




Synthesis of New Chromene Derivatives Targeting Triple-Negative Breast Cancer Cells

 , ,
, ,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Measurement of Cell Viability
2.3. Colony Growth Assay
2.4. Cell Cycle Analysis
2.5. Caspase 3/7 Activity
2.6. Immunofluorescence Staining
2.7. Hematoxylin and Eosin Staining
2.8. Whole-Cell Extraction and Western Blotting Analysis
2.9. Senescence-Associated β-Galactosidase Staining
2.10. Wound-Healing Assay
2.11. HUVEC Tube Formation Assay & Cell Viability
2.12. Tubulin Polymerization
2.13. Molecular Docking
2.14. Statistical Analysis
3. Results
3.1. Synthesis of 2-Amino-3-Cyanochromene C1
3.2. Synthesis of Schiff Base C2
3.3. Chromenes C1 and C2 Selectively Inhibit the Cell Viability of TNBC Cells
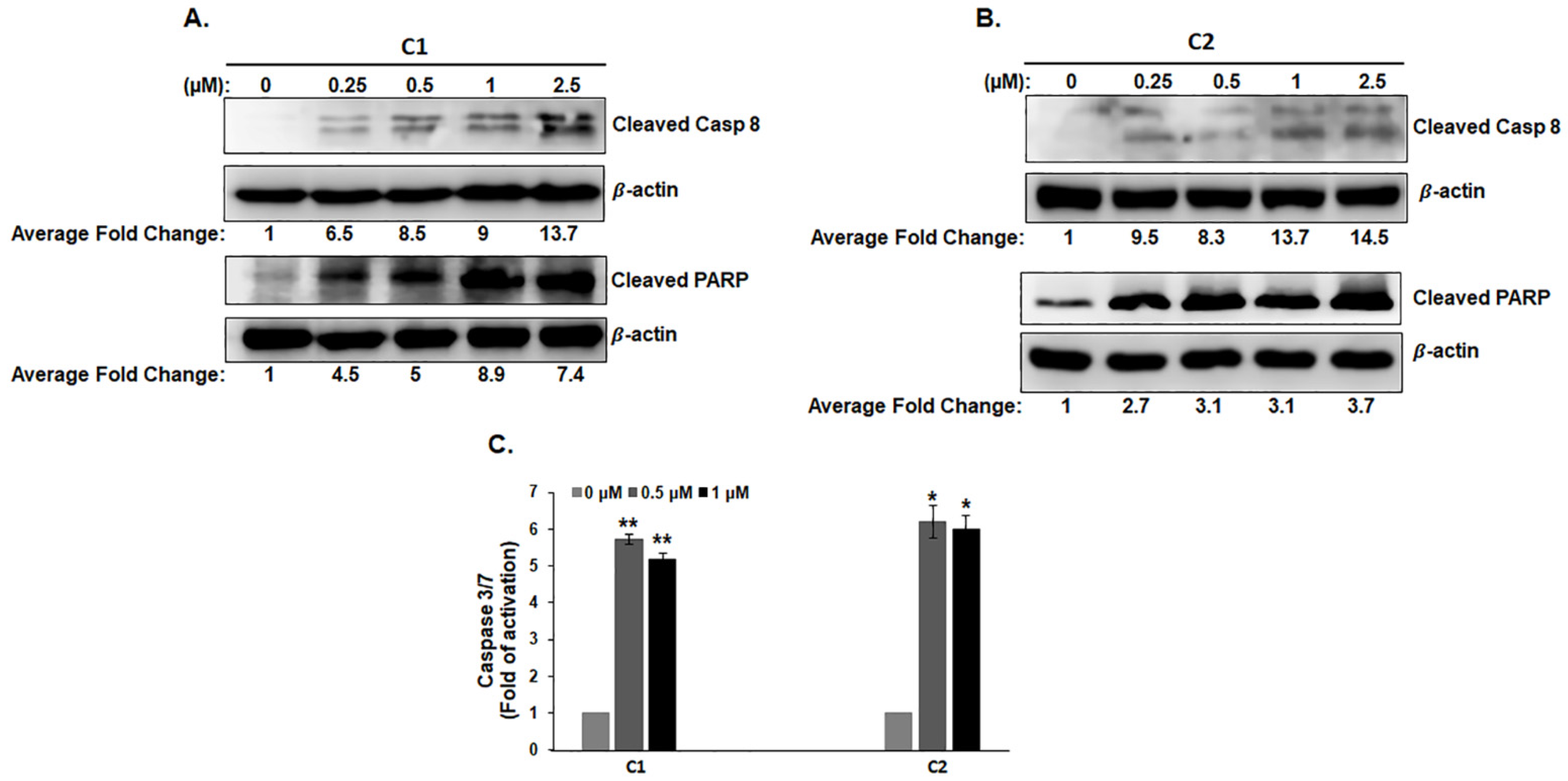
3.4. C1 and C2 Induce Cell Death of TNBC Cells by Activating the Extrinsic Apoptotic Pathway
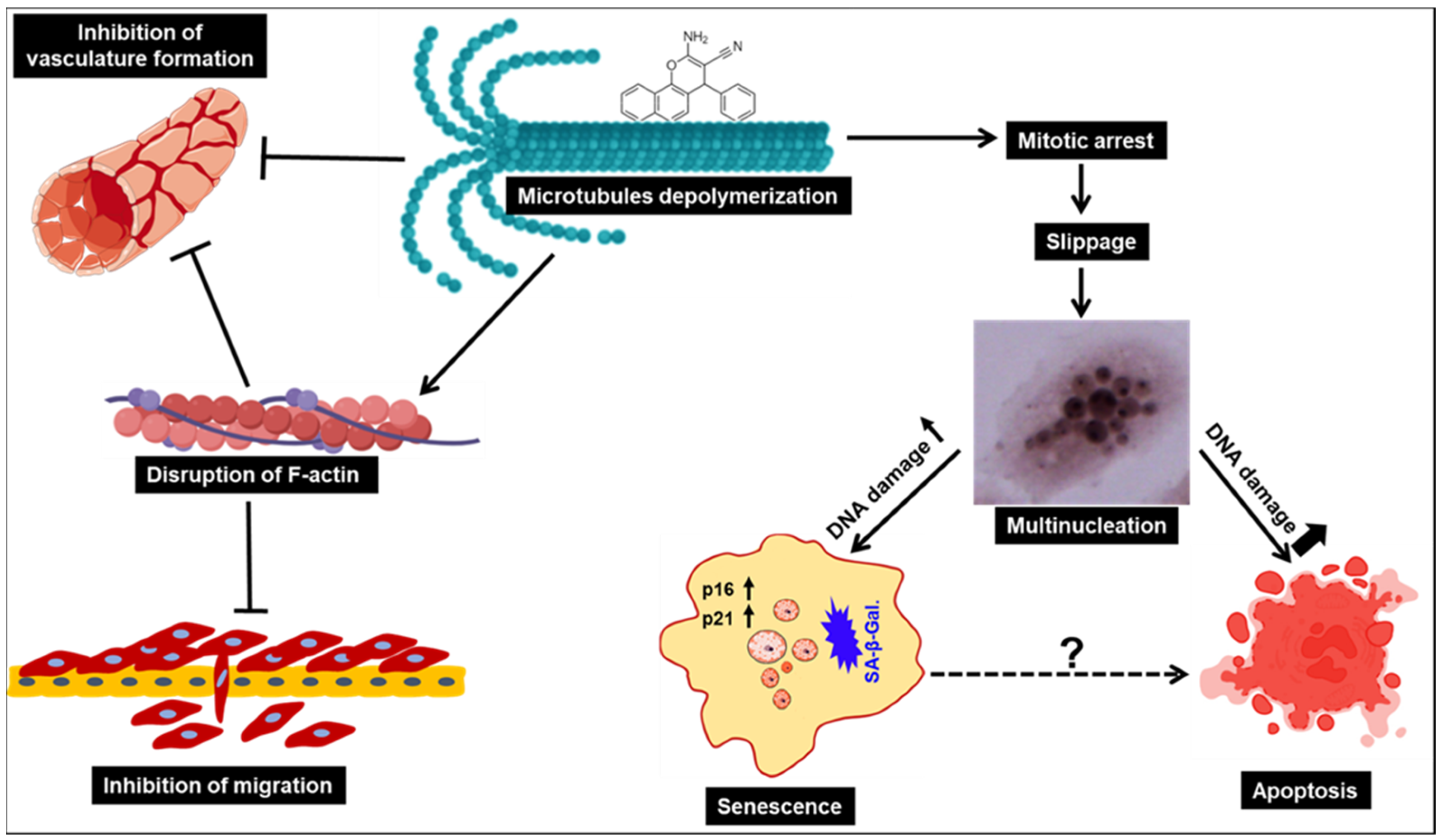
3.5. C1 and C2 Induce Morphological Changes Associated with Massive Multinucleation and Accumulation of Double-Stranded DNA Breaks in TNBC Cells
3.6. C1 and C2 Induce Mitotic Arrest and Senescence in TNBC Cells
3.7. C1 and C2 Impair the Polymerization of Microtubules through Binding to the Colchicine Binding Site of β-Tubulin
3.8. C1 and C2 Cause Disruption of F-Actin Cytoskeleton Organization in TNBC
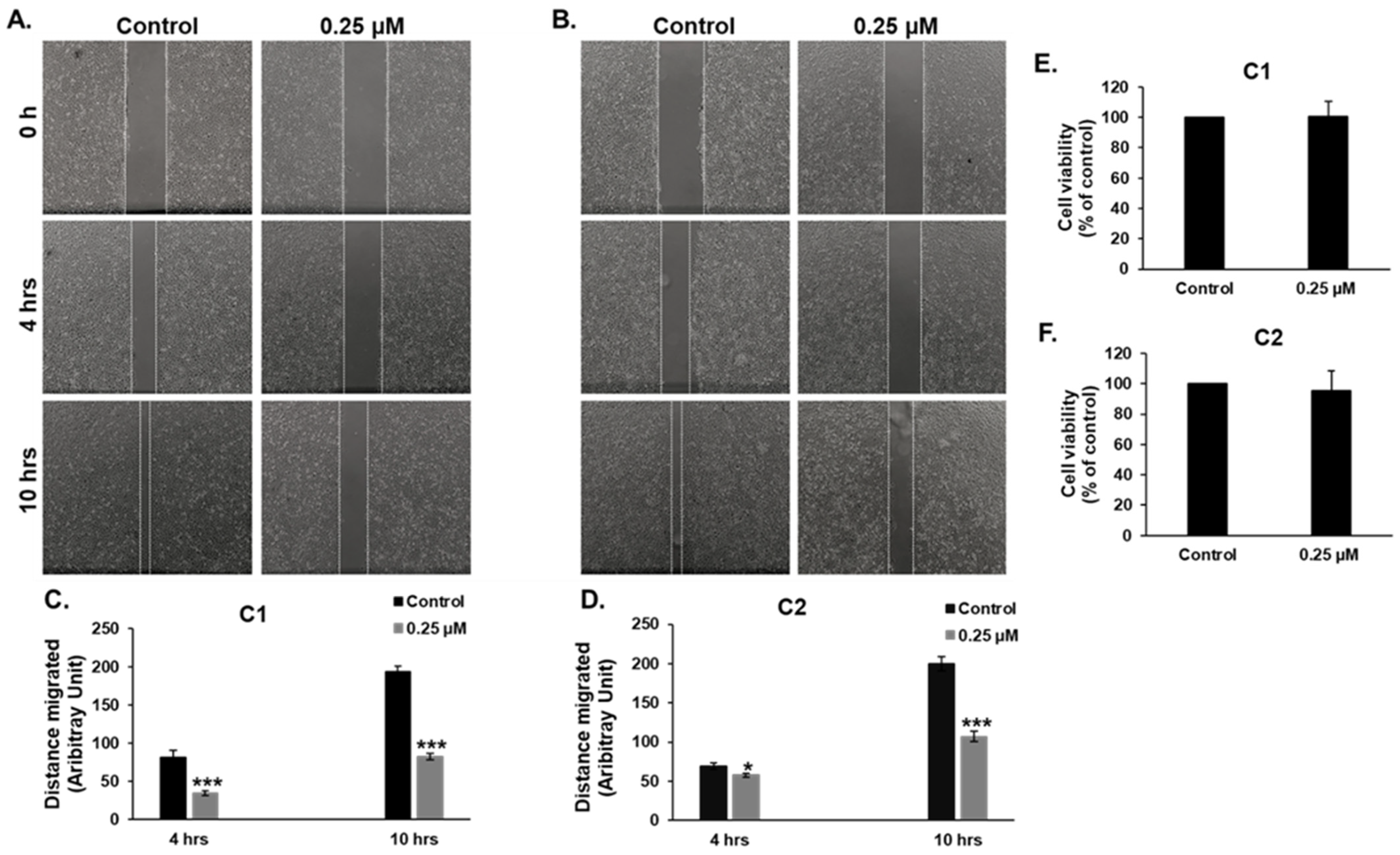
3.9. C1 and C2 Inhibit Cell Migration of MDA-MB-231 Cells
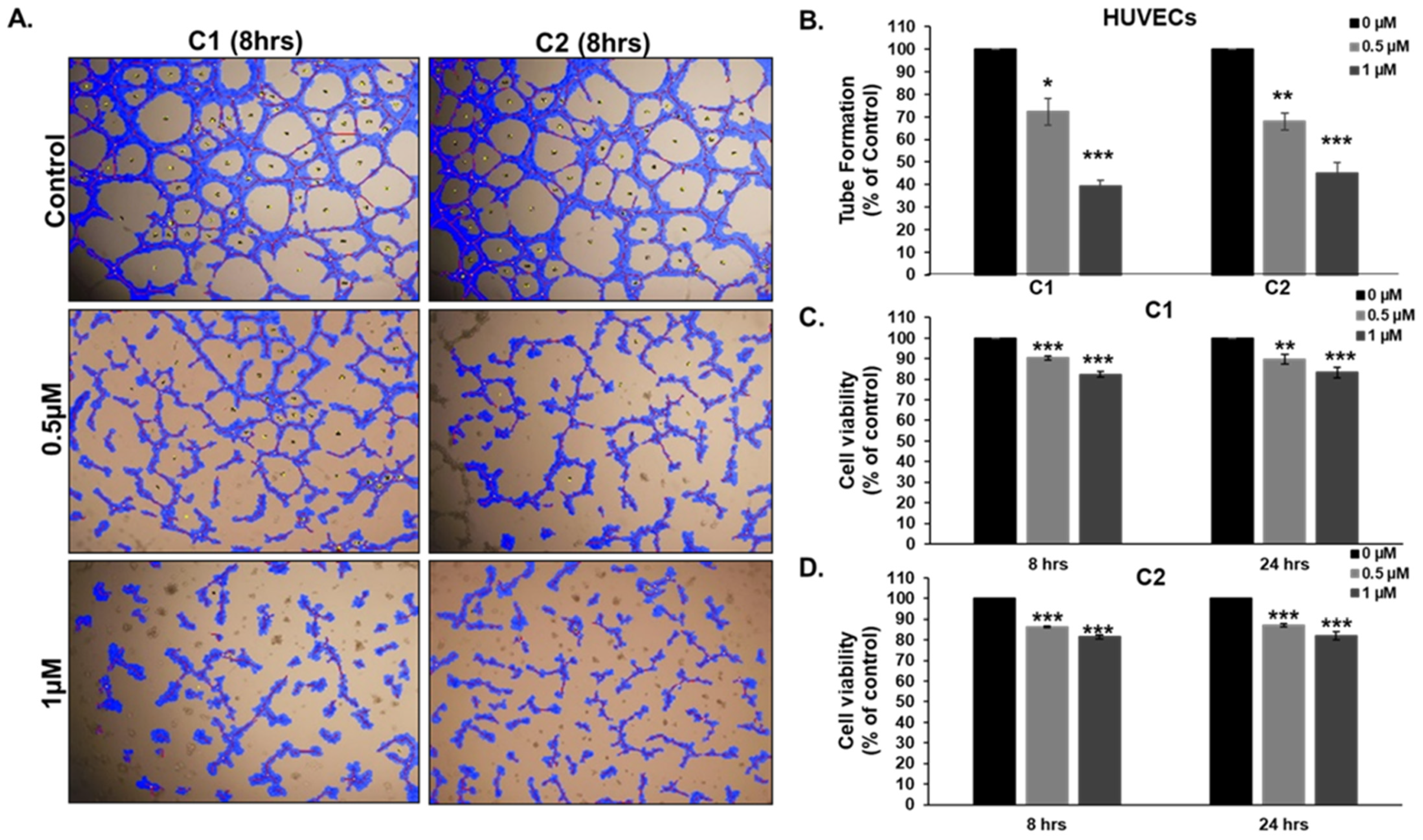
3.10. Chromenes C1 and C2 Inhibit Angiogenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and Future Burden of Breast Cancer: Global Statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 8 February 2023).
