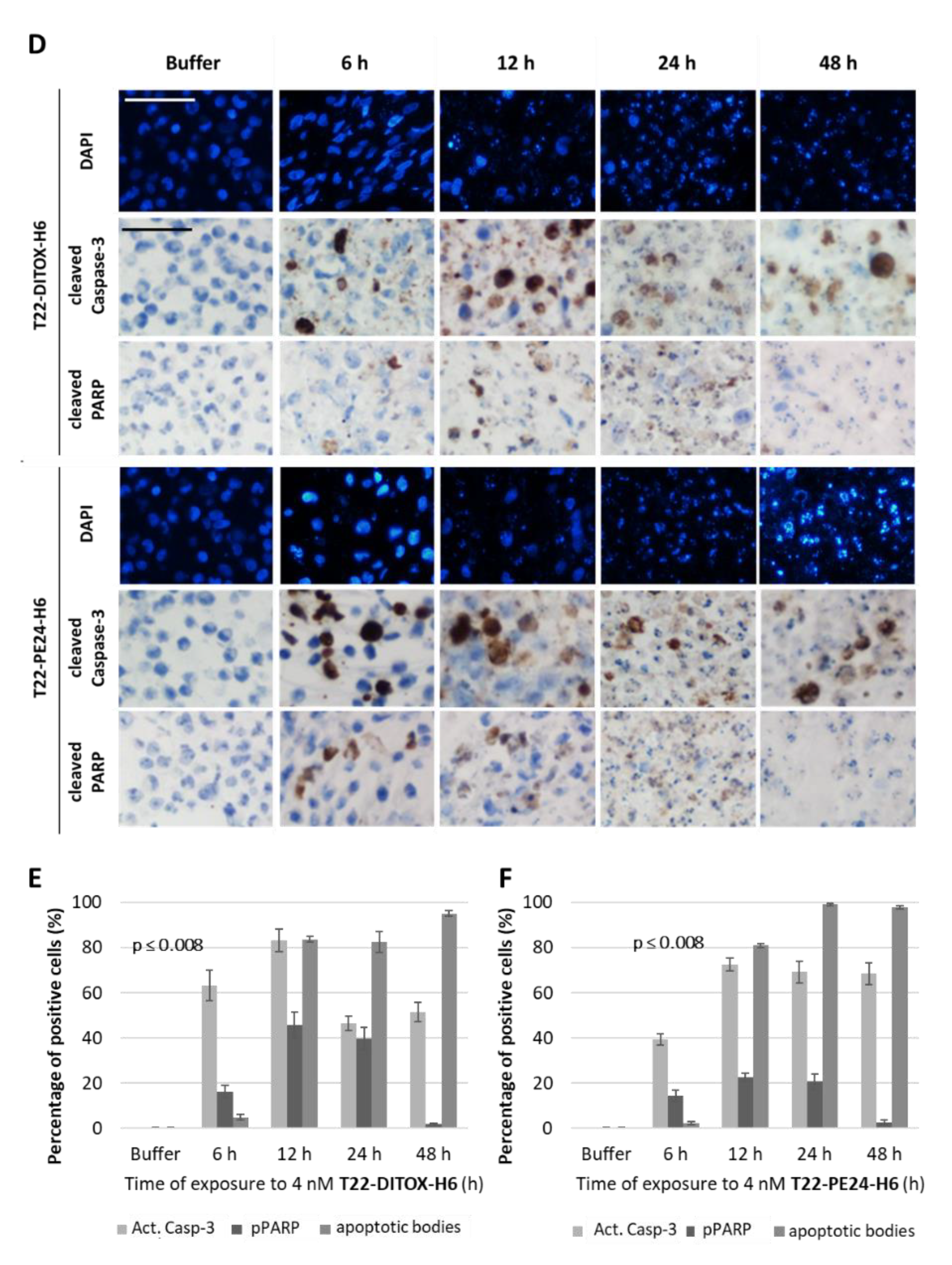
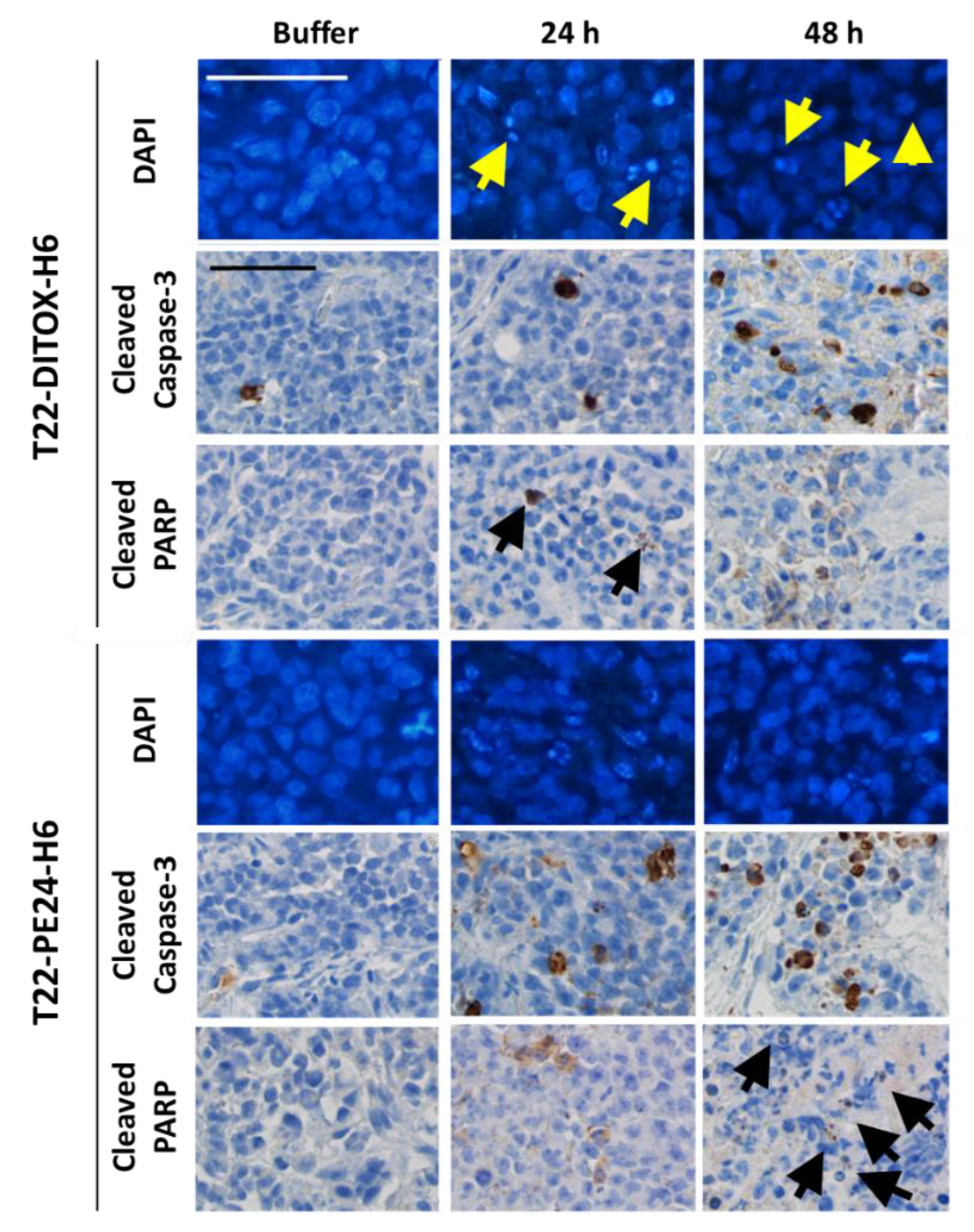
3.2. T22-DITOX-H6 and T22-PE24-H6-Induced EC Cell Death In Vitro Is Mediated by Apoptosis Induction
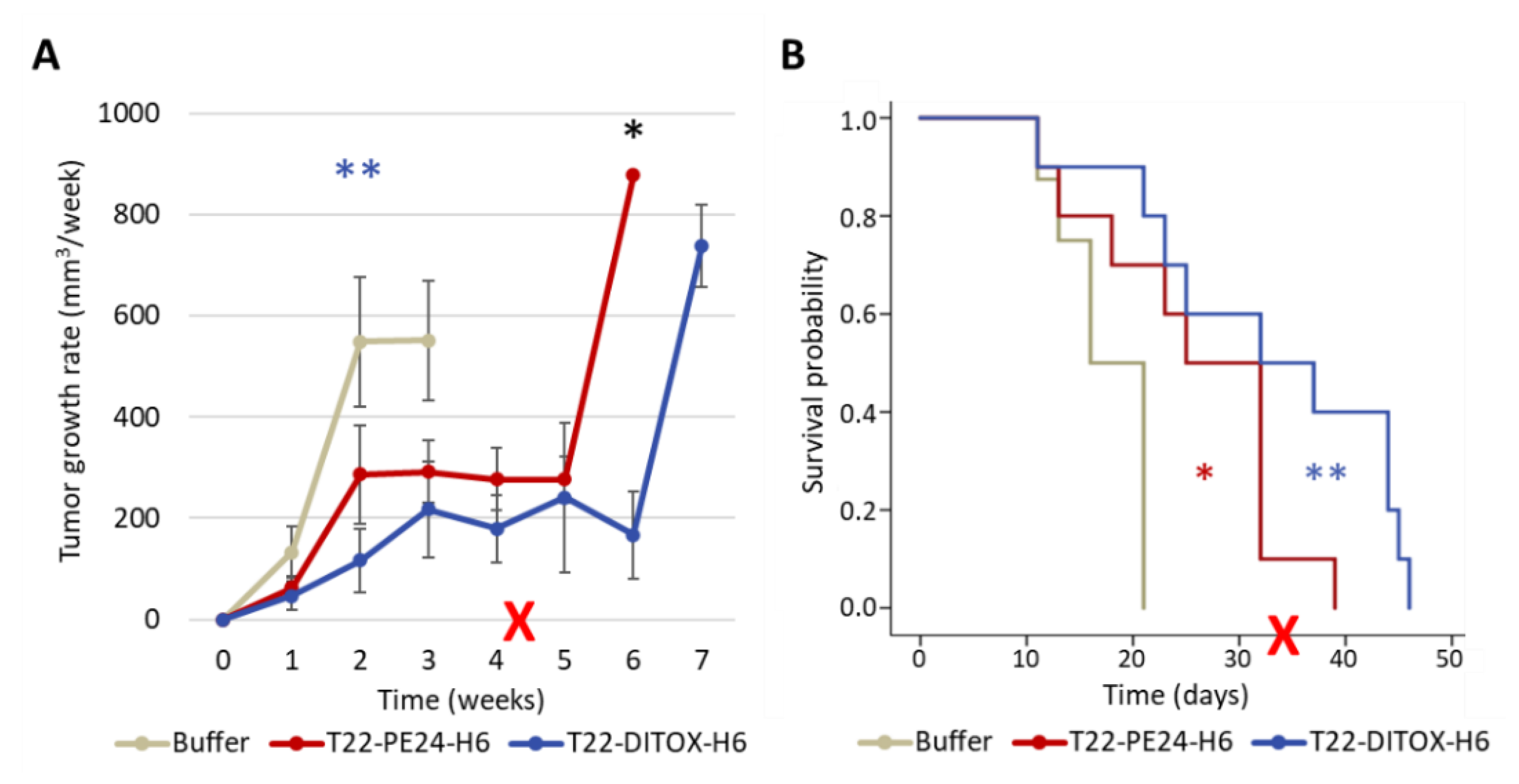
