

Characteristics and Outcome of FLT3-ITD-Positive Pediatric Acute Myeloid Leukemia—Experience of Polish Pediatric Leukemia and Lymphoma Study Group from 2005 to 2022
 , , , , , , , , , , , and add
Show full author list
, , , , , , , , , , , and add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
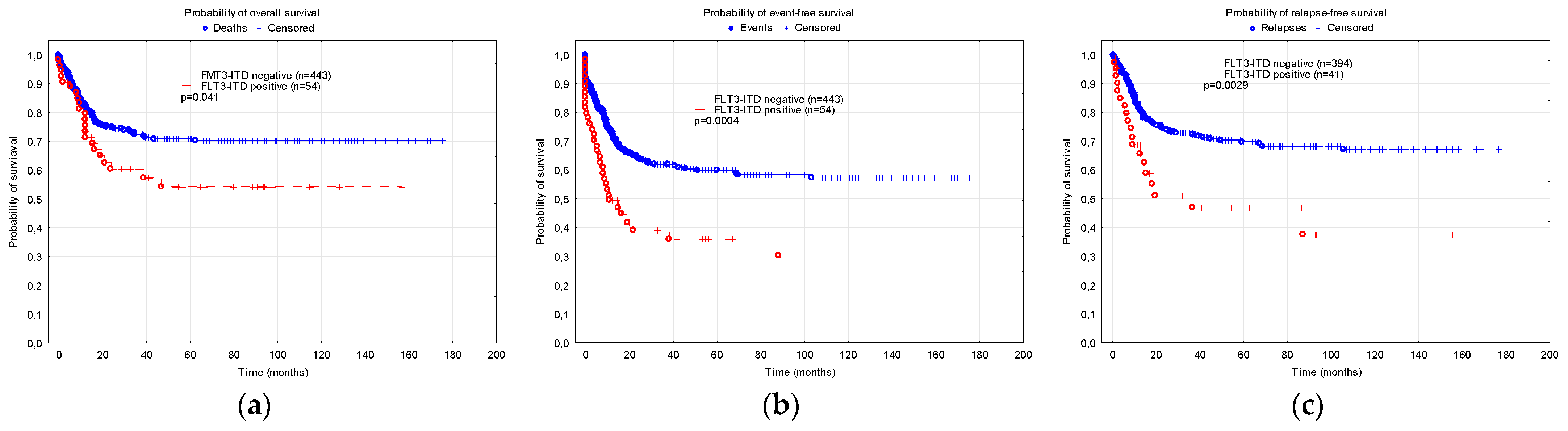
3.1. Comparison of the Patients with and without FLT3-ITD Mutation
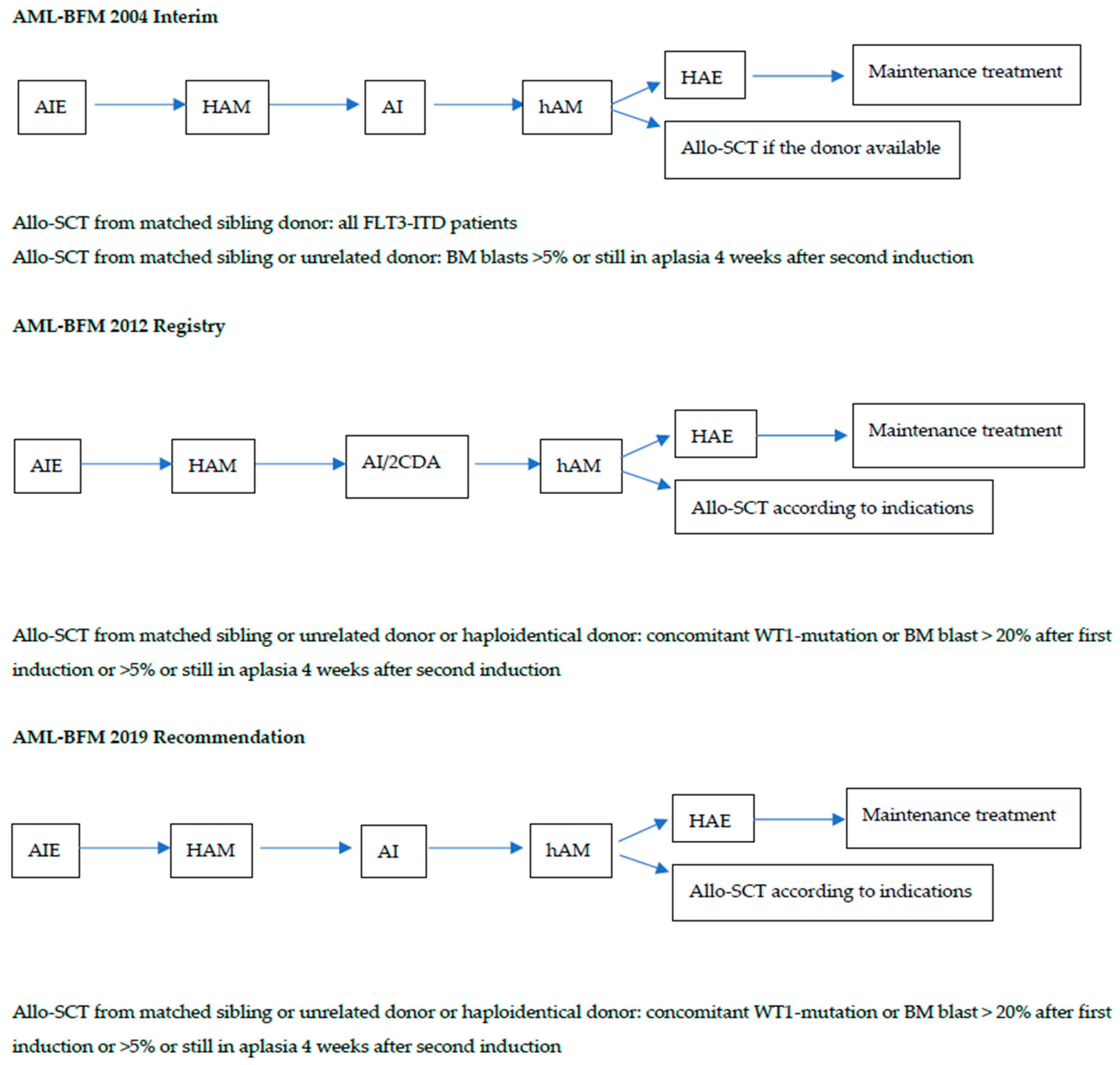
3.2. Characteristics of the Patients and Outcome Depending on the Treatment Protocol
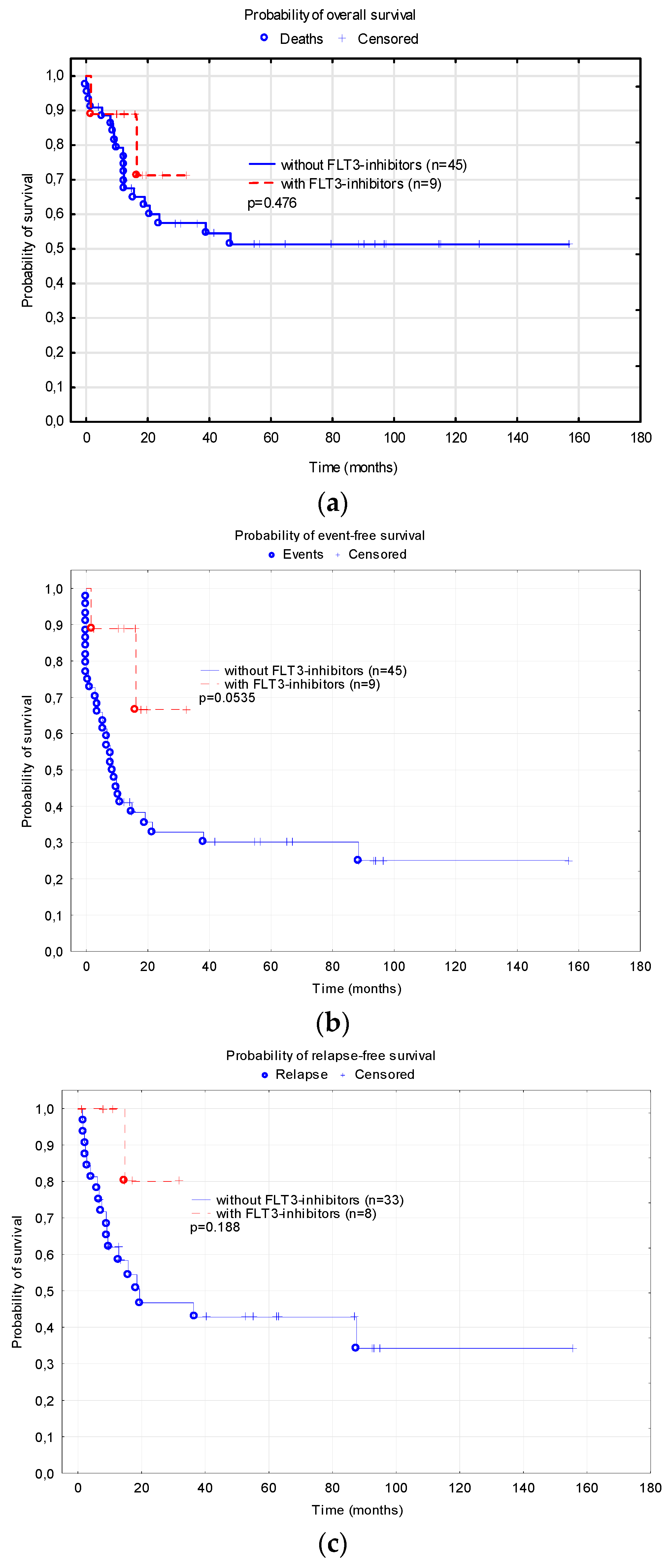
3.3. FLT3-Inhibitors
3.4. Stem Cell Transplantation
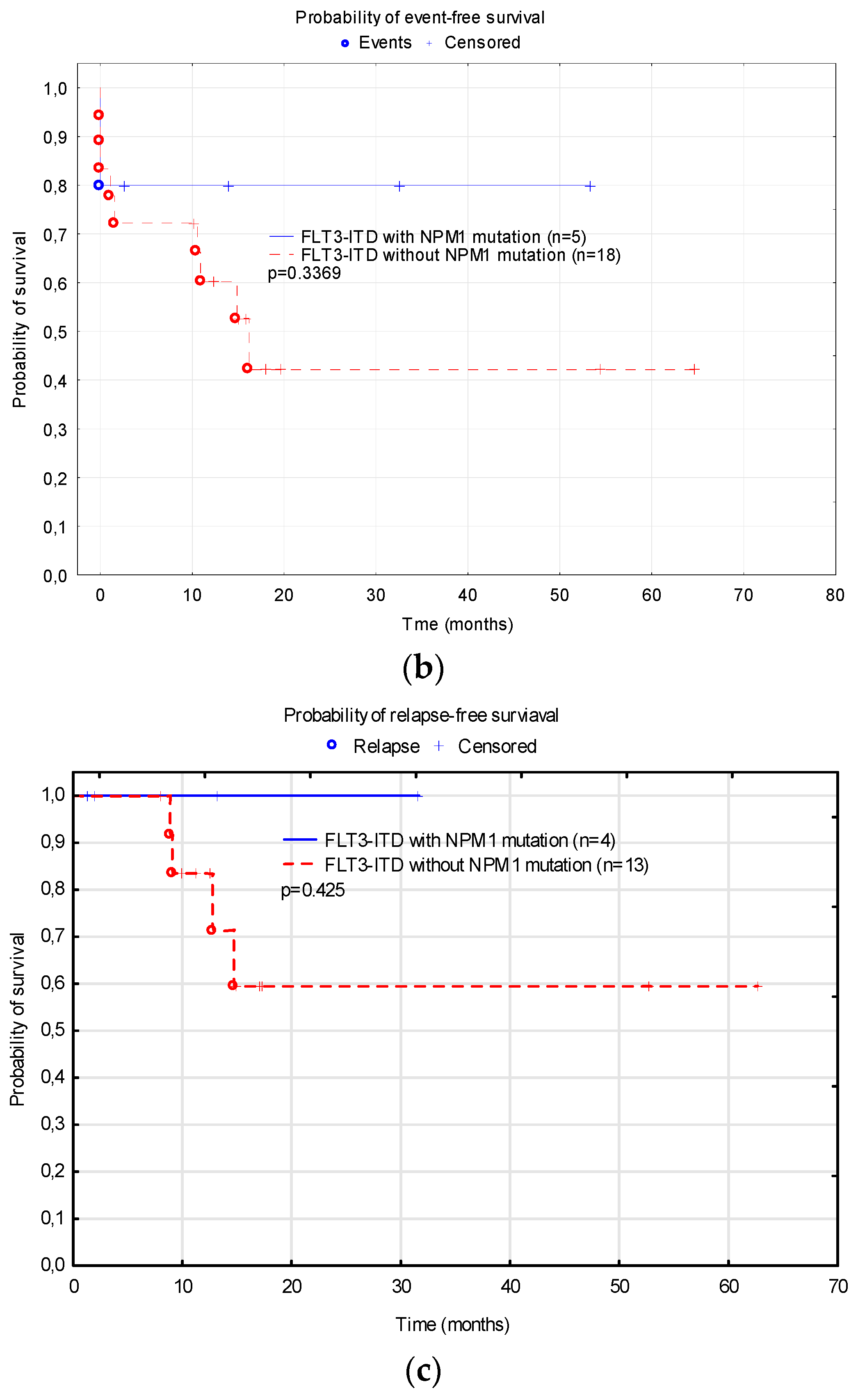
3.5. Treatment Results Depending on Additional Mutation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rasche, M.; Zimmermann, M.; Borschel, L.; Bourquin, J.P.; Dworzak, M.; Klingebiel, T.; Lehrnbecher, T.; Creutzig, U.; Klusmann, J.H.; Reinhardt, D. Successes and challenges in the treatment of pediatric acute myeloid leukemia: A retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia 2018, 32, 2167–2177. [Google Scholar] [CrossRef]
- Reinhardt, D.; Antoniou, E.; Waack, K. Pediatric Acute Myeloid Leukemia-Past, Present, and Future. J. Clin. Med. 2022, 11, 504. [Google Scholar] [CrossRef]
- Griffith, J.; Black, J.; Faerman, C.; Swenson, L.; Wynn, M.; Lu, F.; Lippke, J.; Saxena, K. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol. Cell. 2004, 13, 169–178. [Google Scholar] [CrossRef]
- Small, D.; Levenstein, M.; Kim, E.; Carow, C.; Amin, S.; Rockwell, P.; Witte, L.; Burrow, C.; Ratajczak, M.Z.; Gewirtz, A.M. STK-1, the human homolog of Flk-2/Flt-3, is selectively expressed in CD34+ human bone marrow cells and is involved in the proliferation of early progenitor/stem cells. Proc. Natl. Acad. Sci. USA 1994, 91, 459–463. [Google Scholar] [CrossRef]
