
The Interaction of Mechanics and the Hippo Pathway in Drosophila melanogaster


Abstract
:Simple Summary
Abstract
1. Introduction


2. The Biochemical Regulation of the Hippo Pathway
Model Description
- 1.
- Fat and Ds bind together on adjacent membranes, where the cytosolic Fj acts on Fat to promote the binding and on Ds to inhibit it.
- 2.
- Most protein–protein interactions are described by reversible binding and dissociation steps, except for those between Fat and Dachs, Ds and Riq, and Fj and Fat/Ds and the inhibition of Ex on the active Yki level. For instance, the inhibition by Fat of membrane localization of Dachs is modeled by a reduced binding rate and described by a decreasing Hill function of total membrane-bound Fat. The effect of Ds on facilitating the junctional localization of Riq is modeled in a similar way but with an increasing Hill function.
- 3.
- The activation of Wts by Ex is modeled through direct, reversible binding of the two molecules. The level of the Ex-Wts complex is assumed to be proportional to the active Wts that is involved in Yki phosphorylation. The inhibitory effect of Ex on nuclear Yki activity is modeled as a separate inhibitory term in the governing equation for Yki.
- 4.
- As the interactions between Fat and Ex are still poorly defined, we test the case where Fat stabilizes the junctional localization of Ex and compare it with the case where Fat and Ex act in parallel.
- 5.
- As stated above, the feedback loops through the genetic regulations of Yki on Ex, Wts, and Fj were not incorporated in the current model.
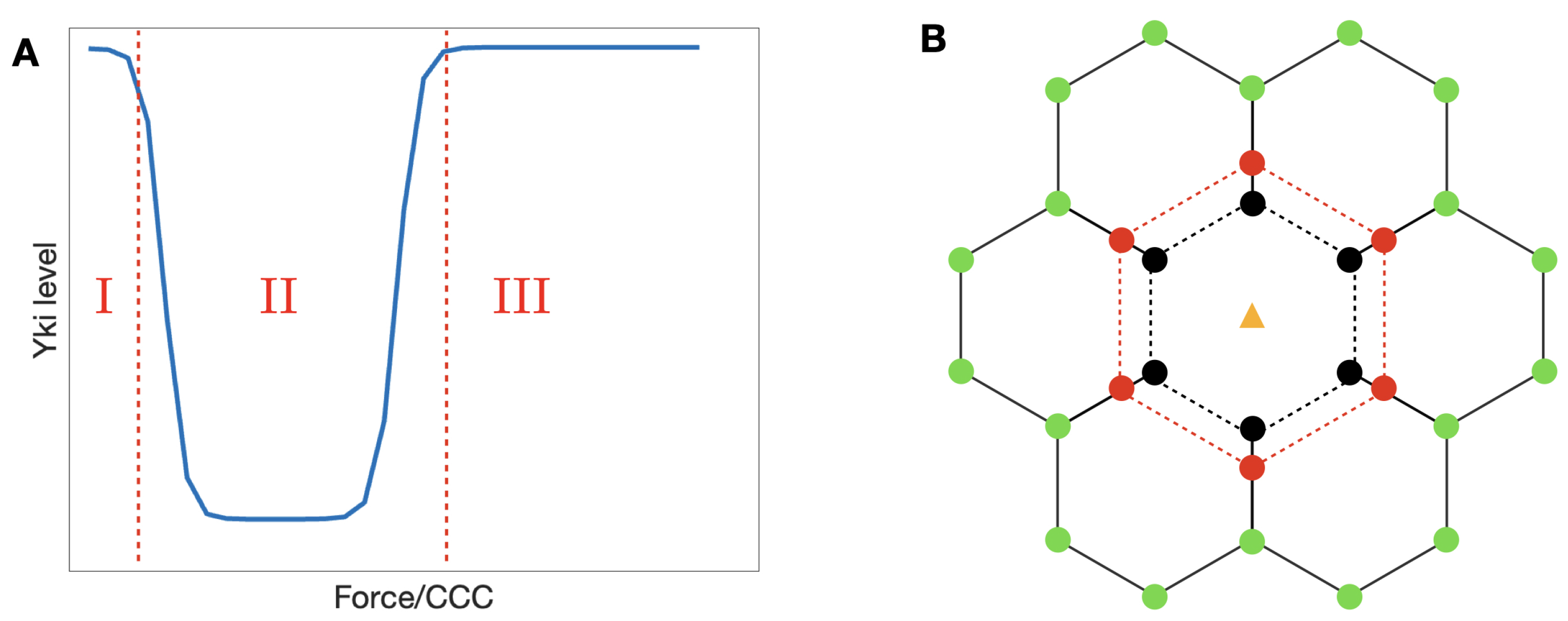
3. Force Transmission within and between Cells
3.1. The Structure of -Catenin
- An N domain which can bind -catenin and participate in the homo-dimerization of non-junctional -catenin;
- An M domain (central modulatory domain) which contains the Vinculin binding site (VBS);
- An ABD domain, namely the C-terminal actin-binding domain.
