
PDAC, the Influencer Cancer: Cross-Talk with Tumor Microenvironment and Connected Potential Therapy Strategies

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
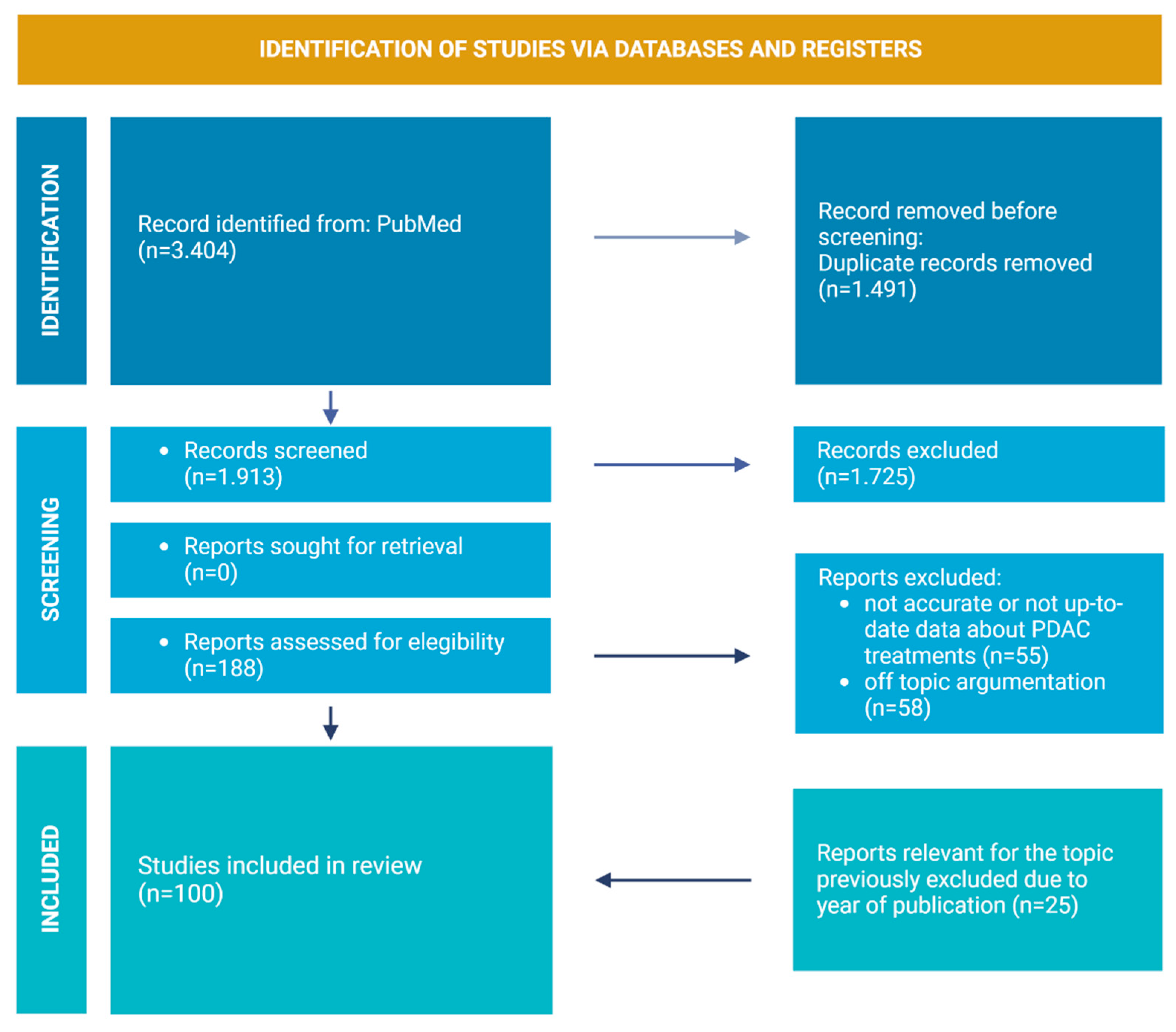
2. Review Strategies and Literature Included
3. PDAC TME
3.1. Acellular Component of PDAC TME
3.2. PSCs and CAFs
3.3. Immune Cells
3.3.1. Macrophages
3.3.2. Lymphocytes
3.3.3. Neutrophils
3.3.4. Mast Cells
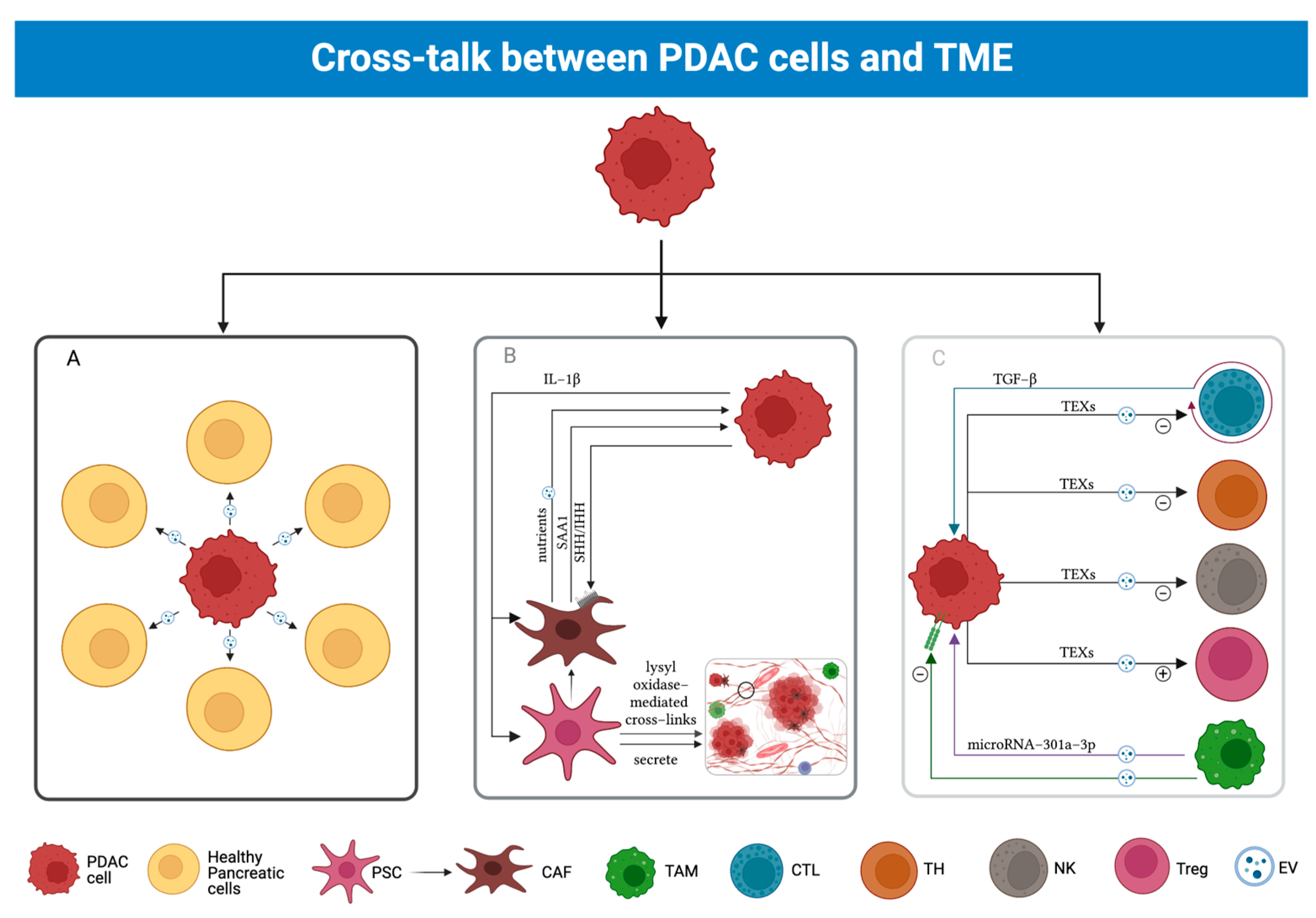
3.4. Cross-Talk between PDAC and TME
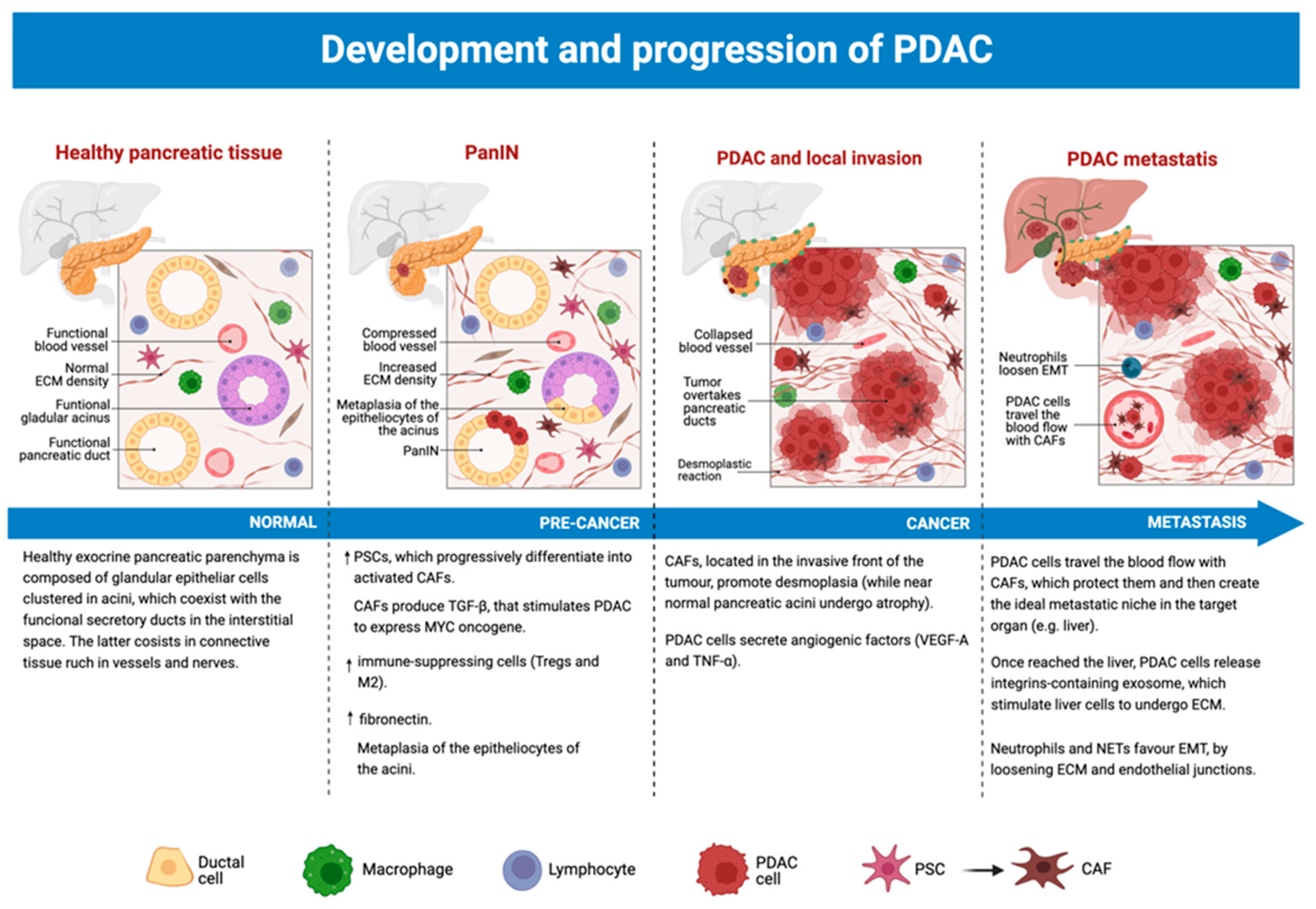
3.5. The Role of TME from Precancerous Lesions to PDAC
4. Conventional Treatment in Resectable Pancreatic Cancers
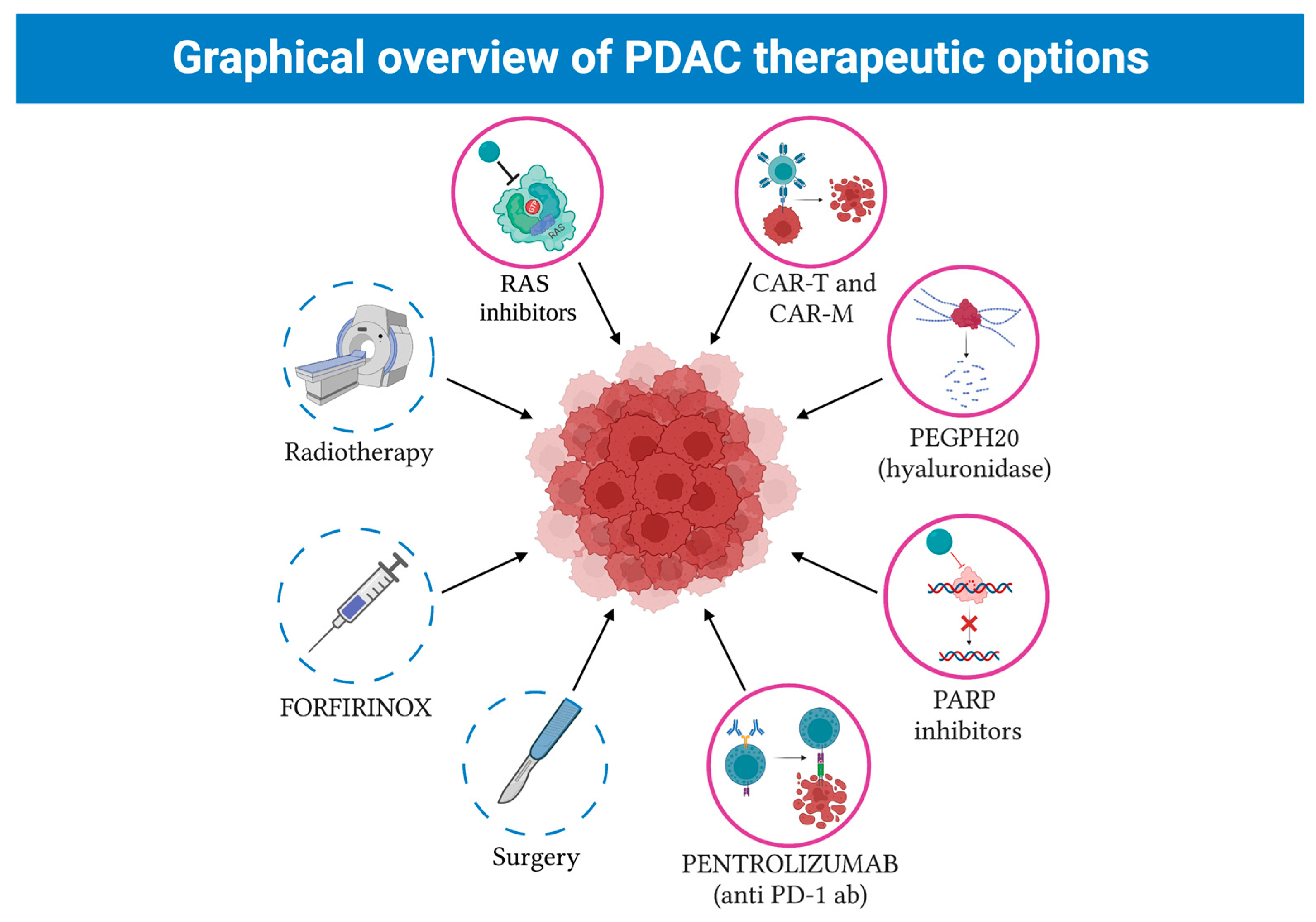
5. New Therapeutical Strategies against PDCA
5.1. Pathway Inhibition
5.2. DNA Repair
5.3. Immunotherapy
Extracellular Tumor Microenvironment
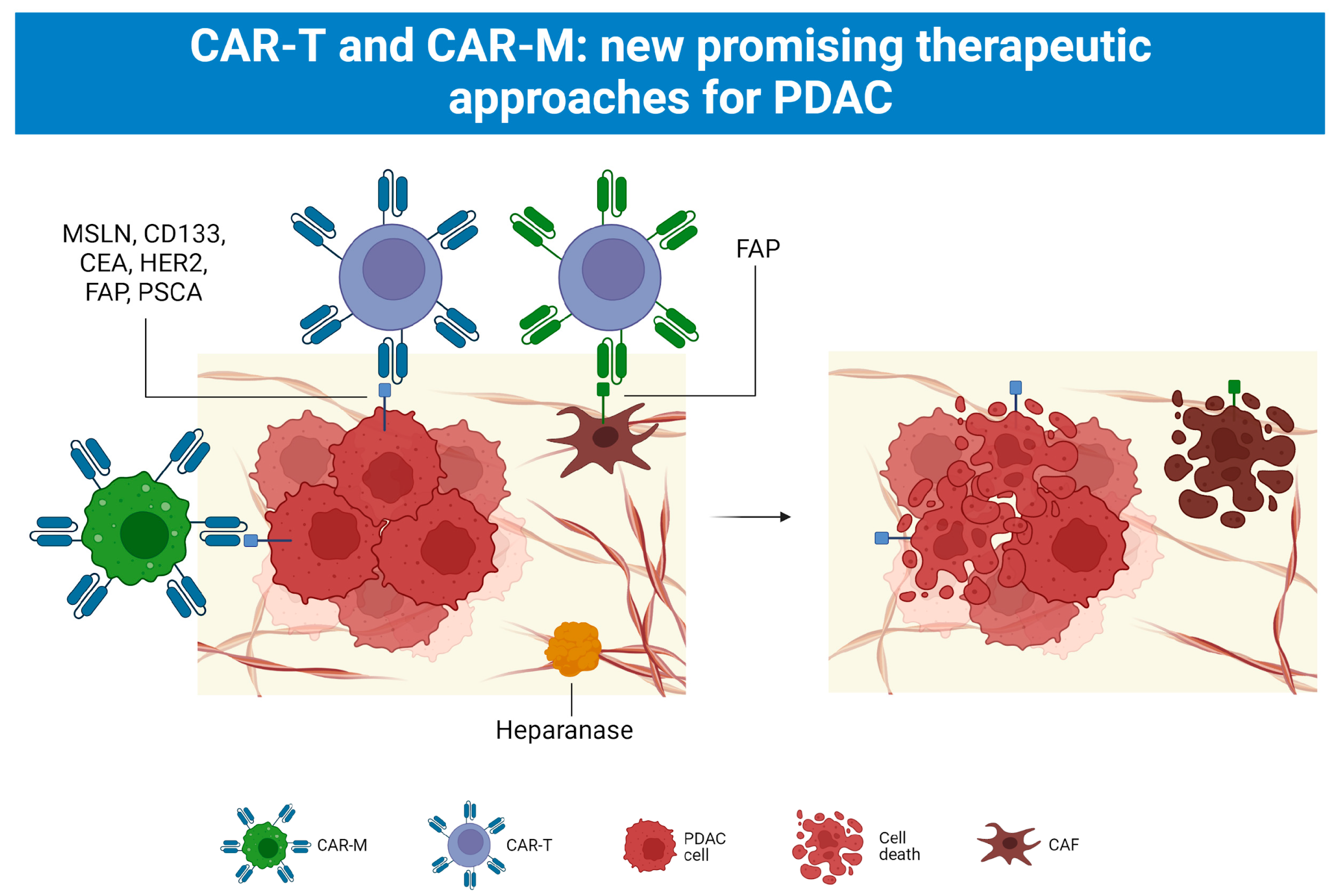
5.4. CAR-T and CAR-M Cell Therapy
5.4.1. First Use of CAR T Cell Therapy
5.4.2. CAR-T Cell Therapy in Solid Tumors: Pancreatic Cancer
5.4.3. Side Effects of CAR-T Cell Therapy
5.4.4. CAR-M Cell Therapy as Another Chance towards Solid Malignancies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADM | Acinar-Ductal Metaplasia |
| αKG | α-ketoglutarate |
| AKT | Protein Kinase B |
| apCAFs | Antigen-Presenting Cancer-Associated Macrophages |
| APCs | Antigen-Presenting Cells |
| ARID1A | AT-rich interactive domain-containing protein 1A |
| α-SMA | α-Smooth Muscle Actin |
| BM | Bone Marrow |
| B-reg | Regulatory B Lymphocytes |
| C1Q | complement component 1q |
| CAFs | Cancer-Associated Fibroblasts |
| CAR-T | chimeric antigen receptor T-cell therapy |
| CAR-M | chimeric antigen receptor macrophages therapy |
| CARTmeso cells | Mesothelin-directed CAR-T cells |
| CCL2 | C-C Chemokine Ligand |
| CD | Cluster of Differentiation |
| CD133 | Prominin1 |
| CDKN2A | cyclin-dependent kinase inhibitor 2A |
| CEA | Carcinoembryonic Antigen |
| CEACAM | CEA-related cell adhesion molecule |
| CICs | Cancer Inducing Cells |
| Col1a1 | Collagen, type I, alpha 1 |
| Co-SMAD | co-mediator SMAD |
| csCAFs | Complement-Secreting Cancer-Associated Macrophages |
| CTLs | Cytotoxic T Lymphocytes |
| CXCL | C-X-C motif Chemokine ligand |
| DCs | Dendritic Cells |
| ECM | Extracellular Matrix |
| EMT | Epithelial-Mesenchymal Transition |
| ERK | Extracellular signal-regulated kinase |
| EVs | Extracellular Vesicles |
| FAP | Fibroblast Activation Protein |
| FGF | Fibroblasts Growth Factor |
| FOXP3 | Forkhead box P3 |
| FSP-1 | Fibroblast Surface Protein 1 |
| GAGs | glycosaminoglycans |
| GM-CSF | Granulocyte-Macrophage Colony Stimulating Factor |
| GREM1 | Gremlin 1 |
| HRAS | Harvey rat sarcoma viral |
| HLA | Human Leukocyte Antigen |
| HLA-DR | Human Leukocyte Antigen-DR isotype |
| iCAFs | Inflammatory Cancer-Associated Fibroblasts |
| IHH | Indian Hedgehog |
| IL | Interleukin |
| IPMN | intraductal papillary mucinous neoplasm |
| KRAS | Kirsten Rat Sarcoma Virus |
| M-CSF | Macrophage-Colony Stimulating Factor |