- Turner, A.M.; Lin, N.L.; Issarachai, S.; Lyman, S.D.; Broudy, V.C. FLT3 receptor expression on the surface of normal and malignant human hematopoietic cells. Blood 1996, 88, 3383–3390. [Google Scholar] [CrossRef]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schäkel, U.; Platzbecker, U.; Wermke, M.; Bornhäuser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002, 99, 4326–4335. [Google Scholar] [CrossRef]
- Meshinchi, S.; Alonzo, T.A.; Stirewalt, D.L.; Zwaan, M.; Zimmerman, M.; Reinhardt, D.; Kaspers, G.J.; Heerema, N.A.; Gerbing, R.; Lange, B.J.; et al. Clinical implications of FLT3 mutations in pediatric AML. Blood 2006, 108, 3654–3661. [Google Scholar] [CrossRef]
- Zwaan, C.M.; Meshinchi, S.; Radich, J.P.; Veerman, A.J.; Huismans, D.R.; Munske, L.; Podleschny, M.; Hählen, K.; Pieters, R.; Zimmermann, M.; et al. FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: Prognostic significance and relation to cellular drug resistance. Blood 2003, 102, 2387–2394. [Google Scholar] [CrossRef]
- Manara, E.; Basso, G.; Zampini, M.; Buldini, B.; Tregnago, C.; Rondelli, R.; Masetti, R.; Bisio, V.; Frison, M.; Polato, K.; et al. Characterization of children with FLT3-ITD acute myeloid leukemia: A report from the AIEOP AML-2002 study group. Leukemia 2017, 31, 18–25. [Google Scholar] [CrossRef]
- Meshinchi, S.; Appelbaum, F.R. Structural and functional alterations of FLT3 in acute myeloid leukemia. Clin. Cancer Res. 2009, 15, 4263–4269. [Google Scholar] [CrossRef]
- Sexauer, A.N.; Tasian, S.K. Targeting FLT3 Signaling in Childhood Acute Myeloid Leukemia. Front. Pediatr. 2017, 5, 248. [Google Scholar] [CrossRef]
