
Genomic Alterations Associated with Estrogen Receptor Pathway Activity in Metastatic Breast Cancer Have a Differential Impact on Downstream ER Signaling

 , , , , , , ,
, , , , , , ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Study Design and Patients
2.2. Prior Endocrine Therapy
2.3. Treatment Outcome and Response to Endocrine Therapies
2.4. WGS and Data Analyses
2.5. RNA Sequencing: RNA Isolation, Library Preparation, and Sequencing
2.6. Processing of RNA Sequencing Data
2.7. Hierarchical Clustering on RNA Expression Levels of ER-Regulated Genes
2.8. Statistical Methods
3. Results
3.1. Patient Cohort
3.2. Hierarchical Clustering Reveals Distinct RNA Expression of ER Target Genes between ESR1 Mutant and ESR1 Wild-Type Samples
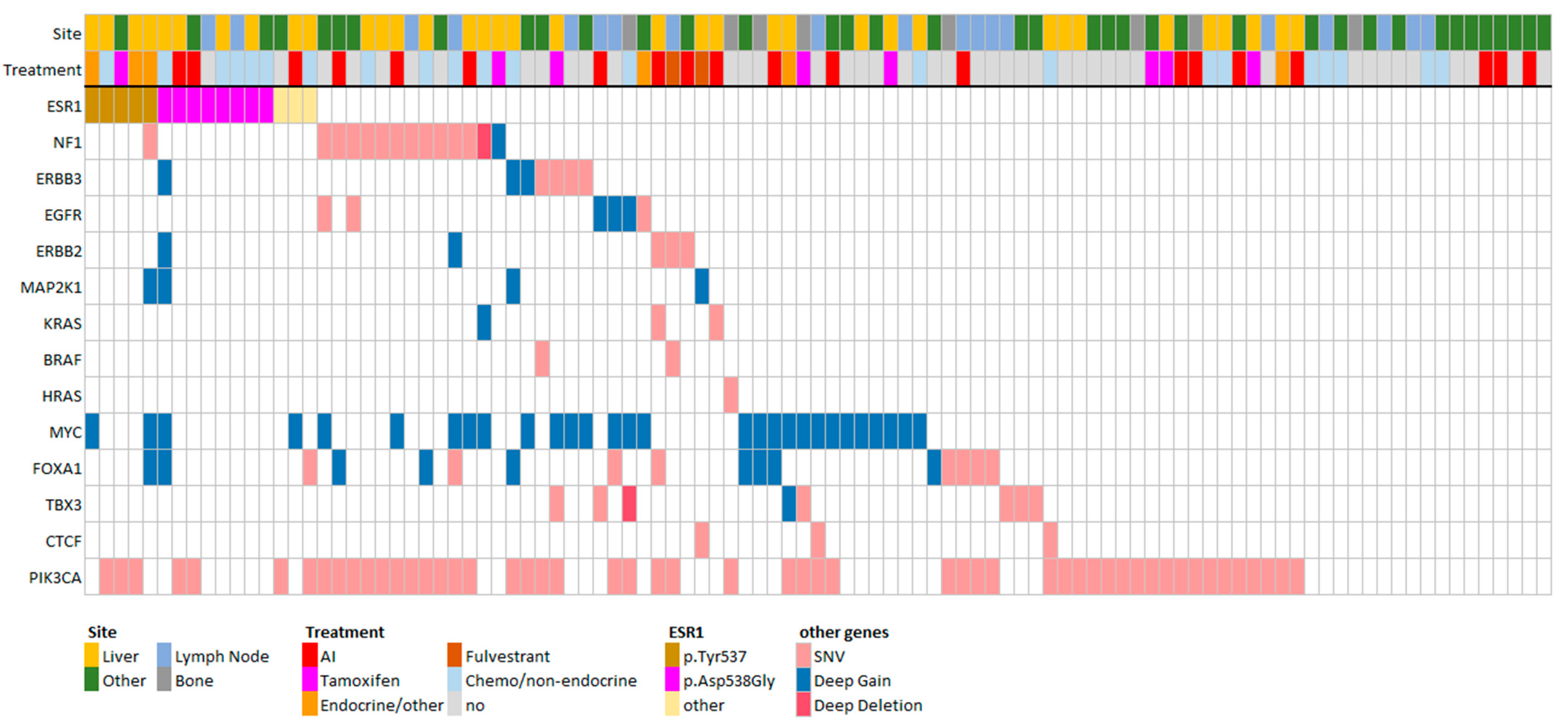
3.3. FGFR1 and TSPYL Amplifications Are Enriched beyond ESR1 Mutations
3.4. Best Response on Endocrine Therapies versus Expression Clusters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Senkus, E.; Costa, A.; Papadopoulos, E.; Aapro, M.; Andre, F.; Harbeck, N.; Aguilar Lopez, B.; Barrios, C.H.; Bergh, J.; et al. 4th ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4). Ann. Oncol. 2018, 29, 1634–1657. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.F.R.; Bondarenko, I.M.; Trishkina, E.; Dvorkin, M.; Panasci, L.; Manikhas, A.; Shparyk, Y.; Cardona-Huerta, S.; Cheung, K.L.; Philco-Salas, M.J.; et al. Fulvestrant 500 mg versus anastrozole 1 mg for hormone receptor-positive advanced breast cancer (FALCON): An international, randomised, double-blind, phase 3 trial. Lancet 2016, 388, 2997–3005. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Petrakova, K.; Blackwell, K.L.; Winer, E.P.; et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann. Oncol. 2018, 29, 1541–1547. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Tredan, O.; Chen, S.C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e426. [Google Scholar] [CrossRef]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef]
- Angus, L.; Smid, M.; Wilting, S.M.; van Riet, J.; Van Hoeck, A.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.G.; Tjan-Heijnen, V.C.G.; Labots, M.; et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat. Genet. 2019, 51, 1450–1458. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol. 2016, 2, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Spoerke, J.M.; Gendreau, S.; Walter, K.; Qiu, J.; Wilson, T.R.; Savage, H.; Aimi, J.; Derynck, M.K.; Chen, M.; Chan, I.T.; et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat. Commun. 2016, 7, 11579. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef]
- Schiavon, G.; Hrebien, S.; Garcia-Murillas, I.; Cutts, R.J.; Pearson, A.; Tarazona, N.; Fenwick, K.; Kozarewa, I.; Lopez-Knowles, E.; Ribas, R.; et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med. 2015, 7, 313ra182. [Google Scholar] [CrossRef]