- Orrantia-Borunda, E.; Anchondo-Nuñez, P.; Acuña-Aguilar, L.E.; Gómez-Valles, F.O.; Ramírez-Valdespino, C.A. Subtypes of Breast Cancer. In Breast Cancer; Mayrovitz, H.N., Ed.; Exon Publications: Brisbane, Australia, 2022; ISBN 978-0-645-33203-2. [Google Scholar]
- Yadav, B.S.; Chanana, P.; Jhamb, S. Biomarkers in Triple Negative Breast Cancer: A Review. World J. Clin. Oncol. 2015, 6, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Geyer, F.C.; Pareja, F.; Weigelt, B.; Rakha, E.; Ellis, I.O.; Schnitt, S.J.; Reis-Filho, J.S. The Spectrum of Triple-Negative Breast Disease: High- and Low-Grade Lesions. Am. J. Pathol. 2017, 187, 2139–2151. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, H.; Guerra, M.R.; Duarte Cintra, J.R.; Fayer, V.A.; Brum, I.V.; Bustamante Teixeira, M.T. Survival Study of Triple-Negative and Non-Triple-Negative Breast Cancer in a Brazilian Cohort. Clin. Med. Insights Oncol. 2018, 12, 1179554918790563. [Google Scholar] [CrossRef] [PubMed]
- Simões-Wüst, A.P.; Schürpf, T.; Hall, J.; Stahel, R.A.; Zangemeister-Wittke, U. Bcl-2/Bcl-XL Bispecific Antisense Treatment Sensitizes Breast Carcinoma Cells to Doxorubicin, Paclitaxel and Cyclophosphamide. Breast Cancer Res. Treat. 2002, 76, 157–166. [Google Scholar] [CrossRef]
- Inao, T.; Iida, Y.; Moritani, T.; Okimoto, T.; Tanino, R.; Kotani, H.; Harada, M. Bcl-2 Inhibition Sensitizes Triple-Negative Human Breast Cancer Cells to Doxorubicin. Oncotarget 2018, 9, 25545–25556. [Google Scholar] [CrossRef]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef]
- Raj, V.; Lee, J. 2H/4H-Chromenes-A Versatile Biologically Attractive Scaffold. Front. Chem. 2020, 8, 623. [Google Scholar] [CrossRef]