- M1 limits Jub recruitment, and loss of M1 causes Jub hyper-recruitment to AJs, promoting tissue tension-independent overgrowth. Although M1 binds Vinculin, Vinculin is not responsible for this effect.
- M1 normally limits junctional localization of Jub, while M2 and M3 normally appear to enhance junctional localization of Jub.
- The N domain is essential for Jub recruitment to AJs, but it was also found that the -catenin-mediated recuitment of Jub to AJs does not seem to have a linear relationship with growth regulation in the wing disc epithelium, as normal growth is compatible with normal or low concentrations of Jub at AJs.
3.2. A Three-State Model for Binding and Unfurling of the AJs
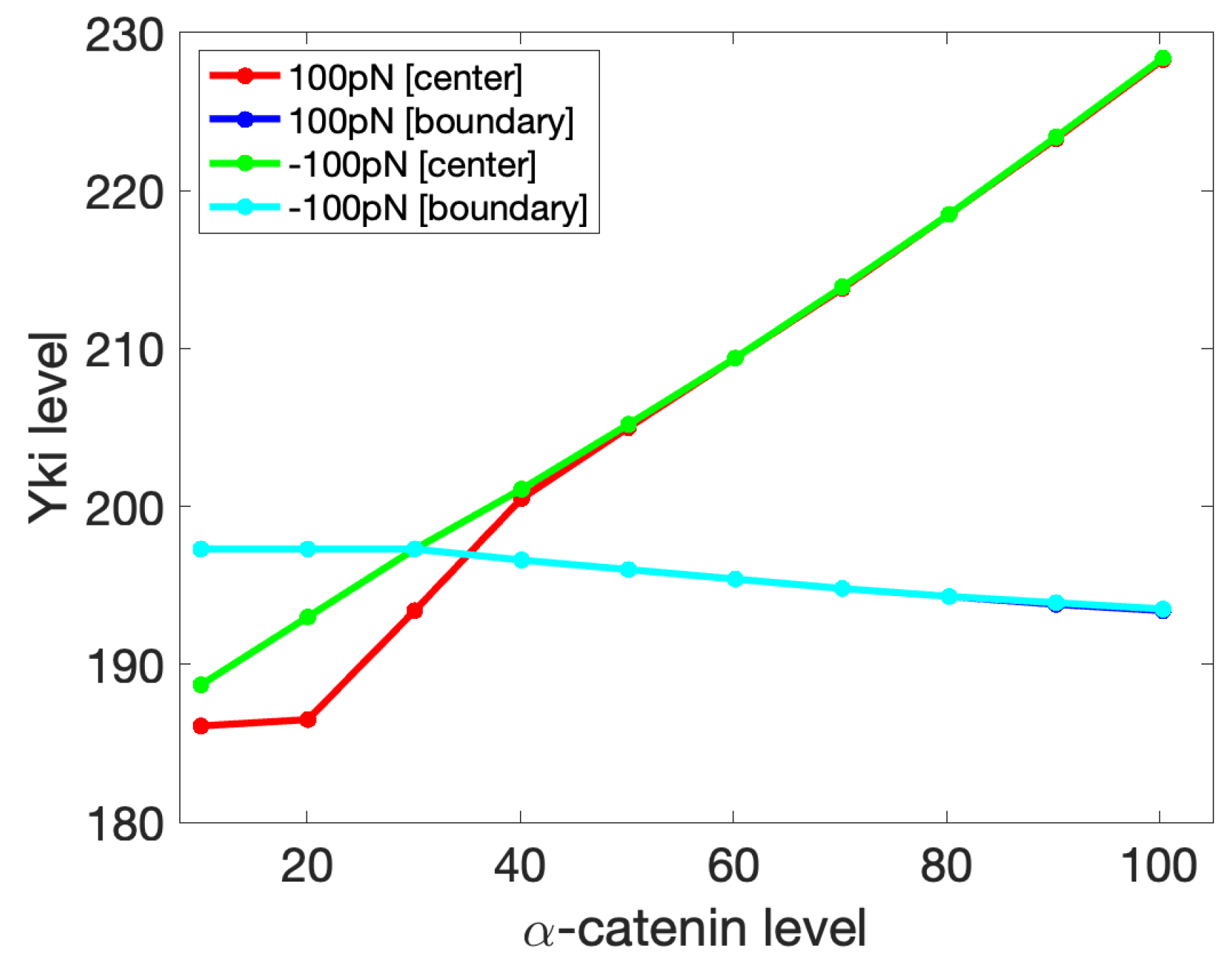
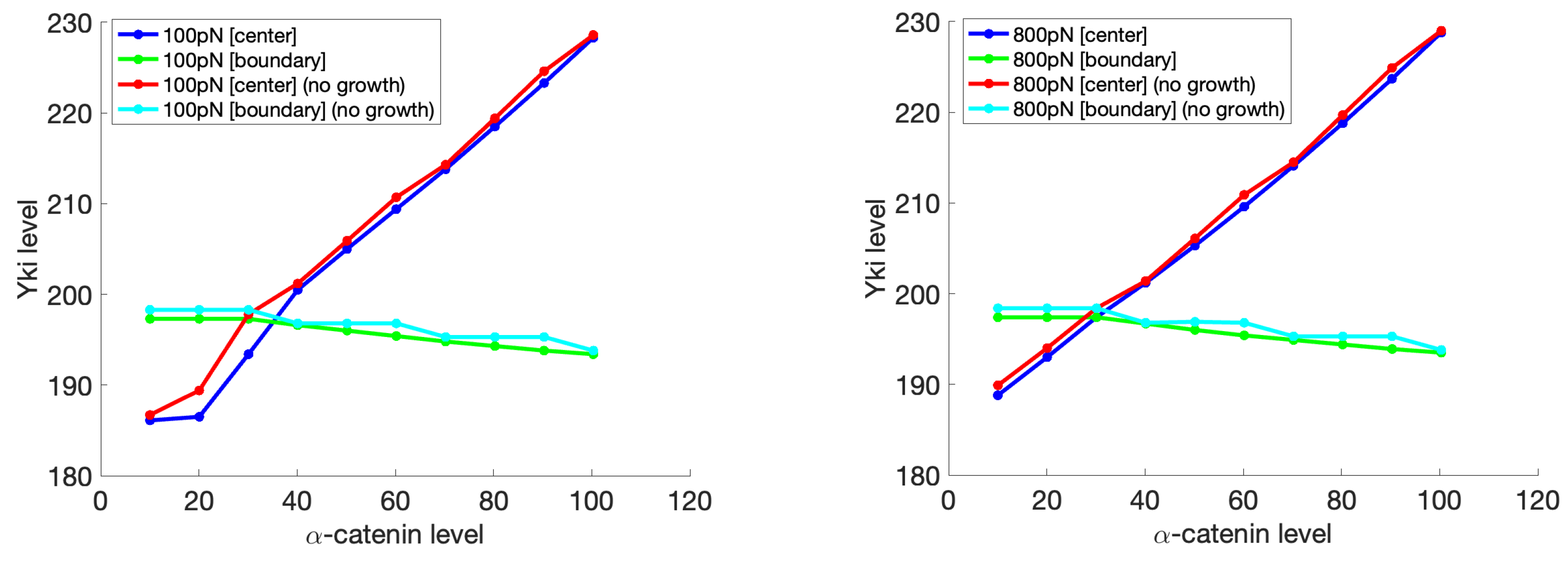
3.3. A Mechanical Model for Tissue-Level Interactions
4. The Interaction of Biochemical Signaling and Mechanics
5. Conclusions
5.1. Overview
5.2. Current Results
5.3. Future Directions
- As stated earlier, in the current model, we ignored the feedback loop in which Yki activity activates the expression of upstream regulators, such as Four-jointed, Ex, Mer, and Kibra [138], and a similar feedback loop exists in that of mammalian cells [139]. On a longer time scale, this feedback may play a significant role in controlling growth.
- A more detailed model of the actomyosin dynamics is needed to more accurately describe the tissue mechanics. While much is known about individual components, a cell-level model that describes the details of the actin belt dynamics and its interaction with the E-cad--catenin system is not at hand. For example, a more detailed model would incorporate spectrin, a contractile protein that forms a membrane-attached skeleton beneath the plasma membrane by crosslinking short F-actin and binding-integral membrane proteins [140]. Spectrin is required for the formation of epithelia, and unlike other regulators such as Crumbs and Merlin, it regulates Hippo signaling by modulating cortical actomyosin activity through non-muscle myosin II [141].
- A more detailed and dynamic model of the mechanics of cell–cell interactions is needed, including the binding of E-cadherin between cells and -catenin binding and production or mobilization, is needed. This is particularly important for understanding relative movement between cells and T1 transitions.
- Because the copy numbers of signaling molecules and other key components are frequently low, stochastic effects should be considered with a view toward understanding if and how the network structure plays a role in adapting to noisy signals. Noise can affect the precision of gene expression in simple networks [142], and key components of the Dpp pathway, for example, are present at nanomolar concentrations [143] in the disc, yet the disc patterning and size are remarkably reproducible. Though it has been argued that even such low concentrations are sufficient to mitigate stochastic noise [144], the network structure may play a role in mitigation. How the functions of signal transduction networks and mechanical regulation are maintained in the presence of fluctuations is still a major question in cellular biology.