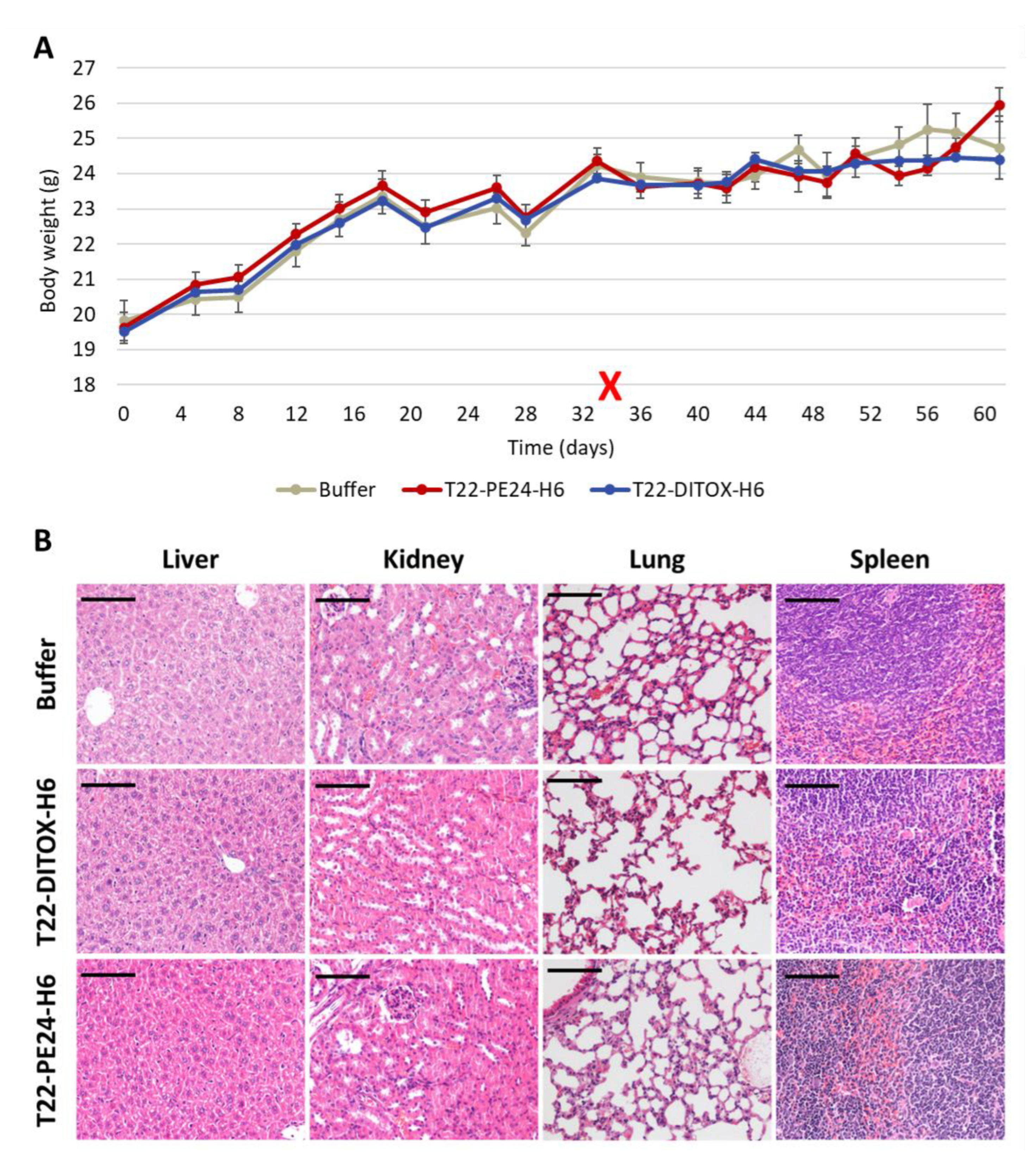
3.3. Repeated T22-DITOX-H6 or T22-PE24-H6 Treatment Inhibit Tumor Growth and Increases Survival without Systemic Toxicity in a CXCR4+ AN3CA Subcutaneous CE Model