- Thomas, N.; Zachariah, S. Pharmacological Activities of Chromene Derivatives: An Overview. Asian J. Pharm. Clin. Res. 2013, 6, 11–15. [Google Scholar]
- Katiyar, M.K.; Dhakad, G.K.; Shivani; Arora, S.; Bhagat, S.; Arora, T.; Kumar, R. Synthetic Strategies and Pharmacological Activities of Chromene and Its Derivatives: An Overview. J. Mol. Struct. 2022, 1263, 133012. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.-L.; Fang, Y.-C.; Wang, C.-Y. Tephrosin-Induced Autophagic Cell Death in A549 Non-Small Cell Lung Cancer Cells. J. Asian Nat. Prod. Res. 2010, 12, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Jiang, F.; Xu, S.-S.; Huang, Z.-F.; Chen, L.-L.; Li, L. Tephrosin Induces Apoptosis of Human Pancreatic Cancer Cells through the Generation of Reactive Oxygen Species. J. Cancer 2021, 12, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Ekowati, H.; Astuti, I.; Mustofa, M. Anticancer Activity Of Calanone On Hela Cell Line. Indones. J. Chem. 2010, 10, 240–244. [Google Scholar] [CrossRef]
- Guilbaud, N.; Kraus-Berthier, L.; Meyer-Losic, F.; Malivet, V.; Chacun, C.; Jan, M.; Tillequin, F.; Michel, S.; Koch, M.; Pfeiffer, B.; et al. Marked Antitumor Activity of a New Potent Acronycine Derivative in Orthotopic Models of Human Solid Tumors. Clin. Cancer Res. 2001, 7, 2573–2580. [Google Scholar]
- Nishino, H.; Okuyama, T.; Takata, M.; Shibata, S.; Tokuda, H.; Takayasu, J.; Hasegawa, T.; Nishino, A.; Ueyama, H.; Iwashima, A. Studies on the Anti-Tumor-Promoting Activity of Naturally Occurring Substances. IV. Pd-II [(+)Anomalin, (+)Praeruptorin B], a Seselin-Type Coumarin, Inhibits the Promotion of Skin Tumor Formation by 12-O-Tetradecanoylphorbol-13-Acetate in 7,12-Dimethylbenz[a]Anthracene-Initiated Mice. Carcinogenesis 1990, 11, 1557–1561. [Google Scholar] [CrossRef]