| MAGUK | Membrane-Associated Guanylate Kinase |
| MCL-1 | myeloid cell leukemia-1 |
| MDSCs | Myeloid-Derived Suppressor Cells |
| MHC | Major Histocompatibility Complex |
| MIF | Macrophage Migration Inhibiting Factor |
| MMPs | Matrix Metalloproteinases |
| MMR | Mismatch Repair |
| MPP6 | MAGUK P55 Subfamily Member |
| MSCs | Mesenchymal Stem Cells |
| MVBs | Multivesicular Bodies |
| MYC | myelocytomatosis oncogene |
| myCAFs | Myofibroblastic Cancer-Associated Macrophages |
| NETs | Neutrophil Extracellular Traps |
| NRAS | (Neuroblasoma rat sarcoma viral |
| NK | Natural Killer |
| NLRP3 | NLR family pyrin domain containing 3 |
| Onf-FN | oncofetal Fibronectin |
| PanIN | Pancreatic Intraepithelial Neoplasia |
| PB | Peripheral Blood |
| PC | Pancreatic Cancer |
| PCCs | Pancreatic Cancer Cells |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| PDGFR | Platelet-derived growth factor receptor |
| PD-L1 | Programmed death-ligand 1 |
| PI3K | phosphatidylinositol-3 kinase |
| pp-GalNAc-T6 | UDP-N-acetyl-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase-6 |
| PSCs | Pancreatic Stellate Cells |
| RAF | rapidly accelerated fibrosarcoma |
| RANTES | Regulated on Activation, Normal T Cell Expressed and Secreted |
| R-SMAD | receptor-regulated SMAD |
| R0 | resections: no cancer cells present microscopically at the primary tumor site |
| ROS | Reactive Oxygen Species |
| Saa3 | Serum Amyloid A 3 |
| SBE | SMAD Binding Element |
| SHH | Sonic Hedgehog |
| SMAD | Small Mother Against Decapentaplegic |
| TAMs | Tumour-Associated Macrophages |
| TCA | Tricarboxylic Acid |
| TET | ten-eleven translocation methylcytosine dioxygenases |
| TEXs | Tumor-Derived Exosomes |
| TGF-β | Transforming Growth Factor β |
| TGFβR | Transforming Growth Factor β Receptor |
| TLR | Toll-Like Receptor |
| TME | Tumour Microenvironment |
| TNF-α | Tumor Necrosis Factor alpha |
| TP53 | Tumour Protein 53 |
| T-reg | Regulatory T Lymphocytes |
| VEGF | Vascular Endothelial Growth Factor |
| YAP | Yes-associated protein |
References
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Canto, M.I.; Jaffee, E.M.; Simeone, D.M. Pancreatic Cancer: Pathogenesis, Screening, Diagnosis, and Treatment. Gastroenterology 2022, 163, 386–402.e1. [Google Scholar] [CrossRef]
- Cai, J.; Chen, H.; Lu, M.; Zhang, Y.; Lu, B.; You, L.; Zhang, T.; Dai, M.; Zhao, Y. Advances in the epidemiology of pancreatic cancer: Trends, risk factors, screening, and prognosis. Cancer Lett. 2021, 520, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Espinet, E.; Klein, L.; Puré, E.; Singh, S.K. Mechanisms of PDAC subtype heterogeneity and therapy response. Trends Cancer 2022, 8, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.X.; Zhao, C.F.; Chen, W.B.; Liu, Q.C.; Li, Q.W.; Lin, Y.Y.; Gao, F. Pancreatic cancer: A review of epidemiology, trend, and risk factors. World J. Gastroenterol. 2021, 27, 4298–4321. [Google Scholar] [CrossRef]