- Pommert, L.; Tarlock, K. The evolution of targeted therapy in pediatric AML: Gemtuzumab ozogamicin, FLT3/IDH/BCL2 inhibitors, and other therapies. Hematol. Am. Soc. Hematol. Educ. Program. 2022, 1, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.M.; Melgar, K.; Bolanos, L.; Hueneman, K.; Walker, M.M.; Jiang, J.K.; Wilson, K.M.; Zhang, X.; Shen, J.; Jiang, F.; et al. Targeting AML-associated FLT3 mutations with a type I kinase inhibitor. J. Clin. Investig. 2020, 130, 2017–2023. [Google Scholar] [CrossRef] [PubMed]
- Conneely, S.E.; Stevens, A.M. Acute Myeloid Leukemia in Children: Emerging Paradigms in Genetics and New Approaches to Therapy. Curr. Oncol. Rep. 2021, 23, 16. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Rubnitz, J.E.; Coustan-Smith, E.; Li, L.; Furmanski, B.D.; Mascara, G.P.; Heym, K.M.; Christensen, R.; Onciu, M.; Shurtleff, S.A.; et al. Phase I pharmacokinetic and pharmacodynamic study of the multikinase inhibitor sorafenib in combination with clofarabine and cytarabine in pediatric relapsed/refractory leukemia. J. Clin. Oncol. 2011, 29, 3293–3300. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Gerbing, R.; Brown, P.; Fox, E.; Choi, J.; Fisher, B.; Hirsch, B.; Kahwash, S.; Getz, K.; et al. Sorafenib in Combination with Standard Chemotherapy for Children With High Allelic Ratio FLT3/ITD+ Acute Myeloid Leukemia: A Report From the Children’s Oncology Group Protocol AAML1031. J. Clin. Oncol. 2022, 40, 2023–2035. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Czogała, M.; Balwierz, W.; Pawińska-Wąsikowska, K.; Książek, T.; Bukowska-Strakova, K.; Czogała, W.; Sikorska-Fic, B.; Matysiak, M.; Skalska-Sadowska, J.; Wachowiak, J.; et al. Advances in the First Line Treatment of Pediatric Acute Myeloid Leukemia in the Polish Pediatric Leukemia and Lymphoma Study Group from 1983 to 2019. Cancers 2021, 13, 4536. [Google Scholar] [CrossRef]