- Lima, C.F.; Costa, M.; Proença, M.F.; Pereira-Wilson, C. Novel Structurally Similar Chromene Derivatives with Opposing Effects on P53 and Apoptosis Mechanisms in Colorectal HCT116 Cancer Cells. Eur. J. Pharm. Sci. 2015, 72, 34–45. [Google Scholar] [CrossRef]
- Asgari, F.; Mahinpour, R.; Moradi, L.; Haghighipour, N. The Chromene Derivative 4-Clpgc Inhibits Cell Proliferation and Induces Apoptosis in the K562 Cell Line. J. Cell Commun. Signal. 2020, 14, 77–91. [Google Scholar] [CrossRef]
- Zhang, W.-H.; Chen, S.; Liu, X.-L.; Bing-Lin, N.; Liu, X.-W.; Zhou, Y. Study on Antitumor Activities of the Chrysin-Chromene-Spirooxindole on Lewis Lung Carcinoma C57BL/6 Mice in vivo. Bioorg. Med. Chem. Lett. 2020, 30, 127410. [Google Scholar] [CrossRef]
- Kulshrestha, A.; Katara, G.K.; Ibrahim, S.A.; Patil, R.; Patil, S.A.; Beaman, K.D. Microtubule Inhibitor, SP-6-27 Inhibits Angiogenesis and Induces Apoptosis in Ovarian Cancer Cells. Oncotarget 2017, 8, 67017–67028. [Google Scholar] [CrossRef]
- Puppala, M.; Zhao, X.; Casemore, D.; Zhou, B.; Aridoss, G.; Narayanapillai, S.; Xing, C. 4H-Chromene-Based Anticancer Agents towards Multi-Drug Resistant HL60/MX2 Human Leukemia: SAR at the 4th and 6th Positions. Bioorg. Med. Chem. 2016, 24, 1292–1297. [Google Scholar] [CrossRef]
- Kulshrestha, A.; Ibrahim, S.A.; Katara, G.K.; Patil, R.; Patil, S.; Beaman, K.D. Abstract 1231: Novel Chromene Analogs as Small-Molecule Microtubule Destabilizers for the Treatment of Chemo-Resistant Ovarian Cancer. Cancer Res. 2016, 76, 1231. [Google Scholar] [CrossRef]