- Connor, A.A.; Gallinger, S. Pancreatic cancer evolution and heterogeneity: Integrating omics and clinical data. Nat. Rev. Cancer 2022, 22, 131–142. [Google Scholar] [CrossRef]
- Singhi, A.D.; Wood, L.D. Early detection of pancreatic cancer using DNA-based molecular approaches. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 457–468. [Google Scholar] [CrossRef]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 10242, 2008–2020. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Martinez-Useros, J.; Martin-Galan, M.; Garcia-Foncillas, J. The Match between Molecular Subtypes, Histology and Microenvironment of Pancreatic Cancer and Its Relevance for Chemoresistance. Cancers 2021, 13, 322. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; He, H. Pancreatic Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1296, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Maddalena, M.; Mallel, G.; Nataraj, N.B.; Shreberk-Shaked, M.; Hassin, O.; Mukherjee, S.; Arandkar, S.; Rotkopf, R.; Kapsack, A.; Lambiase, G.; et al. TP53 missense mutations in PDAC are associated with enhanced fibrosis and an immunosuppressive microenvironment. Proc. Natl. Acad. Sci. USA 2021, 118, e2025631118. [Google Scholar] [CrossRef]
- Reis, J.S.D.; Santos, M.A.R.D.C.; da Costa, K.M.; Freire-de-Lima, C.G.; Morrot, A.; Previato, J.O.; Previato, L.M.; da Fonseca, L.M.; Freire-de-Lima, L. Increased expression of the pathological O-glycosylated form of oncofetal fibronectin in the multidrug resistance phenotype of cancer cells. Matrix Biol. 2023, 118, 47–68. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, C.; Jiang, K.; Werner, J.; Bazhin, A.V.; D’Haese, J.G. The Role of Stellate Cells in Pancreatic Ductal Adenocarcinoma: Targeting Perspectives. Front. Oncol. 2021, 10, 621937. [Google Scholar] [CrossRef]
- Whittle, M.C.; Hingorani, S.R. Fibroblasts in Pancreatic Ductal Adenocarcinoma: Biological Mechanisms and Therapeutic Targets. Gastroenterology 2019, 156, 2085–2096. [Google Scholar] [CrossRef]
- Kerk, S.A.; Papagiannakopoulos, T.; Shah, Y.M.; Lyssiotis, C.A. Metabolic networks in mutant KRAS-driven tumours: Tissue specificities and the microenvironment. Nat. Rev. Cancer 2021, 21, 510–525. [Google Scholar] [CrossRef]
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers 2020, 12, 1347. [Google Scholar] [CrossRef]
- Barrera, L.N.; Evans, A.; Lane, B.; Brumskill, S.; Oldfield, F.E.; Campbell, F.; Andrews, T.; Lu, Z.; Perez-Mancera, P.A.; Lilnoglou, T.; et al. Fibroblasts from Distinct Pancreatic Pathologies Exhibit Disease-Specific Properties. Cancer Res. 2020, 80, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Ren, Y.; Yang, P.; Wang, J.; Zhou, H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022, 13, 897. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, C.; Ohuchida, K.; Shinkawa, T.; Okuda, S.; Otsubo, Y.; Okumura, T.; Sagara, A.; Koikawa, K.; Ando, Y.; Shindo, K.; et al. Bone marrow-derived macrophages converted into cancer-associated fibroblast-like cells promote pancreatic cancer progression. Cancer Lett. 2021, 512, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734, Erratum in Cancer Cell 2015, 28, 831–833. [Google Scholar] [CrossRef]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Chen, K.; Wang, Q.; Li, M.; Guo, H.; Liu, W.; Wang, F.; Tian, X.; Yang, Y. Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. EBioMedicine 2021, 66, 103315. [Google Scholar] [CrossRef]