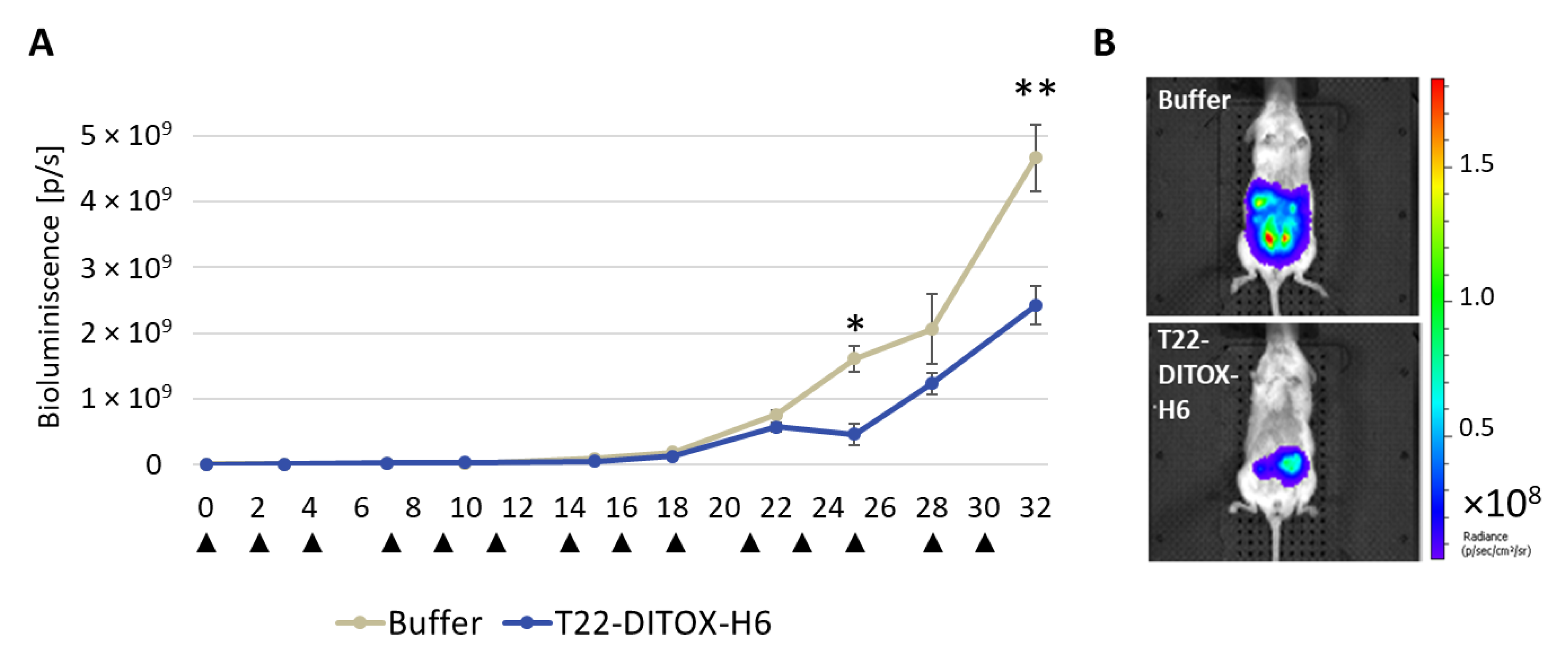
3.4. T22-DITOX-H6 Repeated Administration Reduces Total Tumor Burden as Measured by Bioluminescence in a CXCR4+ AN3CA EC Orthotopic Model
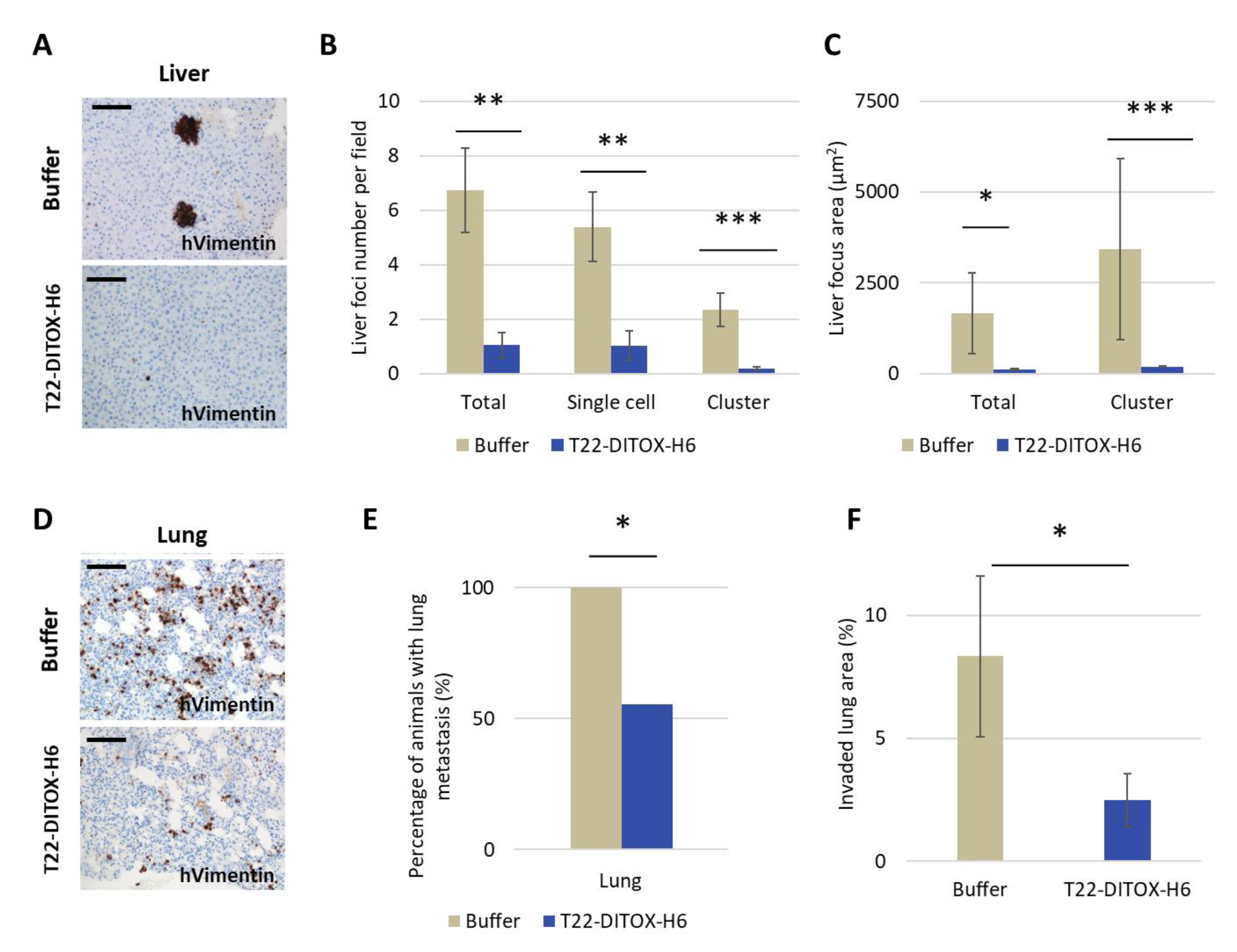