- Allouchery, V.; Beaussire, L.; Perdrix, A.; Sefrioui, D.; Augusto, L.; Guillemet, C.; Sarafan-Vasseur, N.; Di Fiore, F.; Clatot, F. Circulating ESR1 mutations at the end of aromatase inhibitor adjuvant treatment and after relapse in breast cancer patients. Breast Cancer Res. 2018, 20, 40. [Google Scholar] [CrossRef]
- Bidard, F.C.; Pistilli, B.; Dalenc, F.; De la Motte Rouge, T.; Sabatier, R.; Frenel, J.S.; Ladoire, S.; Dubot, C.; Ferrero, J.M.; Levy, C.; et al. Abstract PD2-06: Circulating ESR1 mutation detection rate and early decrease under first line aromatase inhibitor and palbociclib in the PADA-1 trial (UCBG-GINECO). Cancer Res. 2019, 79 (Suppl. 4), PD2-06. [Google Scholar] [CrossRef]
- Sokol, E.S.; Feng, Y.X.; Jin, D.X.; Basudan, A.; Lee, A.V.; Atkinson, J.M.; Chen, J.; Stephens, P.J.; Frampton, G.M.; Gupta, P.B.; et al. Loss of function of NF1 is a mechanism of acquired resistance to endocrine therapy in lobular breast cancer. Ann. Oncol. 2019, 30, 115–123. [Google Scholar] [CrossRef]
- Zheng, Z.Y.; Anurag, M.; Lei, J.T.; Cao, J.; Singh, P.; Peng, J.; Kennedy, H.; Nguyen, N.C.; Chen, Y.; Lavere, P.; et al. Neurofibromin Is an Estrogen Receptor-alpha Transcriptional Co-repressor in Breast Cancer. Cancer Cell 2020, 37, 387–402.e7. [Google Scholar] [CrossRef]
- Priestley, P.; Baber, J.; Lolkema, M.; Steeghs, N.; de Bruijn, E.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; Roepman, P.; et al. Pan-cancer whole genome analyses of metastatic solid tumors. bioRxiv 2018. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Blokzijl, F.; Janssen, R.; van Boxtel, R.; Cuppen, E. MutationalPatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Smid, M.; Coebergh van den Braak, R.R.J.; van de Werken, H.J.G.; van Riet, J.; van Galen, A.; de Weerd, V.; van der Vlugt-Daane, M.; Bril, S.I.; Lalmahomed, Z.S.; Kloosterman, W.P.; et al. Gene length corrected trimmed mean of M-values (GeTMM) processing of RNA-seq data performs similarly in intersample analyses while improving intrasample comparisons. BMC Bioinform. 2018, 19, 236. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Verhaegh, W.; van Ooijen, H.; Inda, M.A.; Hatzis, P.; Versteeg, R.; Smid, M.; Martens, J.; Foekens, J.; van de Wiel, P.; Clevers, H.; et al. Selection of personalized patient therapy through the use of knowledge-based computational models that identify tumor-driving signal transduction pathways. Cancer Res. 2014, 74, 2936–2945. [Google Scholar] [CrossRef] [PubMed]
- de Hoon, M.J.L.; Imoto, S.; Nolan, J.; Miyano, S. Open source clustering software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, A.J. Java Treeview—Extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef] [PubMed]
- Desmedt, C.; Haibe-Kains, B.; Wirapati, P.; Buyse, M.; Larsimont, D.; Bontempi, G.; Delorenzi, M.; Piccart, M.; Sotiriou, C. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin. Cancer Res. 2008, 14, 5158–5165. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yeheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef]