- Chen, Z.G.; Wang, Y.; Fong, W.P.; Hu, M.T.; Liang, J.Y.; Wang, L.; Li, Y.H. A quantitative score of immune cell infiltration predicts the prognosis in pancreatic ductal adenocarcinoma. Int. Immunopharmacol. 2021, 98, 107890. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell-Derived IL1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020, 80, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Liot, S.; Balas, J.; Aubert, A.; Prigent, L.; Mercier-Gouy, P.; Verrier, B.; Bertolino, P.; Hennino, A.; Valcourt, U.; Lambert, E. Stroma Involvement in Pancreatic Ductal Adenocarcinoma: An Overview Focusing on Extracellular Matrix Proteins. Front. Immunol. 2021, 12, 612271. [Google Scholar] [CrossRef] [PubMed]
- Pratt, H.G.; Steinberger, K.J.; Mihalik, N.E.; Ott, S.; Whalley, T.; Szomolay, B.; Boone, B.A.; Eubank, T.D. Macrophage and Neutrophil Interactions in the Pancreatic Tumor Microenvironment Drive the Pathogenesis of Pancreatic Cancer. Cancers 2021, 14, 194. [Google Scholar] [CrossRef] [PubMed]
- Väyrynen, S.A.; Zhang, J.; Yuan, C.; Väyrynen, J.P.; Dias Costa, A.; Williams, H.; Morales-Oyarvide, V.; Lau, M.C.; Rubinson, D.A.; Dunne, R.F.; et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 1069–1081. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, Q.; Peng, J.; Wang, M.; Gao, X.; Liao, Q.; Zhao, Y. Pancreatic cancer-educated macrophages protect cancer cells from complement-dependent cytotoxicity by up-regulation of CD59. Cell Death Dis. 2019, 10, 836. [Google Scholar] [CrossRef]
- Foucher, E.D.; Ghigo, C.; Chouaib, S.; Galon, J.; Iovanna, J.; Olive, D. Pancreatic Ductal Adenocarcinoma: A Strong Imbalance of Good and Bad Immunological Cops in the Tumor Microenvironment. Front. Immunol. 2018, 9, 1044. [Google Scholar] [CrossRef]
- Hegde, S.; Krisnawan, V.E.; Herzog, B.H.; Zuo, C.; Breden, M.A.; Knolhoff, B.L.; Hogg, G.D.; Tang, J.P.; Baer, J.M.; Mpoy, C.; et al. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell 2020, 37, 289–307.e9. [Google Scholar] [CrossRef]
- Goulart, M.R.; Stasinos, K.; Fincham, R.E.A.; Delvecchio, F.R.; Kocher, H.M. T cells in pancreatic cancer stroma. World J. Gastroenterol. 2021, 27, 7956–7968. [Google Scholar] [CrossRef]
- Grunberg, N.; Pevsner-Fischer, M.; Goshen-Lago, T.; Diment, J.; Stein, Y.; Lavon, H.; Mayer, S.; Levi-Galibov, O.; Friedman, G.; Ofir-Birin, Y.; et al. Cancer-Associated Fibroblasts Promote Aggressive Gastric Cancer Phenotypes via Heat Shock Factor 1-Mediated Secretion of Extracellular Vesicles. Cancer Res. 2021, 81, 1639–1653. [Google Scholar] [CrossRef]