3.5. Potent Inhibition of the Development of Liver and Lung Metastases by T22-DITOX-H6 Repeated Dosage in a CXCR4+ AN3CA EC Model
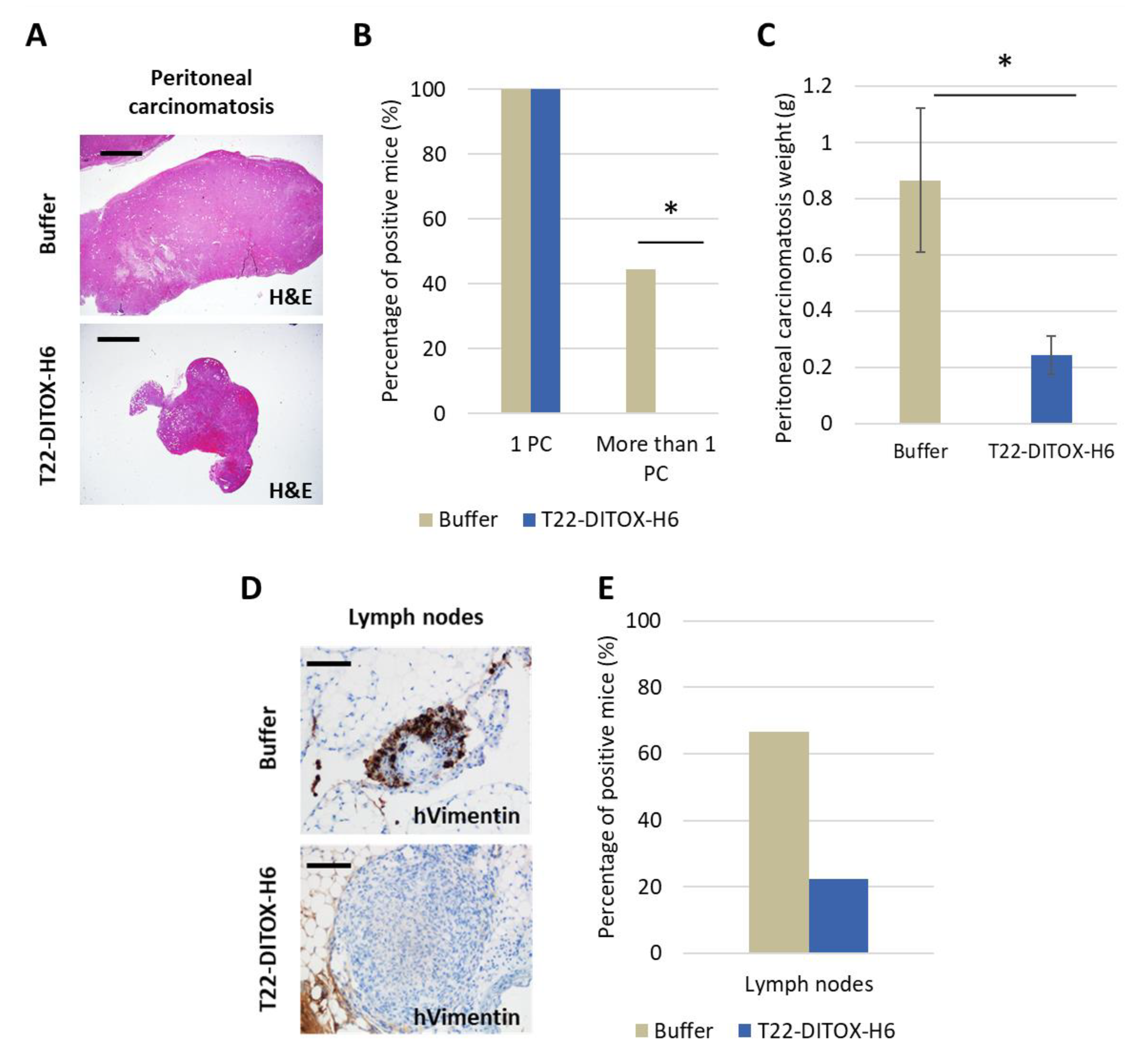
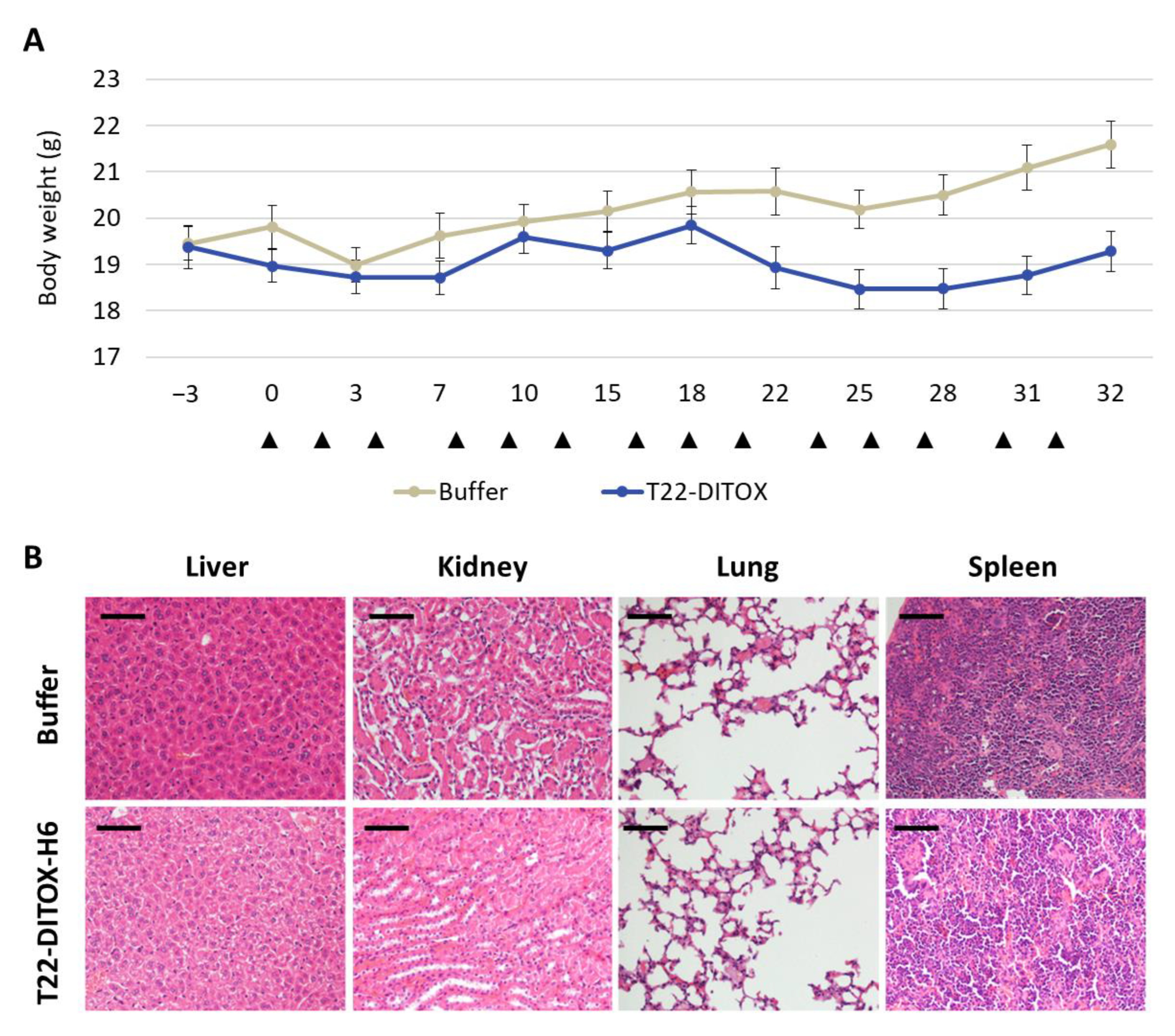
3.6. Inhibition of Lymph Node Metastases and Carcinomatotic Foci and Metastases after T22-DITOX-H6 Repeated Dosage in the Disseminated EC Model in the Absence of Systemic Toxicity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Cancer Research Fund. Endometrial Cancer Statistics 2018. Available online: https://www.wcrf.org/dietandcancer/cancer-trends/endometrial-cancer-statistics (accessed on 4 April 2020).