- Semary, S.F.; Hammad, M.; Soliman, S.; Yassen, D.; Gamal, M.; Albeltagy, D.; Hamdy, N.; Mahmoud, S. Outcome of Childhood Acute Myeloid Leukemia with FLT3-ITD Mutation: The Experience of Children’s Cancer Hospital Egypt, 2007–2017. Clin. Lymphoma Myeloma Leuk. 2020, 20, e529–e541. [Google Scholar] [CrossRef]
- Ostronoff, F.; Othus, M.; Gerbing, R.B.; Loken, M.R.; Raimondi, S.C.; Hirsch, B.A.; Lange, B.J.; Petersdorf, S.; Radich, J.; Appelbaum, F.R.; et al. NUP98/NSD1 and FLT3/ITD coexpression is more prevalent in younger AML patients and leads to induction failure: A COG and SWOG report. Blood 2014, 124, 2400–2407. [Google Scholar] [CrossRef]
- Cucchi, D.G.J.; Denys, B.; Kaspers, G.J.L.; Janssen, J.J.W.M.; Ossenkoppele, G.J.; de Haas, V.; Zwaan, C.M.; van den Heuvel-Eibrink, M.M.; Philippé, J.; Csikós, T.; et al. RNA-based FLT3-ITD allelic ratio is associated with outcome and ex vivo response to FLT3 inhibitors in pediatric AML. Blood 2018, 131, 2485–2489. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A.; Iijima-Yamashita, Y.; Tawa, A.; Tomizawa, D.; Yamada, M.; Norio, S.; Watanabe, T.; Taga, T.; Iwamoto, S.; Terui, K.; et al. Risk-stratified therapy for children with FLT3-ITD-positive acute myeloid leukemia: Results from the JPLSG AML-05 study. Int. J. Hematol. 2018, 107, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Bazarbachi, A.; Bug, G.; Baron, F.; Brissot, E.; Ciceri, F.; Dalle, I.A.; Döhner, H.; Esteve, J.; Floisand, Y.; Giebel, S.; et al. Clinical practice recommendation on hematopoietic stem cell transplantation for acute myeloid leukemia patients with FLT3-internal tandem duplication: A position statement from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Haematologica 2020, 105, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Niktoreh, N.; Walter, C.; Zimmermann, M.; von Neuhoff, C.; von Neuhoff, N.; Rasche, M.; Waack, K.; Creutzig, U.; Hanenberg, H.; Reinhardt, D. Mutated WT1, FLT3-ITD, and NUP98-NSD1 Fusion in Various Combinations Define a Poor Prognostic Group in Pediatric Acute Myeloid Leukemia. J. Oncol. 2019, 2019, 1609128. [Google Scholar] [CrossRef] [PubMed]