- Formisano, L.; Stauffer, K.M.; Young, C.D.; Bhola, N.E.; Guerrero-Zotano, A.L.; Jansen, V.M.; Estrada, M.M.; Hutchinson, K.E.; Giltnane, J.M.; Schwarz, L.J.; et al. Association of FGFR1 with ERalpha Maintains Ligand-Independent ER Transcription and Mediates Resistance to Estrogen Deprivation in ER+ Breast Cancer. Clin. Cancer Res. 2017, 23, 6138–6150. [Google Scholar] [CrossRef]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef]
- Drago, J.Z.; Formisano, L.; Juric, D.; Niemierko, A.; Servetto, A.; Wander, S.A.; Spring, L.M.; Vidula, N.; Younger, J.; Peppercorn, J.; et al. FGFR1 Amplification Mediates Endocrine Resistance but Retains TORC Sensitivity in Metastatic Hormone Receptor-Positive (HR+) Breast Cancer. Clin. Cancer Res. 2019, 25, 6443–6451. [Google Scholar] [CrossRef]
- Mouron, S.; Manso, L.; Caleiras, E.; Rodriguez-Peralto, J.L.; Rueda, O.M.; Caldas, C.; Colomer, R.; Quintela-Fandino, M.; Bueno, M.J. FGFR1 amplification or overexpression and hormonal resistance in luminal breast cancer: Rationale for a triple blockade of ER, CDK4/6, and FGFR1. Breast Cancer Res. 2021, 23, 21. [Google Scholar] [CrossRef]
- Hayes, E.L.; Lewis-Wambi, J.S. Mechanisms of endocrine resistance in breast cancer: An overview of the proposed roles of noncoding RNA. Breast Cancer Res. 2015, 17, 40. [Google Scholar] [CrossRef]
- Musolino, A.; Campone, M.; Neven, P.; Denduluri, N.; Barrios, C.H.; Cortes, J.; Blackwell, K.; Soliman, H.; Kahan, Z.; Bonnefoi, H.; et al. Phase II, randomized, placebo-controlled study of dovitinib in combination with fulvestrant in postmenopausal patients with HR+, HER2− breast cancer that had progressed during or after prior endocrine therapy. Breast Cancer Res. 2017, 19, 18. [Google Scholar] [CrossRef]
- Campone, M.; Bachelot, T.; Penault-Llorca, F.; Pallis, A.; Agrapart, V.; Pierrat, M.J.; Poirot, C.; Dubois, F.; Xuereb, L.; Bossard, C.J.; et al. A phase Ib dose allocation study of oral administration of lucitanib given in combination with fulvestrant in patients with estrogen receptor-positive and FGFR1-amplified or non-amplified metastatic breast cancer. Cancer Chemother. Pharmacol. 2019, 83, 743–753. [Google Scholar] [CrossRef]
- Quintela-Fandino, M.; Apala, J.V.; Malon, D.; Mouron, S.; Hornedo, J.; Gonzalez-Cortijo, L.; Colomer, R.; Guerra, J. Nintedanib plus letrozole in early breast cancer: A phase 0/I pharmacodynamic, pharmacokinetic, and safety clinical trial of combined FGFR1 and aromatase inhibition. Breast Cancer Res. 2019, 21, 69. [Google Scholar] [CrossRef]
- Hui, R.; Pearson, A.; Cortes, J.; Campbell, C.; Poirot, C.; Azim, H.A., Jr.; Fumagalli, D.; Lambertini, M.; Daly, F.; Arahmani, A.; et al. Lucitanib for the Treatment of HR+/HER2− Metastatic Breast Cancer: Results from the Multicohort Phase II FINESSE Study. Clin. Cancer Res. 2020, 26, 354–363. [Google Scholar] [CrossRef]