- Braun, M.M. Diagnosis and Management of Endometrial Cancer. Endometrial Cancer 2016, 93, 7. [Google Scholar]
- Santaballa, A.; Matías-Guiu, X.; Redondo, A.; Carballo, N.; Gil, M.; Gómez, C.; Gorostidi, M.; Gutierrez, M.; Gónzalez-Martín, A. SEOM clinical guidelines for endometrial cancer (2017). Clin. Transl. Oncol. 2018, 20, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, S.M.; Cohn, D.E. Treatment of Metastatic Endometrial Cancer; UpToDate: Waltham, MA, USA, 2021; Volume 26. [Google Scholar]
- Moxley, K.M.; McMeekin, D.S. Endometrial carcinoma: A review of chemotherapy, drug resistance, and the search for new agents. Oncologist 2010, 15, 1026–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawar, P.V.; Domb, A.J.; Kumar, N. Systemic Targeting Systems-EPR Effect, Ligand Targeting Systems. In Focal Controlled Drug Delivery; Advances in Delivery Science and Technology; Domb, A.J., Khan, W., Eds.; Springer: Boston, MA, USA, 2014; pp. 61–91. [Google Scholar] [CrossRef]
- Unzueta, U.; Céspedes, M.V.; Vázquez, E.; Ferrer-Miralles, N.; Mangues, R.; Villaverde, A. Towards protein-based viral mimetics for cancer therapies. Trends Biotechnol. 2015, 33, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Sabroso, C.; Lozza, I.; Torres-Suárez, A.I.; Fraguas-Sánchez, A.I. Antibody-Antineoplastic Conjugates in Gynecological Malignancies: Current Status and Future Perspectives. Pharmaceutics 2021, 13, 1705. [Google Scholar] [CrossRef]
- Dean, A.Q.; Luo, S.; Twomey, J.D.; Zhang, B. Targeting cancer with antibody-drug conjugates: Promises and challenges. mAbs 2021, 13, 1951427. [Google Scholar] [CrossRef]
- Akbari, B.; Farajnia, S.; Ahdi Khosroshahi, S.; Safari, F.; Yousefi, M.; Dariushnejad, H.; Rahbarnia, L. Immunotoxins in cancer therapy: Review and update. Int. Rev. Immunol. 2017, 36, 207–219. [Google Scholar] [CrossRef]
- Khirehgesh, M.R.; Sharifi, J.; Safari, F.; Akbari, B. Immunotoxins and nanobody-based immunotoxins: Review and update. J. Drug Target. 2021, 29, 848–862. [Google Scholar] [CrossRef]
- Sánchez-García, L. Self-assembling toxin-based nanoparticles as self-delivered antitumoral drugs. J. Control. Release 2018, 274, 81–92. [Google Scholar] [CrossRef]
- Sánchez-García, L.; Sala, R.; Serna, N.; Álamo, P.; Parladé, E.; Alba-Castellón, L.; Voltà-Durán, E.; Sánchez-Chardi, A.; Unzueta, U.; Vázquez, E.; et al. A refined cocktailing of pro-apoptotic nanoparticles boosts anti-tumor activity. Acta Biomater. 2020, 113, 584–596. [Google Scholar] [CrossRef]
- Alexa, M.; Hasenburg, A.; Battista, M.J. The TCGA Molecular Classification of Endometrial Cancer and Its Possible Impact on Adjuvant Treatment Decisions. Cancers 2021, 13, 1478. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Long, P.; Huang, Y.; Sun, F.; Wang, Z. CXCL12/CXCR4 axis induces proliferation and invasion in human endometrial cancer. Am. J. Transl. Res. 2016, 8, 1719. [Google Scholar] [PubMed]
- Medina-Gutiérrez, E.; Céspedes, M.V.; Gallardo, A.; Rioja-Blanco, E.; Pavón, M.À.; Asensio-Puig, L.; Farré, L.; Alba-Castellón, L.; Unzueta, U.; Villaverde, A.; et al. Novel Endometrial Cancer Models Using Sensitive Metastasis Tracing for CXCR4-Targeted Therapy in Advanced Disease. Biomedicines 2022, 10, 1680. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; Pastan, I. Immunogenicity of Immunotoxins Containing Pseudomonas Exotoxin A: Causes, Consequences, and Mitigation. Front. Immunol. 2020, 11, 1261. [Google Scholar] [CrossRef]
- Falgàs, A.; Pallarès, V.; Unzueta, U.; Céspedes, M.V.; Arroyo-Solera, I.; Moreno, M.J.; Sierra, J.; Gallardo, A.; Mangues, M.A.; Vázquez, E.; et al. A CXCR4-targeted nanocarrier achieves highly selective tumor uptake in diffuse large B-cell lymphoma mouse models. Haematologica 2020, 105, 741–753. [Google Scholar] [CrossRef] [Green Version]
- Sala, R.; Rioja-Blanco, E.; Serna, N.; Sánchez-García, L.; Álamo, P.; Alba-Castellón, L.; Casanova, I.; López-Pousa, A.; Unzueta, U.; Céspedes, M.V.; et al. GSDMD-dependent pyroptotic induction by a multivalent CXCR4-targeted nanotoxin blocks colorectal cancer metastases. Drug delivery 2020, 29, 1384–1397. [Google Scholar] [CrossRef]
- Rioja-Blanco, E.; Arroyo-Solera, I.; Álamo, P.; Casanova, I.; Gallardo, A.; Unzueta, U.; Serna, N.; Sánchez-García, L.; Quer, M.; Villaverde, A.; et al. Self-assembling protein nanocarrier for selective delivery of cytotoxic polypeptides to CXCR4+ head and neck squamous cell carcinoma tumors. Acta Pharm. Sin. B 2021, 12, 2578–2591. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]