- Campbell, J.R.; McDonald, B.R.; Mesko, P.B.; Siemers, N.O.; Singh, P.B.; Selby, M.; Sproul, T.W.; Korman, A.J.; Vlach, L.M.; Houser, J.; et al. Fc-Optimized Anti-CCR8 Antibody Depletes Regulatory T Cells in Human Tumor Models. Cancer Res. 2021, 81, 2983–2994. [Google Scholar] [CrossRef] [PubMed]
- Daley, D.; Zambirinis, C.P.; Seifert, L.; Akkad, N.; Mohan, N.; Werba, G.; Barilla, R.; Torres-Hernandez, A.; Hundeyin, M.; Mani, V.R.K.; et al. γδ T Cells Support Pancreatic Oncogenesis by Restraining αβ T Cell Activation. Cell 2016, 166, 1485–1499.e15. [Google Scholar] [CrossRef] [PubMed]
- McAllister, F.; Bailey, J.M.; Alsina, J.; Nirschl, C.J.; Sharma, R.; Fan, H.; Rattigan, Y.; Roeser, J.C.; Lankapalli, R.H.; Zhang, H.; et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 2014, 25, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, S.; Micke, P.; Elebro, J.; Heby, M.; Hrynchyk, I.; Nodin, B.; Leandersson, K.; Mezheyeuski, A.; Jirström, K. Topographical Distribution and Spatial Interactions of Innate and Semi-Innate Immune Cells in Pancreatic and Other Periampullary Adenocarcinoma. Front. Immunol. 2020, 11, 558169. [Google Scholar] [CrossRef]
- Hiroshima, Y.; Kasajima, R.; Kimura, Y.; Komura, D.; Ishikawa, S.; Ichikawa, Y.; Bouvet, M.; Yamamoto, N.; Oshima, T.; Morinaga, S.; et al. Novel targets identified by integrated cancer-stromal interactome analysis of pancreatic adenocarcinoma. Cancer Lett. 2020, 469, 217–227. [Google Scholar] [CrossRef]
- Dardare, J.; Witz, A.; Merlin, J.L.; Gilson, P.; Harlé, A. SMAD4 and the TGFβ Pathway in Patients with Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 3534. [Google Scholar] [CrossRef]
- Garcia, P.E.; Scales, M.K.; Allen, B.L.; Pasca di Magliano, M. Pancreatic Fibroblast Heterogeneity: From Development to Cancer. Cells 2020, 9, 2464. [Google Scholar] [CrossRef]
- Djurec, M.; Graña, O.; Lee, A.; Troulé, K.; Espinet, E.; Cabras, L.; Navas, C.; Blasco, M.T.; Martín-Díaz, L.; Burdiel, M.; et al. Saa3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. Proc. Natl. Acad. Sci. USA 2018, 115, E1147–E1156. [Google Scholar] [CrossRef]
- Jang, J.E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef]
- Zhang, W.; Campbell, D.H.; Walsh, B.J.; Packer, N.H.; Liu, D.; Wang, Y. Cancer-derived small extracellular vesicles: Emerging biomarkers and therapies for pancreatic ductal adenocarcinoma diagnosis/prognosis and treatment. J. Nanobiotechnol. 2022, 20, 446. [Google Scholar] [CrossRef]
- Yin, Z.; Ma, T.; Huang, B.; Lin, L.; Zhou, Y.; Yan, J.; Zou, Y.; Chen, S. Macrophage-derived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-β signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 310. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.E.; Xiao, W.; Hill, R.; Usc Pancreas Research Team. Cancer-Associated Fibroblasts Confer Gemcitabine Resistance to Pancreatic Cancer Cells through PTEN-Targeting miRNAs in Exosomes. Cancers 2022, 14, 2812. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Papademetrio, D.L.; Garcia, M.N.; Grasso, D.; Alvarez, É. Autophagy-Mediated Exosomes as Immunomodulators of Natural Killer Cells in Pancreatic Cancer Microenvironment. Front. Oncol. 2021, 10, 622956. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Pahlavanneshan, M.; Eun, C.Y.; Zhang, X.; DeKalb, C.; Mahgoub, B.; Knaneh-Monem, H.; Shah, S.; Sohrabi, A.; Seidlits, S.K.; et al. Matrix stiffness mediates pancreatic cancer chemoresistance through induction of exosome hypersecretion in a cancer associated fibroblasts-tumor organoid biomimetic model. Matrix Biol. Plus 2022, 14, 100111. [Google Scholar] [CrossRef]
- Opitz, F.V.; Haeberle, L.; Daum, A.; Esposito, I. Tumor Microenvironment in Pancreatic Intraepithelial Neoplasia. Cancers 2021, 13, 6188. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.I.; Jang, S.I.; Hong, J.; Kim, C.H.; Kwon, S.S.; Park, J.S.; Lim, J.B. Cancer-initiating cells in human pancreatic cancer organoids are maintained by interactions with endothelial cells. Cancer Lett. 2021, 498, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kang, K.N.; Shin, Y.S.; Byun, Y.; Han, Y.; Kwon, W.; Kim, C.W.; Jang, J.Y. Biomarker Panel for the Diagnosis of Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 1443. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.P.; Walsh, R.M.; Ramanathan, R.K.; Khorana, A.A. Pancreatic adenocarcinoma: Treating a systemic disease with systemic therapy. J. Natl. Cancer Inst. 2014, 106, dju011. [Google Scholar] [CrossRef] [PubMed]
- Motoi, F.; Kosuge, T.; Ueno, H.; Yamaue, H.; Satoi, S.; Sho, M.; Honda, G.; Matsumoto, I.; Wada, K.; Furuse, J.; et al. Randomized phase II/III trial of neoadjuvant chemotherapy with gemcitabine and S-1 versus upfront surgery for resectable pancreatic cancer (Prep-02/JSAP05). Jpn. J. Clin. Oncol. 2019, 49, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Han, Y.; Lee, H.; Kim, S.W.; Kwon, W.; Lee, K.H.; Oh, D.Y.; Chie, E.K.; Lee, J.M.; Heo, J.S.; et al. Oncological Benefits of Neoadjuvant Chemoradiation With Gemcitabine Versus Upfront Surgery in Patients With Borderline Resectable Pancreatic Cancer: A Prospective, Randomized, Open-label, Multicenter Phase 2/3 Trial. Ann. Surg. 2018, 268, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2020, 17, 108–123. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Davide Melisi, D.; Teresa Macarulla, T.; Pazo-Cid, T.; Chandana, S.R.; De La Fouchardiere, C.; Dean, A.P.; Igor Kiss, I.; Lee, W.; Goetze, T.O.; et al. NAPOLI-3: A randomized, open-label phase 3 study of liposomal irinotecan + 5-fluorouracil/leucovorin + oxaliplatin (NALIRIFOX) versus nab-paclitaxel + gemcitabine in treatment-naïve patients with metastatic pancreatic ductal adenocarcinoma (mPDAC). J. Clin. Oncol. 2023, 41 (Suppl. S4), LBA661. [Google Scholar] [CrossRef]