- Tarlock, K.; Sulis, M.L.; Chewning, J.H.; Pollard, J.A.; Cooper, T.; Gamis, A.; Shenoy, S.; Kutny, M.; Horan, J.; Meshinchi, S.; et al. Hematopoietic Cell Transplantation in the Treatment of Pediatric Acute Myelogenous Leukemia and Myelodysplastic Syndromes: Guidelines from the American Society of Transplantation and Cellular Therapy. Transplant. Cell Ther. 2022, 28, 530–545. [Google Scholar] [CrossRef]
- Tarlock, K.; Chang, B.; Cooper, T.; Gross, T.; Gupta, S.; Neudorf, S.; Adlard, K.; Ho, P.A.; McGoldrick, S.; Watt, T.; et al. Sorafenib treatment following hematopoietic stem cell transplant in pediatric FLT3/ITD acute myeloid leukemia. Pediatr. Blood Cancer 2015, 62, 1048–1054. [Google Scholar] [CrossRef]
- McCall, D.; Roth, M.; Mahadeo, K.M.; Toepfer, L.; Nunez, C.; Short, N.J.; Daver, N.; Kadia, T.M.; DiNardo, C.; Yi, J.S.; et al. Gilteritinib combination therapies in pediatric patients with FLT3-mutated acute myeloid leukemia. Blood Adv. 2021, 5, 5215–5219. [Google Scholar] [CrossRef]
- Bolouri, H.; Farrar, J.E.; Triche, T., Jr.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef]
- Hollink, I.H.; van den Heuvel-Eibrink, M.M.; Zimmermann, M.; Balgobind, B.V.; Arentsen-Peters, S.T.; Alders, M.; Willasch, A.; Kaspers, G.J.; Trka, J.; Baruchel, A.; et al. Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood 2009, 113, 5951–5960. [Google Scholar] [CrossRef]
- Tarlock, K.; Alonzo, T.A.; Gerbing, R.B.; Ries, R.E.; Gibson, B.; Niktoreh, N.; Noort, S.; Van den Heuvel-Eibrink, M.M.; Zwaan, M.; Meshinchi, S. Distinct Co-Occurring Mutational Profiles in Acute Myeloid Leukemia Confers Prognostic Significance in Children and Young Adults with FLT3/ITD Mutations. Blood 2018, 132 (Suppl. S1), 443. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FLT3 WT | FLT3-ITD | p | |