- Patil, S.A.; Patil, R.; Pfeffer, L.M.; Miller, D.D. Chromenes: Potential New Chemotherapeutic Agents for Cancer. Future Med. Chem. 2013, 5, 1647–1660. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.X.; Drewe, J.; Kemnitzer, W. Discovery of 4-Aryl-4H-Chromenes as Potent Apoptosis Inducers Using a Cell- and Caspase-Based Anti-Cancer Screening Apoptosis Program (ASAP): SAR Studies and the Identification of Novel Vascular Disrupting Agents. Anticancer Agents Med. Chem. 2009, 9, 437–456. [Google Scholar] [CrossRef] [PubMed]
- Fallah-Tafti, A.; Tiwari, R.; Shirazi, A.N.; Akbarzadeh, T.; Mandal, D.; Shafiee, A.; Parang, K.; Foroumadi, A. 4-Aryl-4H-Chromene-3-Carbonitrile Derivatives: Evaluation of Src Kinase Inhibitory and Anticancer Activities. Med. Chem. 2011, 7, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Benhalilou, N.; Alsamri, H.; Alneyadi, A.; Athamneh, K.; Alrashedi, A.; Altamimi, N.; Al Dhaheri, Y.; Eid, A.H.; Iratni, R. Origanum Majorana Ethanolic Extract Promotes Colorectal Cancer Cell Death by Triggering Abortive Autophagy and Activation of the Extrinsic Apoptotic Pathway. Front. Oncol. 2019, 9, 795. [Google Scholar] [CrossRef]
- Al Dhaheri, Y.; Attoub, S.; Arafat, K.; Abuqamar, S.; Eid, A.; Al Faresi, N.; Iratni, R. Salinomycin Induces Apoptosis and Senescence in Breast Cancer: Upregulation of P21, Downregulation of Survivin and Histone H3 and H4 Hyperacetylation. Biochim. Biophys. Acta 2013, 1830, 3121–3135. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Hayashi, M.T.; Cesare, A.J.; Fitzpatrick, J.A.J.; Lazzerini-Denchi, E.; Karlseder, J. A Telomere-Dependent DNA Damage Checkpoint Induced by Prolonged Mitotic Arrest. Nat. Struct. Mol. Biol. 2012, 19, 387–394. [Google Scholar] [CrossRef]