- Springfeld, C.; Jäger, D.; Büchler, M.W.; Strobel, O.; Hackert, T.; Palmer, D.H.; Neoptolemos, J.P. Chemotherapy for pancreatic cancer. Presse Med. 2019, 48, e159–e174. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Dreyer, S.B.; Chang, D.K.; Bailey, P.; Biankin, A.V. Pancreatic Cancer Genomes: Implications for Clinical Management and Therapeutic Development. Clin. Cancer Res. 2017, 23, 1638–1646. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef]
- Fakih, M.; Durm, G.A.; Govindan, R.; Falchook, G.S.; Soman, N.; Henary, H.A.; David, S. HongTrial in progress: A phase Ib study of AMG 510, a specific and irreversible KRAS G12C inhibitor, in combination with other anticancer therapies in patients with advanced solid tumors harboring KRAS p.G12C mutation (CodeBreak 101). J. Clin. Oncol. 2020, 38 (Suppl. S15), TPS3661. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Golan, T.; Varadhachary, G.R.; Sela, T.; Fogelman, D.R.; Halperin, N.; Shroff, R.T.; Halparin, S.; Xiao, L.; Aderka, D.; Maitra, A.; et al. Phase II study of olaparib for BRCAness phenotype in pancreatic cancer. JCO 2018, 36 (Suppl. S4), 297. [Google Scholar] [CrossRef]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef]
- Timmer, F.E.F.; Geboers, B.; Nieuwenhuizen, S.; Dijkstra, M.; Schouten, E.A.C.; Puijk, R.S.; de Vries, J.J.J.; van den Tol, M.P.; Bruynzeel, A.M.E.; Streppel, M.M.; et al. Pancreatic Cancer and Immunotherapy: A Clinical Overview. Cancers 2021, 13, 4138. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Patel, K.; Siraj, S.; Smith, C.; Nair, M.; Vishwanatha, J.K.; Basha, R. Pancreatic Cancer: An Emphasis on Current Perspectives in Immunotherapy. Crit. Rev. Oncog. 2019, 24, 105–118. [Google Scholar] [CrossRef]
- Sohal, D.P.S.; Kennedy, E.B.; Khorana, A.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Krishnamurthi, S.; Moravek, C.; O’Reilly, E.M.; Philip, P.A.; et al. Metastatic Pancreatic Cancer: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2018, 36, 2545–2556. [Google Scholar] [CrossRef]
- Eroglu, Z.; Zaretsky, J.M.; Hu-Lieskovan, S.; Kim, D.W.; Algazi, A.; Johnson, D.B.; Liniker, E.; Kong, B.; Munhoz, R.; Rapisuwon, S.; et al. High response rate to PD-1 blockade in desmoplastic melanomas. Nature 2018, 553, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Singh, V.; Ricca, A.; Lee, P. Survival Benefit of Pembrolizumab for Patients With Pancreatic Adenocarcinoma: A Case Series. J. Med. Cases 2022, 13, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor- modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Locke, F.L.; Neelapu, S.S.; Bartlett, N.L.; Siddiqi, T.; Chavez, J.C.; Hosing, C.M.; Ghobadi, A.; Budde, L.E.; Bot, A.; Rossi, J.M.; et al. Phase 1 results of ZUMA-1: A multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol. Ther. 2017, 25, 285–295. [Google Scholar] [CrossRef]
- Akce, M.; Zaidi, M.; Waller, E.K.; El-Rayes, B.F.; Lesinski, G.B. The Potential of CAR T Cell Therapy in Pancreatic Cancer. Front. Immunol. 2018, 9, 2166. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Lv, J.; Zhao, R.; Wu, D.; Zheng, D.; Wu, Z.; Shi, J.; Wei, X.; Wu, Q.; Long, Y.; Lin, S.; et al. Mesothelin is a target of chimeric antigen receptor T cells for treating gastric cancer. J. Hematol. Oncol. 2019, 12, 18. [Google Scholar] [CrossRef]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J. Carcinoembryonic antigen-based vaccines. Semin Oncol. 2003, 30 (Suppl. S8), 30–36. [Google Scholar] [CrossRef]