- Epping, M.T.; Meijer, L.A.; Krijgsman, O.; Bos, J.L.; Pandolfi, P.P.; Bernards, R. TSPYL5 suppresses p53 levels and function by physical interaction with USP7. Nat. Cell Biol. 2011, 13, 102–108. [Google Scholar] [CrossRef]
- Liu, M.; Ingle, J.N.; Fridley, B.L.; Buzdar, A.U.; Robson, M.E.; Kubo, M.; Wang, L.; Batzler, A.; Jenkins, G.D.; Pietrzak, T.L.; et al. TSPYL5 SNPs: Association with plasma estradiol concentrations and aromatase expression. Mol. Endocrinol. 2013, 27, 657–670. [Google Scholar] [CrossRef]
- Fanning, S.W.; Greene, G.L. Next-Generation ERalpha Inhibitors for Endocrine-Resistant ER+ Breast Cancer. Endocrinology 2019, 160, 759–769. [Google Scholar] [CrossRef]
- Aftimos, P.; Oliveira, M.; Irrthum, A.; Fumagalli, D.; Sotiriou, C.; Gal-Yam, E.N.; Robson, M.E.; Ndozeng, J.; Di Leo, A.; Ciruelos, E.M.; et al. Genomic and Transcriptomic Analyses of Breast Cancer Primaries and Matched Metastases in AURORA, the Breast International Group (BIG) Molecular Screening Initiative. Cancer Discov. 2021, 11, 2796–2811. [Google Scholar] [CrossRef]
- Horwitz, K.B.; Koseki, Y.; McGuire, W.L. Estrogen control of progesterone receptor in human breast cancer: Role of estradiol and antiestrogen. Endocrinology 1978, 103, 1742–1751. [Google Scholar] [CrossRef]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Paish, E.C.; Powe, D.G.; Gee, J.; Nicholson, R.I.; Lee, A.H.; Robertson, J.F.; Ellis, I.O. Biologic and clinical characteristics of breast cancer with single hormone receptor positive phenotype. J. Clin. Oncol. 2007, 25, 4772–4778. [Google Scholar] [CrossRef]
- Colditz, G.A.; Rosner, B.A.; Chen, W.Y.; Holmes, M.D.; Hankinson, S.E. Risk factors for breast cancer according to estrogen and progesterone receptor status. J. Natl. Cancer Inst. 2004, 96, 218–228. [Google Scholar] [CrossRef]
- Lindstrom, L.S.; Karlsson, E.; Wilking, U.M.; Johansson, U.; Hartman, J.; Lidbrink, E.K.; Hatschek, T.; Skoog, L.; Bergh, J. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. J. Clin. Oncol. 2012, 30, 2601–2608. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Patients (n = 101) | Specification of Prior Treatments | |||||
|---|---|---|---|---|---|---|
| Age | N | % | N | % | ||
| Median (interquartile range) | 59 (52–64) | Aromatase inhibitor | 53 | 52.5 | ||
| Gender | Tamoxifen | 57 | 56.4 | |||
| Female | 101 | 100 | Fulvestrant | 16 | 15.8 | |
| Male | 0 | 0 | Everolimus | 15 | 14.9 | |
| Prior systemic therapy | CDK4/6 | 6 | 5.9 | |||
| Yes | 5-FU | 35 | 34.7 | |||
| Endocrine therapy only | 14 | 13.7 | Taxanes | 44 | 43.6 | |
| Chemotherapy only | 11 | 10.9 | Platinum/Parp | 8 | 7.9 | |
| Endocrine and chemotherapy | 51 | 50.5 | Anthracyclines | 52 | 51.5 | |
| Nr of lines (median, IQR) | 3 (2-5) | Cyclophosphamide | 50 | 49.5 | ||
| Nr of drugs (median, IQR) | 5 (4-8) | Eribulin | 3 | 3.0 | ||