- Dikovskaya, D.; Cole, J.J.; Mason, S.M.; Nixon, C.; Karim, S.A.; McGarry, L.; Clark, W.; Hewitt, R.N.; Sammons, M.A.; Zhu, J.; et al. Mitotic Stress Is an Integral Part of the Oncogene-Induced Senescence Program That Promotes Multinucleation and Cell Cycle Arrest. Cell Rep. 2015, 12, 1483–1496. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An Overview of Tubulin Inhibitors That Interact with the Colchicine Binding Site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, R.; Holmes, K.C. Actin Structure and Function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Perrin, B.J.; Ervasti, J.M. The Actin Gene Family: Function Follows Isoform. Cytoskeleton 2010, 67, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Bayless, K.J.; Johnson, G.A. Role of the Cytoskeleton in Formation and Maintenance of Angiogenic Sprouts. J. Vasc. Res. 2011, 48, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, E.O.; Deal, A.M.; Anders, C.K.; Prat, A.; Perou, C.M.; Carey, L.A.; Muss, H.B. Age-Specific Changes in Intrinsic Breast Cancer Subtypes: A Focus on Older Women. Oncologist 2014, 19, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Tzikas, A.-K.; Nemes, S.; Linderholm, B.K. A Comparison between Young and Old Patients with Triple-Negative Breast Cancer: Biology, Survival and Metastatic Patterns. Breast Cancer Res. Treat. 2020, 182, 643–654. [Google Scholar] [CrossRef]
- Gulley, J. A Phase I/II Trial of Crolibulin (EPC2407) Plus Cisplatin in Adults with Solid Tumors with a Focus on Anaplastic Thyroid Cancer (ATC). 2017. Available online: https://clinicaltrials.gov/ (accessed on 1 March 2023).