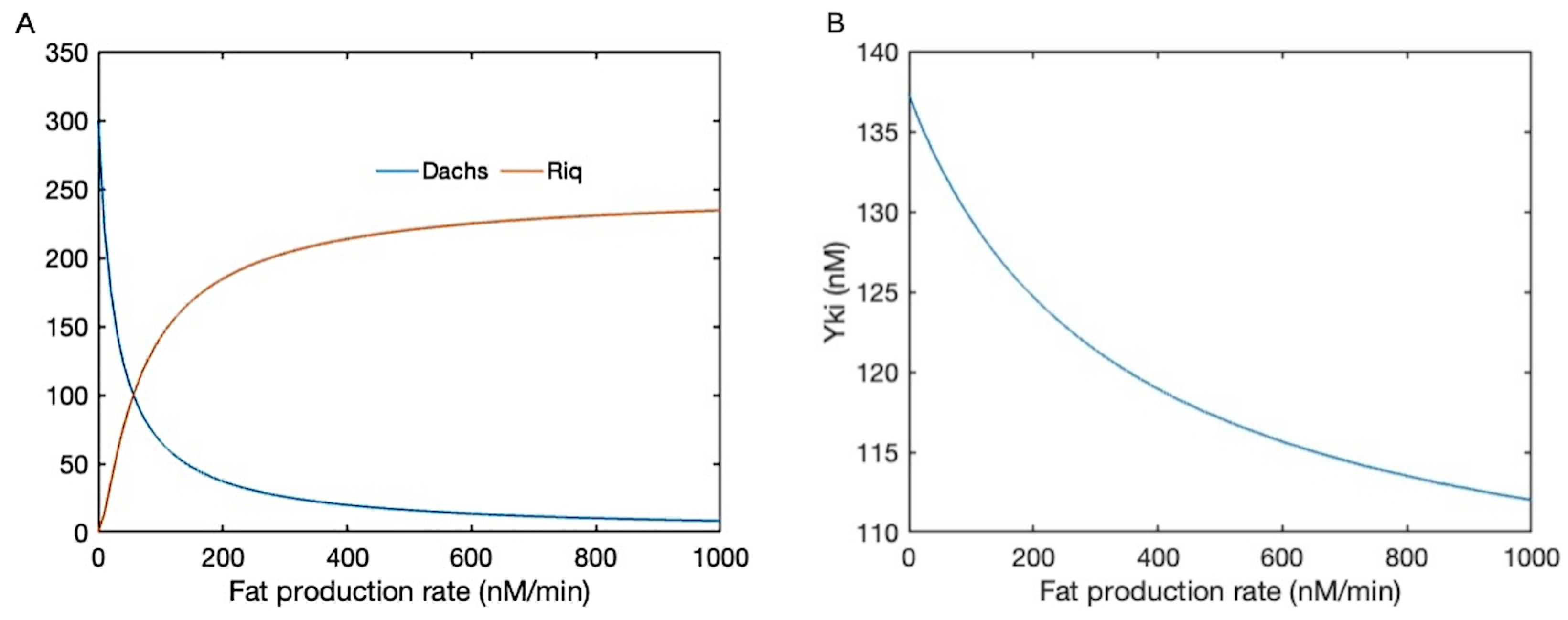
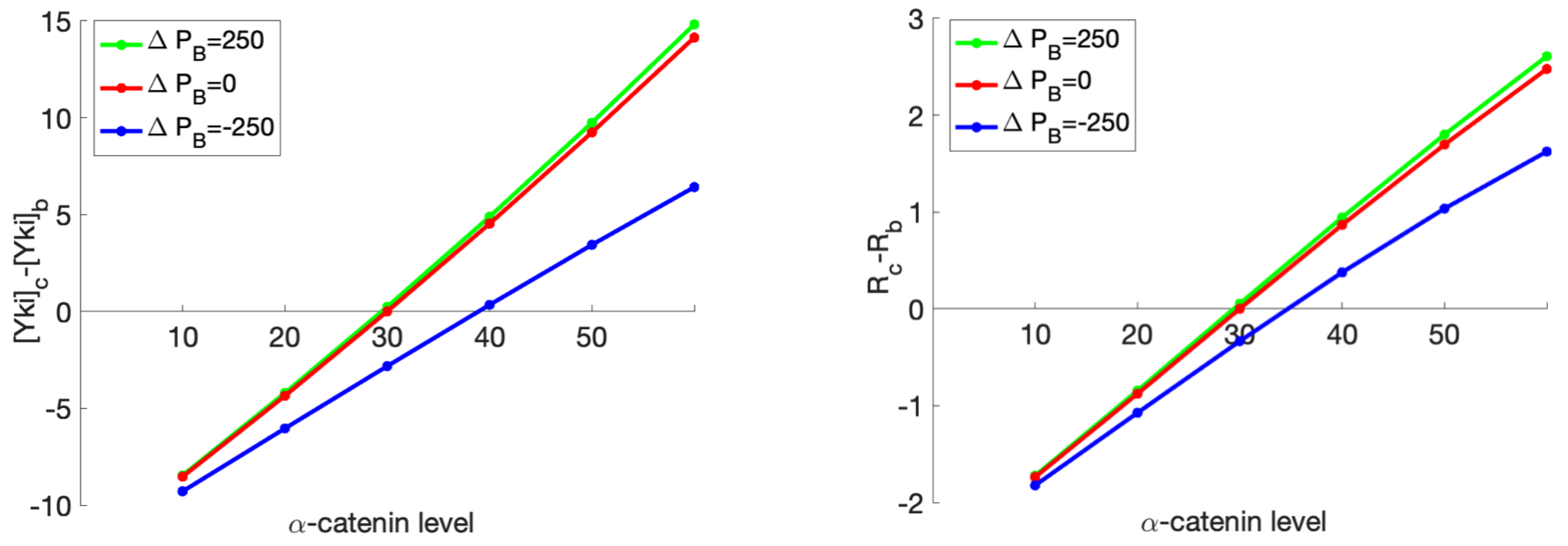
- The current model involves regulation of both the Hippo pathway and the mechanical pathway, and in both components many of the parameters used in the model are unknown or difficult to measure in experiments. Therefore we selected the parameters within biologically-meaningful ranges, but did not do a detailed sensitivity analysis. In the previous version of the biochemical model [9], of which the current biochemical model is a small variation, both local and global sensitivity analysis was performed to analyze the impact of the parameters in the results, and it was found that a small number (6–8) of parameters were very important, and the majority were much less important. However, the Hippo signaling network in both cases has a top-down structure and feedback loops are not considered, which may affect the results. For instance, it was found earlier that the variations of parameters close to the downstream output – the cytosolic Yki concentration – have a greater effect on the variations of the output than do the upstream parameters.A similar sensitivity issue applies to the mechanical component of the model, and when it has been done, the sensitivity analysis for the combined model has to be done. This has the potential to identify the key steps in the combined model and lead to a reduced model that can be used in tissue-level computations, but this is a major project in its own right, in part because how the parameters affect the qualitative responses, for instance, the Yki profile as a function of Fat production as shown in Figure 3, is difficult to analyze and required a method that differs from the traditional global and local sensitivity analysis.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. The Biochemical Model

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Notation |
|---|---|
| Membrane-Bound Species | |
| Fat | |
| Dacsous(Ds) | |
| Fat-Ds complex | |
| Dachs | |
| Warts-Dachs complex | |
| Expanded | |
| E-Cad--cat--cat complex (N, M1, M2+M3) | , , |
| E-Cad--cat--cat-Vinculin complex | , |
| E-Cad--cat--cat-Jub complex | , , |
| E-Cad--cat--cat-Jub-Vinculin complex | , |
| E-Cad--cat--cat-Jub-Warts complex | , , |
| E-Cad--cat--cat-Jub-Vinculin-Warts complex | , |
| Cytosolic Species | |
| Fat | |
| Ds | |
| Dachs | |
| Warts | |
| Yki | |
| Four-jointed | |
| Jub | |
| Vinculin | |
Appendix A.1.1. The Module for Membrane-Bound Species
Fat and Dacsous
Dachs
Riq
Expanded and Expanded Binding with Warts
E-Cad-β-Cat-α-Cat Complex
E-Cad-β-Cat-α-Cat-Vinculin Complex
E-Cad-β-Cat-α-Cat-Jub Complex
E-Cad-β-Cat-α-Cat-Jub-Vinculin Complex
E-Cad-β-Cat-α-Cat-Jub-Warts Complex
E-Cad-β-Cat-α-Cat-Jub-Vinculin-Warts Complex
Appendix A.1.2. The Cytosolic Module
Fat and Dacsous
Dachs
Riq
Warts
Yorki
Four-Jointed
Jub
Vinculin
| Name | Description | Values | Units | Reference |
|---|---|---|---|---|
| Basic parameters | ||||
| Parameters in , , and folding and unfolding module | ||||
| Transition rate of with no load | 3.30 × 10 | - | [87] | |
| Transition distance from to | nm | [87] | ||
| Transition rate of with no load | 7.00 × 10 | [87] | ||
| Transition distance from to | 7.40 | nm | [87] | |
| Transition rate of with no load | 9.40 × 10 | - | [87] | |
| Transition distance from to | nm | [87] | ||
| Transition rate of with no load | 3.00 × 10 | s | [87] | |