|---|---|---|---|
| Number of patients | 443 | 54 | |
| Age median (range) years | 8.8 (0.1–18.0) | 13.4 (3.0–18.0) | <0.0001 |
| Gender male/female | 229/214 | 29/25 | NS |
| WBC at diagnosis—median (range) ×109/L | 19 (0.9–979) | 92 (6.5–573) | <0.0001 |
| FAB types | |||
| M0 | 23 (5.2%) | 4 (7.4%) | NS |
| M1 | 58 (13.1%) | 9 (16.7%) | NS |
| M2 | 109 (24.6%) | 20 (37.0%) | NS |
| M4 | 71 (16.0%) | 12 (22.2%) | NS |
| M5 | 118 (26.6%) | 8 (14.8%) | 0.044 |
| M6 | 6 (1.3%) | 0 | NS |
| M7 | 35 (7.9%) | 0 | 0.029 |
| Non defined | 23 (5.2%) | 1 (1.8%) | |
| Fusion genes analysis | N = 419 | N = 48 | |
| No fusion genes | 216 (51.5%) | 36 (75%) | 0.002 |
| RUNX1::RUNX1T1 | 76 (18.1%) | 0 | 0.0013 |
| CBFB::MYH11 | 28 (6.7%) | 3 (6.2%) | NS |
| MLL::MLLT3 | 27 (6.4%) | 0 | NS |
| MLL::MLLT10 | 27 (6.4%) | 0 | NS |
| MLL::MLLT1 | 9 (2.1%) | 0 | NS |
| MLL::MLLT4 | 4 (0.9%) | 1 (2%) | NS |
| MLL::MLLT6 | 3 (0.7%) | 0 | NS |
| DEK::NUP214 | 6 (1.4%) | 4 (8.3%) | 0.0018 |
| BCR::ABL | 3 (0.7%) | 0 | NS |
| Others | 20 (4.8%) | 2 (4.2%) | NS |
| Additional mutations | |||
| WT1 | 15/189 (7.9) | 13/28 (46.4) | <0.0001 |
| NPM1 | 4/180 (2.2) | 5/23 (21.7) | 0.0002 |
| Complex karyotype | 40/370 (10.8%) | 2/40 (5%) | NS |
| FLT3 WT | FLT3-ITD | p | |
|---|---|---|---|
| Number of patients | 443 | 54 | |
| CR (%) | 394 (88.9) | 41 (76) | 0.0063 |
| Early deaths (%) | 10 (2.2) | 2 (3.7) | NS |
| Deaths in aplasia before CR (%) | 7 (1.6) | 3 (5.5) | 0.049 |
| Non-responders (%) | 24 (5.4) | 8 (11) | 0.008 |
| Relapses (%) | 101 (22.7) | 19 (35) | 0.045 |
| Deaths in CR (%) | 18 (4.1) | 1 (1.8) | NS |
| Deaths from disease progression (%) | 58 (13.1) | 16 (29.6) | 0.013 |