- Shapira, A.; Benhar, I. Toxin-Based Therapeutic Approaches. Toxins 2010, 2, 2519–2583. [Google Scholar] [CrossRef] [Green Version]
- Mahmoudi, R.; Dianat-Moghadam, H.; Poorebrahim, M.; Siapoush, S.; Poortahmasebi, V.; Salahlou, R.; Rahmati, M. Recombinant immunotoxins development for HER2-based targeted cancer therapies. Cancer Cell Int. 2021, 21, 470. [Google Scholar] [CrossRef]
- Jia, L.-T.; Zhang, L.-H.; Yu, C.-J.; Zhao, J.; Xu, Y.-M.; Gui, J.-H.; Jin, M.; Ji, Z.-L.; Wen, W.-H.; Wang, C.-J.; et al. Specific Tumoricidal Activity of a Secreted Proapoptotic Protein Consisting of HER2 Antibody and Constitutively Active Caspase-3. Cancer Res. 2003, 63, 3257–3262. [Google Scholar] [PubMed]
- Wang, L.-F.; Zhou, Y.; Xu, Y.-M.; Qiu, X.-C.; Zhou, B.-G.; Wang, F.; Long, H.; Chen, X.; Yang, T.-T.; Ma, B.-A.; et al. A Caspase-6 and Anti-HER2 Antibody Chimeric Tumor-Targeted Proapoptotic Molecule Decreased Metastasis of Human Osteosarcoma. Cancer Invest. 2009, 27, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-M.; Wang, L.-F.; Jia, L.-T.; Qiu, X.-C.; Zhao, J.; Yu, C.-J.; Zhang, R.; Zhu, F.; Wang, C.-J.; Jin, B.-Q.; et al. A Caspase-6 and Anti-Human Epidermal Growth Factor Receptor-2 (HER2) Antibody Chimeric Molecule Suppresses the Growth of HER2-Overexpressing Tumors. J. Immunol. 2004, 173, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konno, T.; Kohno, T.; Kikuchi, S.; Shimada, H.; Satohisa, S.; Saito, T.; Kondoh, M.; Kojima, T. Epithelial barrier dysfunction and cell migration induction via JNK/cofilin/actin by angubindin-1. Tissue Barriers 2020, 8, 1695475. [Google Scholar] [CrossRef]
- Saito, K.; Konno, T.; Kohno, T.; Shimada, H.; Matsuura, M.; Okada, T.; Kura, A.; Ishii, D.; Kondoh, M.; Saito, T.; et al. LSR antibody promotes apoptosis and disrupts epithelial barriers via signal pathways in endometrial cancer. Tissue Barriers 2022, 2106113. Available online: https://www.tandfonline.com/doi/abs/10.1080/21688370.2022.2106113 (accessed on 15 May 2021). [CrossRef]
- Chung, H.C.; Ros, W.; Delord, J.-P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef]
- Oaknin, A.; Tinker, A.V.; Gilbert, L.; Samouëlian, V.; Mathews, C.; Brown, J.; Barretina-Ginesta, M.-P.; Moreno, V.; Gravina, A.; Abdeddaim, C.; et al. Clinical Activity and Safety of the Anti-Programmed Death 1 Monoclonal Antibody Dostarlimab for Patients With Recurrent or Advanced Mismatch Repair-Deficient Endometrial Cancer: A Nonrandomized Phase 1 Clinical Trial. JAMA Oncol. 2020, 6, 1766–1772. [Google Scholar] [CrossRef]
- Postow, M. Toxicities Associated with Checkpoint Inhibitor Immunotherapy; UpToDate: Waltham, MA, USA, 2021; Volume 38. [Google Scholar]
- Lambert, J.M. Antibody–Drug Conjugates (ADCs): Magic Bullets at Last! Mol. Pharm. 2015, 12, 1701–1702. [Google Scholar] [CrossRef]
- Van Nyen, T.; Moiola, C.P.; Colas, E.; Annibali, D.; Amant, F. Modeling Endometrial Cancer: Past, Present, and Future. Int. J. Mol. Sci. 2018, 19, 2348. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medina-Gutiérrez, E.; García-León, A.; Gallardo, A.; Álamo, P.; Alba-Castellón, L.; Unzueta, U.; Villaverde, A.; Vázquez, E.; Casanova, I.; Mangues, R. Potent Anticancer Activity of CXCR4-Targeted Nanostructured Toxins in Aggressive Endometrial Cancer Models. Cancers 2023, 15, 85. https://doi.org/10.3390/cancers15010085
Medina-Gutiérrez E, García-León A, Gallardo A, Álamo P, Alba-Castellón L, Unzueta U, Villaverde A, Vázquez E, Casanova I, Mangues R. Potent Anticancer Activity of CXCR4-Targeted Nanostructured Toxins in Aggressive Endometrial Cancer Models. Cancers. 2023; 15(1):85. https://doi.org/10.3390/cancers15010085
Chicago/Turabian StyleMedina-Gutiérrez, Esperanza, Annabel García-León, Alberto Gallardo, Patricia Álamo, Lorena Alba-Castellón, Ugutz Unzueta, Antonio Villaverde, Esther Vázquez, Isolda Casanova, and Ramon Mangues. 2023. "Potent Anticancer Activity of CXCR4-Targeted Nanostructured Toxins in Aggressive Endometrial Cancer Models" Cancers 15, no. 1: 85. https://doi.org/10.3390/cancers15010085
APA StyleMedina-Gutiérrez, E., García-León, A., Gallardo, A., Álamo, P., Alba-Castellón, L., Unzueta, U., Villaverde, A., Vázquez, E., Casanova, I., & Mangues, R. (2023). Potent Anticancer Activity of CXCR4-Targeted Nanostructured Toxins in Aggressive Endometrial Cancer Models. Cancers, 15(1), 85. https://doi.org/10.3390/cancers15010085