| Transition distance from to | 5.60 | nm | [87] | |
| Production rates | ||||
| Fat production rate | 0.4 | M·min | [9] | |
| Dacsous production rate | 0.2 | M·min | [9] | |
| Dachs production rate | 0.25 | M·min | [9] | |
| Riq production rate | 0.1 | M·min | [9] | |
| Yki production rate | 0.5 | M·min | [9] | |
| Jub production rate | 0.25 | M·min | This paper | |
| Vinculin production rate | 0.1 | M·min | This paper | |
| Parameters in Jub binding with CCC complex | ||||
| , , | Binding rate of Jub and -catenin | 10 | M·min | This paper |
| Dissociation rate of Jub and | 4.5 | M·min | This paper | |
| Dissociation rate of Jub and | 500 | M·min | This paper | |
| Dissociation rate of Jub and | 4 | M·min | This paper | |
| Parameters in Vh binding with M1 region | ||||
| Binding rate of Vh and M1 region | (2.88 ± 0.42) × 10 | M min | [145,146] | |
| Dissociation rate of Vh and M1 region | min | [145,146] | ||
| Degradation rates | ||||
| , , , | Ft, Ds, Dachs, Riq degradation rate | 0.1 | min | [9] |
| , | Yki and Jub degradation rate | 0.01 | min | This paper |
| Vinculin degradation rate | 0.05 | min | This paper | |
| Parameters in Fat-Ds binding | ||||
| , | Forward mem localization rate ofcytosolic Ft and Ds | 4 | min | [9] |
| , | Backward mem localization rate ofcytosolic Ft and Ds | 3 | min | [9] |
| Binding rate of Ft-Ds | 4 | M·min | [9] | |
| Unbinding rate of Ft-Ds | 3 | min | [9] | |
| Parameters in function | ||||
| Max localization rate of Dachs to mem | 0.21 | min | This paper | |
| Unbinding rate of Dachs from mem | 0.2 | min | This paper | |
| Max localization rate of Riq to mem | 0.3 | min | This paper | |
| Unbinding rate of Riq from mem | 0.5 | min | This paper | |
| , , , | Scaling parameters | 1, 2, 1, 1 | - | This paper |
| , , , | Scaling parameters | 1, 2, 1, 1 | - | This paper |
| Miscellaneous | ||||
| Binding rates between Dachs and Warts | 0.1 | min | This paper | |
| Release rates from Dachs and Warts | 0.05 | min | This paper | |
| Maximum inhibition rate of Expanded on Yki | 0.1 | min | This paper | |
| Scaling parameter | 0.05 | min | This paper | |
| Binding rate between Expanded and Warts | 0.1 | min | This paper | |
| Unbinding rate between Expanded and Warts | 0.05 | min | This paper | |
| Binding rate between Wts and mem localized Jub | 10 | min | This paper | |
| Unbinding rate between Wts and mem localized Jub | 1 | min | This paper | |
| Maximum inhibition rate of Expanded-Warts on Yki | 1 | min | This paper | |
| Scaling parameter | 0.05 | min | This paper | |
Appendix A.2. The Three States of α-Catenin
Appendix A.3. The Detailed Model of the Tissue Mechanics
| Name | Description | Values | Units | Ref. |
|---|---|---|---|---|
| Parameter | ||||
| Density of the cell | 1 | g/cm | ||
| Area of the cell | m | |||
| Elastic stiffness coefficient of a single stress fiber | N/m | [149] | ||
| Timescale constant () | 2 | s | [150] | |
| Maximum value of polymerization velocity | m/s | [151] | ||
| Stall force of a single polymerizing actin filament | 1 (1–10) | pN | [152,153] | |
Appendix A.3.1. Interaction with the Biochemical Pathway
Force Applied on the α-Catenin and Expanded

Yki-Controlled Cell Growth
References
- Strassburger, K.; Lutz, M.; Müller, S.; Teleman, A.A. Ecdysone regulates Drosophila wing disc size via a TORC1 dependent mechanism. Nat. Commun. 2021, 12, 6684. [Google Scholar] [CrossRef]