| Continuous CR after first-line therapy (%) | 275 (62.1) | 21 (38.9) | 0.001 |
| 5-year OS | 0.71 ± 0.03 | 0.54 ± 0.07 | 0.041 |
| 5-year EFS | 0.59 ± 0.03 | 0.36 ± 0.07 | 0.0004 |
| 5-year RFS | 0.70 ± 0.04 | 0.47 ± 0.09 | 0.0029 |
| Protocol | AML-BFM 2004 Interim | AML-BFM 2012 Registry AML-BFM 2019 Recommendations | p |
|---|---|---|---|
| Period | 2004–2014 | 2015–2022 | - |
| Follow-up end-point | 31 December 2020 | 31 May 2023 | - |
| Median observation time (range) months | 93.5 (29–156.8) | 28.7 (10.2–88.6) | <0.0001 |
| Number of patients | 30 | 24 | - |
| Age median (range) years | 12.8 (3.0–17.7) | 13.4 (6.9–17.9) | NS |
| Gender male/female | 18/12 | 11/13 | NS |
| FAB types (%) | - | ||
| M0 | 3 (10) | 1 (4.2) | NS |
| M1 | 8 (26.7) | 1 (4.2) | 0.029 |
| M2 | 9 (30) | 12 (50) | NS |
| M4 | 7 (23.3) | 5 (20.8) | NS |
| M5 | 3 (10) | 5 (20.8) | NS |
| M6 | 0 | 0 | - |
| M7 | 0 | 0 | - |
| Fusion genes | N = 24 | N = 23 | |
| CBFB::MYH11 | 2 (8.3) | 1 (4.3) | NS |
| MLL::MLLT4 | 0 | 1 (4.3) | NS |
| DEK:NUP214 | 0 | 4 (17.4) | NS |
| Risk groups (%) | - | ||
| SR | 0 | 0 | - |
| IR | - | 8 (33.3) | - |
| HR | 30 (100) | 16 (66.7) | NS |
| Treatment results | |||
| CR | 23 (76.7) | 18 (75) | NS |
| Early deaths (%) | 2 (6.7) | 0 | NS |
| Deaths in aplasia before CR (%) | 1 (3.3) | 2 (8.3) | NS |
| Non-responders (%) | 4 (13.3) | 4 (16.7) | NS |
| Relapses (%) | 15 (50) | 4 (16.7) | 0.012 |
| Deaths in CR (%) | 1 (3.3) | 0 | NS |
| Deaths from disease (%) | 11 (36.7) | 5 (20.8) | NS |
| Continuous CR (%) | 7 (23.3) | 14 (58.3) | 0.009 |
| SCT (%) | 12 (40) | 14 (58.3) | NS |
| 5-year OS | 0.49 ± 0.09 | 0.65 ± 0.11 | NS |
| 5-year EFS | 0.27 ± 0.08 | 0.52 ± 0.12 | 0.069 |