- Pontes, O.; Costa, M.; Santos, F.; Sampaio-Marques, B.; Dias, T.; Ludovico, P.; Baltazar, F.; Proença, F. Exploitation of New Chalcones and 4H-Chromenes as Agents for Cancer Treatment. Eur. J. Med. Chem. 2018, 157, 101–114. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Actin and Microtubules in Cell Motility: Which One Is in Control? Traffic 2004, 5, 470–477. [Google Scholar] [CrossRef]
- Mohan, R.; John, A. Microtubule-Associated Proteins as Direct Crosslinkers of Actin Filaments and Microtubules. IUBMB Life 2015, 67, 395–403. [Google Scholar] [CrossRef]
- Bates, D.; Eastman, A. Microtubule Destabilising Agents: Far More than Just Antimitotic Anticancer Drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef]
- Pletjushkina, O.J.; Ivanova, O.J.; Kaverina, I.N.; Vasiliev, J.M. Taxol-Treated Fibroblasts Acquire an Epithelioid Shape and a Circular Pattern of Actin Bundles. Exp. Cell Res. 1994, 212, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, M.D.; Shivers, R.R. “Rings” of F-Actin Form around the Nucleus in Cultured Human MCF7 Adenocarcinoma Cells upon Exposure to Both Taxol and Taxotere. Comp. Biochem. Physiol. Part C Pharmacol. Toxicol. Endocrinol. 2000, 125, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Bijman, M.N.A.; van Nieuw Amerongen, G.P.; Laurens, N.; van Hinsbergh, V.W.M.; Boven, E. Microtubule-Targeting Agents Inhibit Angiogenesis at Subtoxic Concentrations, a Process Associated with Inhibition of Rac1 and Cdc42 Activity and Changes in the Endothelial Cytoskeleton. Mol. Cancer Ther. 2006, 5, 2348–2357. [Google Scholar] [CrossRef] [PubMed]
- Kemnitzer, W.; Drewe, J.; Jiang, S.; Zhang, H.; Wang, Y.; Zhao, J.; Jia, S.; Herich, J.; Labreque, D.; Storer, R.; et al. Discovery of 4-Aryl-4H-Chromenes as a New Series of Apoptosis Inducers Using a Cell- and Caspase-Based High-Throughput Screening Assay. 1. Structure-Activity Relationships of the 4-Aryl Group. J. Med. Chem. 2004, 47, 6299–6310. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.A.; Wang, J.; Li, X.S.; Chen, J.; Jones, T.S.; Hosni-Ahmed, A.; Patil, R.; Seibel, W.L.; Li, W.; Miller, D.D. New Substituted 4H-Chromenes as Anticancer Agents. Bioorg. Med. Chem. Lett. 2012, 22, 4458–4461. [Google Scholar] [CrossRef] [PubMed]
- Weinert, T.; Olieric, N.; Cheng, R.; Brünle, S.; James, D.; Ozerov, D.; Gashi, D.; Vera, L.; Marsh, M.; Jaeger, K.; et al. Serial Millisecond Crystallography for Routine Room-Temperature Structure Determination at Synchrotrons. Nat. Commun. 2017, 8, 542. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Miller, D.D.; Li, W. Molecular Interactions at the Colchicine Binding Site in Tubulin: An X-ray Crystallography Perspective. Drug Discov. Today 2022, 27, 759–776. [Google Scholar] [CrossRef] [PubMed]
- Di Rorà, A.G.L.; Martinelli, G.; Simonetti, G. The Balance between Mitotic Death and Mitotic Slippage in Acute Leukemia: A New Therapeutic Window? J. Hematol. Oncol. 2019, 12, 123. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Crasta, K. Consequences of Mitotic Slippage for Antimicrotubule Drug Therapy. Endocr. Relat. Cancer 2017, 24, T97–T106. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Orth, J.D.; Mitchison, T. Cell Type Variation in Responses to Antimitotic Drugs That Target Microtubules and Kinesin-5. Cancer Res. 2008, 68, 3269–3276. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, Y.; Shi, J. Post-Slippage Multinucleation Renders Cytotoxic Variation in Anti-Mitotic Drugs That Target the Microtubules or Mitotic Spindle. Cell Cycle 2014, 13, 1756–1764. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.; Adams, S.D.; Draviam, V.M. Multinucleation Associated DNA Damage Blocks Proliferation in P53-Compromised Cells. Commun. Biol. 2021, 4, 451. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Dong, R.; Ying, M.; He, Q.; Cao, J.; Yang, B. Cellular Senescence and Anti-Cancer Therapy. Curr. Drug Targets 2019, 20, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Prata, L.G.P.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent Cell Clearance by the Immune System: Emerging Therapeutic Opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential Roles for Cyclin-Dependent Kinase Inhibitors P21 and P16 in the Mechanisms of Senescence and Differentiation in Human Fibroblasts. Mol. Cell Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef]