- Ye, X.; Deng, Y.; Lai, Z.C. Akt is negatively regulated by Hippo signaling for growth inhibition in Drosophila. Dev. Biol. 2012, 369, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Borreguero-Muñoz, N.; Fletcher, G.C.; Aguilar-Aragon, M.; Elbediwy, A.; Vincent-Mistiaen, Z.I.; Thompson, B.J. The Hippo pathway integrates PI3K–Akt signals with mechanical and polarity cues to control tissue growth. PLoS Biol. 2019, 17, e3000509. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 1–19. [Google Scholar] [CrossRef]
- Paton, E.L.; Turner, J.A.; Schlaepfer, I.R. Overcoming resistance to therapies targeting the MAPK pathway in BRAF-mutated tumours. J. Oncol. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.L.; et al. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell 2017, 171, 1397–1410. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, I.; McCollum, D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J. Biol. Chem. 2019, 294, 17693–17706. [Google Scholar] [CrossRef]
- Kaszak, I.; Witkowska-Piłaszewicz, O.; Niewiadomska, Z.; Dworecka-Kaszak, B.; Ngosa Toka, F.; Jurka, P. Role of cadherins in cancer—A review. Int. J. Mol. Sci. 2020, 21, 7624. [Google Scholar] [CrossRef]
- Gou, J.; Lin, L.; Othmer, H.G. A Model for the Hippo Pathway in the Drosophila Wing Disc. Biophys. J. 2018, 115, 737–747, PMID: 30041810. [Google Scholar] [CrossRef]
- Csibi, A.; Blenis, J. Hippo–YAP and mTOR pathways collaborate to regulate organ size. Nat. Cell Biol. 2012, 14, 1244–1245. [Google Scholar] [CrossRef]
- Mo, J.S.; Park, H.W.; Guan, K.L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Shraiman, B.I. Mechanicall feedback as a possible regulator of tissue growth. Proc. Nat. Acad. Sci. USA 2005, 102, 3318–3323. [Google Scholar] [CrossRef] [PubMed]
- Aegerter-Wilmsen, T.; Aegerter, C.M.; Bisseling, T. Model for the robust establishment of precise proportions in the early Drosophila embryo. J. Theor. Biol. 2005, 234, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Irvine, K.D.; Shraiman, B.I. Mechanical control of growth: Ideas, facts and challenges. Development 2017, 144, 4238–4248. [Google Scholar] [CrossRef]
- Aegerter-Wilmsen, T.; Heimlicher, M.B.; Smith, A.C.; de Reuille, P.B.; Smith, R.S.; Aegerter, C.M.; Basler, K. Integrating force-sensing and signaling pathways in a model for the regulation of wing imaginal disc size. Development 2012, 139, 3221–3231. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. Hippo signaling in organ size control. Genes Dev. 2007, 21, 886–897. [Google Scholar] [CrossRef]
- Ikmi, A.; Gaertner, B.; Seidel, C.; Srivastava, M.; Zeitlinger, J.; Gibson, M.C. Molecular evolution of the Yap/Yorkie proto-oncogene and elucidation of its core transcriptional program. Mol. Biol. Evol. 2014, 31, 1375–1390. [Google Scholar] [CrossRef]
- Willecke, M.; Hamaratoglu, F.; Kango-Singh, M.; Udan, R.; Chen, C.l.; Tao, C.; Zhang, X.; Halder, G. The fat cadherin acts through the hippo tumor-suppressor pathway to regulate tissue size. Curr. Biol. 2006, 16, 2090–2100. [Google Scholar] [CrossRef]
- Andl, T. miRNAs: Miracle or Mirage? The Limes Against the Barbaric Floods of Leaky and Undesired Transcripts. Organogenesis 2007, 3, 25–33. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Matakatsu, H.; Fehon, R.; Blair, S.S. The novel SH3 domain protein Dlish/CG10933 mediates fat signaling in Drosophila by binding and regulating Dachs. Elife 2016, 5, e16624. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Feng, Y.; Rauskolb, C.; Maitra, S.; Fehon, R.; Irvine, K.D. Delineation of a Fat tumor suppressor pathway. Nat. Genet. 2006, 38, 1142. [Google Scholar] [CrossRef]