- Berinstein, N.L. Carcinoembryonic antigen as a target for therapeutic anticancer vaccines: A review. J. Clin. Oncol. 2002, 20, 2197–2207. [Google Scholar] [CrossRef]
- Katz, S.C.; Burga, R.A.; McCormack, E.; Wang, L.J.; Mooring, W.; Point, G.R.; Khare, P.D.; Thorn, M.; Ma, Q.; Stainken, B.F.; et al. Phase I Hepatic Immunotherapy for Metastases Study of Intra-Arterial Chimeric Antigen Receptor-Modified T-cell Therapy for CEA+ Liver Metastases. Clin. Cancer Res. 2015, 21, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Varghese, A.M. Chimeric antigen receptor (CAR) T and other T cell strategies for pancreas adenocarcinoma. Chin. Clin. Oncol. 2017, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef]
- Tran, E.; Chinnasamy, D.; Yu, Z.; Morgan, R.A.; Lee, C.C.; Restifo, N.P.; Rosenberg, S.A. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 2013, 210, 1125–1135. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.; Wesolowski, R.; Ahn, D.H.; Wu, C.; Mortazavi, A.; Lustberg, M.; Ramaswamy, B.; Fowler, J.; Wei, L.; Overholser, J.; et al. Phase I Immunotherapy Trial with Two Chimeric HER-2 B-Cell Peptide Vaccines Emulsified in Montanide ISA 720VG and Nor-MDP Adjuvant in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 3495–3507. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, Z.; Tan, X.; Jiang, H.; Xu, Z.; Fang, Y.; Han, D.; Hong, W.; Wei, W.; Tu, J. CAR-macrophage: A new immunotherapy candidate against solid tumors. Biomed. Pharmacother. 2021, 139, 111605. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mercanti, L.; Sindaco, M.; Mazzone, M.; Di Marcantonio, M.C.; Piscione, M.; Muraro, R.; Mincione, G. PDAC, the Influencer Cancer: Cross-Talk with Tumor Microenvironment and Connected Potential Therapy Strategies. Cancers 2023, 15, 2923. https://doi.org/10.3390/cancers15112923
Mercanti L, Sindaco M, Mazzone M, Di Marcantonio MC, Piscione M, Muraro R, Mincione G. PDAC, the Influencer Cancer: Cross-Talk with Tumor Microenvironment and Connected Potential Therapy Strategies. Cancers. 2023; 15(11):2923. https://doi.org/10.3390/cancers15112923
Chicago/Turabian StyleMercanti, Leonardo, Maria Sindaco, Mariangela Mazzone, Maria Carmela Di Marcantonio, Mariagrazia Piscione, Raffaella Muraro, and Gabriella Mincione. 2023. "PDAC, the Influencer Cancer: Cross-Talk with Tumor Microenvironment and Connected Potential Therapy Strategies" Cancers 15, no. 11: 2923. https://doi.org/10.3390/cancers15112923
APA StyleMercanti, L., Sindaco, M., Mazzone, M., Di Marcantonio, M. C., Piscione, M., Muraro, R., & Mincione, G. (2023). PDAC, the Influencer Cancer: Cross-Talk with Tumor Microenvironment and Connected Potential Therapy Strategies. Cancers, 15(11), 2923. https://doi.org/10.3390/cancers15112923