| No prior treatment | 25 | 27.8 | Vinorelbine | 3 | 3.0 | |
| Last treatment before biopsy | Anti-HER2 | 3 | 3.0 | |||
| Tamoxifen | 8 | 7.9 | ||||
| Aromatase inhibitor | 20 | 19.8 | ||||
| Fulvestrant | 2 | 2.0 | ||||
| Combination endocrine therapy | 6 | 5.9 | ||||
| Chemotherapy or non-containing endocrine therapy | 22 | 21.8 | ||||
| No treatment within one year before biopsy | 43 | 42.6 | ||||
| Prior radiotherapy | ||||||
| Yes | 62 | 61.4 | ||||
| No | 39 | 38.6 | ||||
| Biopsy site | ||||||
| Liver | 36 | 35.6 | ||||
| Bone | 8 | 7.9 | ||||
| Lymph node | 20 | 19.8 | ||||
| Breast | 14 | 13.9 | ||||
| Other | 7 | 6.9 | ||||
| Unknown | 3 | 3.0 | ||||
| Gene | p-Value FDR Hochberg | Fold Change * |
|---|---|---|
| TPBG | 1.88 × 10−8 | 2.6 |
| IGF1R | 4.30 × 10−5 | 3.2 |
| CYP2T1P | 5.02 × 10−5 | 4.4 |
| SIAH2 | 0.00011 | 2.3 |
| FMN1 | 0.00039 | 2.1 |
| THSD4 | 0.00043 | 3.1 |
| AC0647992 | 0.00141 | 3.1 |
| CUEDC1 | 0.00213 | 2.0 |
| SUSD3 | 0.00402 | 5.3 |
| PARD6B | 0.00715 | 3.5 |
| ZNF516 | 0.00746 | 2.2 |
| PREX1 | 0.01012 | 2.6 |
| IL6ST | 0.01655 | 2.3 |
| STC2 | 0.01764 | 4.6 |
| MYBL1 | 0.02001 | 2.5 |
| EGLN2 | 0.02175 | 2.0 |
| COX6C | 0.03034 | 3.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angus, L.; Smid, M.; Wilting, S.M.; Bos, M.K.; Steeghs, N.; Konings, I.R.H.M.; Tjan-Heijnen, V.C.G.; van Riel, J.M.G.H.; van de Wouw, A.J.; CPCT Consortium; et al. Genomic Alterations Associated with Estrogen Receptor Pathway Activity in Metastatic Breast Cancer Have a Differential Impact on Downstream ER Signaling. Cancers 2023, 15, 4416. https://doi.org/10.3390/cancers15174416
Angus L, Smid M, Wilting SM, Bos MK, Steeghs N, Konings IRHM, Tjan-Heijnen VCG, van Riel JMGH, van de Wouw AJ, CPCT Consortium, et al. Genomic Alterations Associated with Estrogen Receptor Pathway Activity in Metastatic Breast Cancer Have a Differential Impact on Downstream ER Signaling. Cancers. 2023; 15(17):4416. https://doi.org/10.3390/cancers15174416
Chicago/Turabian StyleAngus, Lindsay, Marcel Smid, Saskia M. Wilting, Manouk K. Bos, Neeltje Steeghs, Inge R. H. M. Konings, Vivianne C. G. Tjan-Heijnen, Johanna M. G. H. van Riel, Agnes J. van de Wouw, CPCT Consortium, and et al. 2023. "Genomic Alterations Associated with Estrogen Receptor Pathway Activity in Metastatic Breast Cancer Have a Differential Impact on Downstream ER Signaling" Cancers 15, no. 17: 4416. https://doi.org/10.3390/cancers15174416
APA StyleAngus, L., Smid, M., Wilting, S. M., Bos, M. K., Steeghs, N., Konings, I. R. H. M., Tjan-Heijnen, V. C. G., van Riel, J. M. G. H., van de Wouw, A. J., CPCT Consortium, Cuppen, E., Lolkema, M. P., Jager, A., Sleijfer, S., & Martens, J. W. M. (2023). Genomic Alterations Associated with Estrogen Receptor Pathway Activity in Metastatic Breast Cancer Have a Differential Impact on Downstream ER Signaling. Cancers, 15(17), 4416. https://doi.org/10.3390/cancers15174416