- Cho, E.; Irvine, K.D. Action of fat, four-jointed, dachsous and dachs in distal-to-proximal wing signaling. Development 2004, 131, 4489–4500. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Rauskolb, C.; Cho, E.; Hu, W.L.; Hayter, H.; Minihan, G.; Katz, F.N.; Irvine, K.D. Dachs: An unconventional myosin that functions downstream of Fat to regulate growth, affinity and gene expression in Drosophila. Development 2006, 133, 2539–2551. [Google Scholar] [CrossRef] [PubMed]
- Degoutin, J.L.; Milton, C.C.; Yu, E.; Tipping, M.; Bosveld, F.; Yang, L.; Bellaiche, Y.; Veraksa, A.; Harvey, K.F. Riquiqui and minibrain are regulators of the hippo pathway downstream of Dachsous. Nat. Cell Biol. 2013, 15, 1176. [Google Scholar] [CrossRef] [PubMed]
- Misra, J.R.; Irvine, K.D. Vamana couples fat signaling to the hippo pathway. Developmental Cell 2016, 39, 254–266. [Google Scholar] [CrossRef]
- Ishikawa, H.O.; Takeuchi, H.; Haltiwanger, R.S.; Irvine, K.D. Four-jointed is a Golgi kinase that phosphorylates a subset of cadherin domains. Science 2008, 321, 401–404. [Google Scholar] [CrossRef]
- Simon, M.A.; Xu, A.; Ishikawa, H.O.; Irvine, K.D. Modulation of fat: Dachsous binding by the cadherin domain kinase four-jointed. Curr. Biol. 2010, 20, 811–817. [Google Scholar] [CrossRef]
- Matakatsu, H.; Blair, S.S. Interactions between Fat and Dachsous and the regulation of planar cell polarity in the Drosophila wing. Development 2004, 131, 3785–3794. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Irvine, K.D. Processing and phosphorylation of the Fat receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 11989–11994. [Google Scholar] [CrossRef]
- Rogulja, D.; Rauskolb, C.; Irvine, K.D. Morphogen control of wing growth through the Fat signaling pathway. Dev. Cell 2008, 15, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Hamaratoglu, F.; Willecke, M.; Kango-Singh, M.; Nolo, R.; Hyun, E.; Tao, C.; Jafar-Nejad, H.; Halder, G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat. Cell Biol. 2006, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zheng, Y.; Dong, J.; Klusza, S.; Deng, W.M.; Pan, D. Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev. Cell 2010, 18, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Reddy, B.; Irvine, K.D. Phosphorylation-independent repression of Yorkie in Fat-Hippo signaling. Dev. Biol. 2009, 335, 188–197. [Google Scholar] [CrossRef]
- Ling, C.; Zheng, Y.; Yin, F.; Yu, J.; Huang, J.; Hong, Y.; Wu, S.; Pan, D. The apical transmembrane protein Crumbs functions as a tumor suppressor that regulates Hippo signaling by binding to Expanded. Proc. Natl. Acad. Sci. USA 2010, 107, 10532–10537. [Google Scholar] [CrossRef]
- Ribeiro, P.; Holder, M.; Frith, D.; Snijders, A.P.; Tapon, N. Crumbs promotes expanded recognition and degradation by the SCFSlimb/β-TrCP ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2014, 111, E1980–E1989. [Google Scholar] [CrossRef]
- Sun, S.; Reddy, B.; Irvine, K.D. Localization of Hippo signalling complexes and Warts activation in vivo. Nat. Commun. 2015, 6, 8402. [Google Scholar] [CrossRef] [PubMed]