| 5-year RFS | 0.37 ± 0.10 | 0.68 ± 0.13 | 0.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czogała, M.; Czogała, W.; Pawińska-Wąsikowska, K.; Książek, T.; Bukowska-Strakova, K.; Sikorska-Fic, B.; Łaguna, P.; Fałkowska, A.; Drabko, K.; Muszyńska-Rosłan, K.; et al. Characteristics and Outcome of FLT3-ITD-Positive Pediatric Acute Myeloid Leukemia—Experience of Polish Pediatric Leukemia and Lymphoma Study Group from 2005 to 2022. Cancers 2023, 15, 4557. https://doi.org/10.3390/cancers15184557
Czogała M, Czogała W, Pawińska-Wąsikowska K, Książek T, Bukowska-Strakova K, Sikorska-Fic B, Łaguna P, Fałkowska A, Drabko K, Muszyńska-Rosłan K, et al. Characteristics and Outcome of FLT3-ITD-Positive Pediatric Acute Myeloid Leukemia—Experience of Polish Pediatric Leukemia and Lymphoma Study Group from 2005 to 2022. Cancers. 2023; 15(18):4557. https://doi.org/10.3390/cancers15184557
Chicago/Turabian StyleCzogała, Małgorzata, Wojciech Czogała, Katarzyna Pawińska-Wąsikowska, Teofila Książek, Karolina Bukowska-Strakova, Barbara Sikorska-Fic, Paweł Łaguna, Anna Fałkowska, Katarzyna Drabko, Katarzyna Muszyńska-Rosłan, and et al. 2023. "Characteristics and Outcome of FLT3-ITD-Positive Pediatric Acute Myeloid Leukemia—Experience of Polish Pediatric Leukemia and Lymphoma Study Group from 2005 to 2022" Cancers 15, no. 18: 4557. https://doi.org/10.3390/cancers15184557
APA StyleCzogała, M., Czogała, W., Pawińska-Wąsikowska, K., Książek, T., Bukowska-Strakova, K., Sikorska-Fic, B., Łaguna, P., Fałkowska, A., Drabko, K., Muszyńska-Rosłan, K., Krawczuk-Rybak, M., Kozłowska, M., Irga-Jaworska, N., Zielezińska, K., Urasiński, T., Bartoszewicz, N., Styczyński, J., Skalska-Sadowska, J., Wachowiak, J., ... Skoczeń, S. (2023). Characteristics and Outcome of FLT3-ITD-Positive Pediatric Acute Myeloid Leukemia—Experience of Polish Pediatric Leukemia and Lymphoma Study Group from 2005 to 2022. Cancers, 15(18), 4557. https://doi.org/10.3390/cancers15184557