- Aseervatham, J. Cytoskeletal Remodeling in Cancer. Biology 2020, 9, 385. [Google Scholar] [CrossRef]
- Fischer, R.S.; Sun, X.; Baird, M.A.; Hourwitz, M.J.; Seo, B.R.; Pasapera, A.M.; Mehta, S.B.; Losert, W.; Fischbach, C.; Fourkas, J.T.; et al. Contractility, Focal Adhesion Orientation, and Stress Fiber Orientation Drive Cancer Cell Polarity and Migration along Wavy ECM Substrates. Proc. Natl. Acad. Sci. USA 2021, 118, e2021135118. [Google Scholar] [CrossRef]
- Čermák, V.; Dostál, V.; Jelínek, M.; Libusová, L.; Kovář, J.; Rösel, D.; Brábek, J. Microtubule-Targeting Agents and Their Impact on Cancer Treatment. Eur. J. Cell Biol. 2020, 99, 151075. [Google Scholar] [CrossRef]
- Geoffroy, M.; Lemesle, M.; Kleinclauss, A.; Mazerbourg, S.; Batista, L.; Barberi-Heyob, M.; Bastogne, T.; Boireau, W.; Rouleau, A.; Dupommier, D.; et al. AB186 Inhibits Migration of Triple-Negative Breast Cancer Cells and Interacts with α-Tubulin. Int. J. Mol. Sci. 2022, 23, 6859. [Google Scholar] [CrossRef]
- Wang, P.-S.; Chou, F.-S.; Porchia, L.; Saji, M.; Pinzone, J.J. Troglitazone Inhibits Cell Migration, Adhesion, and Spreading by Modulating Cytoskeletal Rearrangement in Human Breast Cancer Cells. Mol. Carcinog. 2008, 47, 905–915. [Google Scholar] [CrossRef]
- Yang, Y.-C.; Ho, T.-C.; Chen, S.-L.; Lai, H.-Y.; Wu, J.-Y.; Tsao, Y.-P. Inhibition of Cell Motility by Troglitazone in Human Ovarian Carcinoma Cell Line. BMC Cancer 2007, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, A.; Subramani, R.; Lakshmanaswamy, R. Involvement of Actin Cytoskeletal Modifications in the Inhibition of Triple-Negative Breast Cancer Growth and Metastasis by Nimbolide. Mol. Ther. Oncolytics 2021, 20, 596–606. [Google Scholar] [CrossRef]
- Pasquier, E.; André, N.; Braguer, D. Targeting Microtubules to Inhibit Angiogenesis and Disrupt Tumour Vasculature: Implications for Cancer Treatment. Curr. Cancer Drug Targets 2007, 7, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, A.; Yang, H.; Zhang, H.; Cabral, F.; Patel, K.D. Microtubule Dynamics Control Tail Retraction in Migrating Vascular Endothelial Cells. Mol. Cancer Ther. 2013, 12, 2837–2846. [Google Scholar] [CrossRef] [PubMed]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alneyadi, A.; Nizami, Z.N.; Aburawi, H.E.; Hisaindee, S.; Nawaz, M.; Attoub, S.; Ramadan, G.; Benhalilou, N.; Al Azzani, M.; Elmahi, Y.; et al. Synthesis of New Chromene Derivatives Targeting Triple-Negative Breast Cancer Cells. Cancers 2023, 15, 2682. https://doi.org/10.3390/cancers15102682
Alneyadi A, Nizami ZN, Aburawi HE, Hisaindee S, Nawaz M, Attoub S, Ramadan G, Benhalilou N, Al Azzani M, Elmahi Y, et al. Synthesis of New Chromene Derivatives Targeting Triple-Negative Breast Cancer Cells. Cancers. 2023; 15(10):2682. https://doi.org/10.3390/cancers15102682
Chicago/Turabian StyleAlneyadi, Aysha, Zohra Nausheen Nizami, Hanan E. Aburawi, Soleiman Hisaindee, Muhammad Nawaz, Samir Attoub, Gaber Ramadan, Nehla Benhalilou, Mazoun Al Azzani, Yassine Elmahi, and et al. 2023. "Synthesis of New Chromene Derivatives Targeting Triple-Negative Breast Cancer Cells" Cancers 15, no. 10: 2682. https://doi.org/10.3390/cancers15102682
APA StyleAlneyadi, A., Nizami, Z. N., Aburawi, H. E., Hisaindee, S., Nawaz, M., Attoub, S., Ramadan, G., Benhalilou, N., Al Azzani, M., Elmahi, Y., Almeqbali, A., Muhammad, K., Eid, A. H., Vijayan, R., & Iratni, R. (2023). Synthesis of New Chromene Derivatives Targeting Triple-Negative Breast Cancer Cells. Cancers, 15(10), 2682. https://doi.org/10.3390/cancers15102682