- Badouel, C.; Gardano, L.; Amin, N.; Garg, A.; Rosenfeld, R.; Le Bihan, T.; McNeill, H. The FERM-domain protein Expanded regulates Hippo pathway activity via direct interactions with the transcriptional activator Yorkie. Dev. Cell 2009, 16, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Ludwig, M.Z.; Xu, J.; Fehon, R.G. Kibra and merlin activate the hippo pathway spatially distinct from and independent of expanded. Dev. Cell 2017, 40, 478–490. [Google Scholar] [CrossRef]
- Baumgartner, R.; Poernbacher, I.; Buser, N.; Hafen, E.; Stocker, H. The WW domain protein Kibra acts upstream of Hippo in Drosophila. Dev. Cell 2010, 18, 309–316. [Google Scholar] [CrossRef]
- Yin, F.; Yu, J.; Zheng, Y.; Chen, Q.; Zhang, N.; Pan, D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 2013, 154, 1342–1355. [Google Scholar] [CrossRef]
- Bennett, F.C.; Harvey, K.F. Fat cadherin modulates organ size in Drosophila via the Salvador/Warts/Hippo signaling pathway. Curr. Biol. 2006, 16, 2101–2110. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.; Tsatskis, Y.; Gardano, L.; Tapon, N.; McNeill, H. The tumor-suppressor gene fat controls tissue growth upstream of expanded in the hippo signaling pathway. Curr. Biol. 2006, 16, 2081–2089. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Blair, S.S. Fat-regulated adaptor protein Dlish binds the growth suppressor Expanded and controls its stability and ubiquitination. Proc. Natl. Acad. Sci. USA 2019, 116, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Fulford, A.D.; Enderle, L.; Rusch, J.; Hodzic, D.; Holder, M.V.; Earl, A.; Oh, R.H.; Tapon, N.; McNeill, H. Expanded directly binds conserved regions of Fat to restrain growth via the Hippo pathway. J. Cell Biol. 2023, 222, e202204059. [Google Scholar] [CrossRef]
- Feng, Y.; Irvine, K.D. Fat and expanded act in parallel to regulate growth through warts. Proc. Natl. Acad. Sci. USA 2007, 104, 20362–20367. [Google Scholar] [CrossRef]
- Campbell, H.K.; Maiers, J.L.; DeMali, K.A. Interplay between tight junctions & adherens junctions. Exp. Cell Res. 2017, 358, 39–44. [Google Scholar]
- Thomas, C.; Strutt, D. The roles of the cadherins Fat and Dachsous in planar polarity specification in Drosophila. Dev. Dyn. 2012, 241, 27–39. [Google Scholar] [CrossRef]
- Leckband, D.E.; de Rooij, J. Cadherin Adhesion and Mechanotransduction. Annu. Rev. Cell Dev. Biol. 2014, 30, 291–315. [Google Scholar] [CrossRef]
- Pinheiro, D.; Bellaiche, Y. Mechanical force-driven adherens junction remodeling and epithelial dynamics. Dev. Cell 2018, 47, 3–19. [Google Scholar] [CrossRef]
- Tang, V.W.; Brieher, W.M. α-Actinin-4/FSGS1 is required for Arp2/3-dependent actin assembly at the adherens junction. J. Cell Biol. 2012, 196, 115–130. [Google Scholar] [CrossRef]
- Pinheiro, D.; Hannezo, E.; Herszterg, S.; Bosveld, F.; Gaugue, I.; Balakireva, M.; Wang, Z.; Cristo, I.; Rigaud, S.U.; Markova, O.; et al. Transmission of cytokinesis forces via E-cadherin dilution and actomyosin flows. Nature 2017, 545, 103. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Alégot, H.; Rauskolb, C.; Irvine, K.D. The dynamics of hippo signaling during Drosophila wing development. Development 2018, 145, dev165712. [Google Scholar] [CrossRef] [PubMed]
- Major, R.J.; Irvine, K.D. Localization and requirement for Myosin II at the dorsal-ventral compartment boundary of the Drosophila wing. Dev. Dyn. 2006, 235, 3051–3058. [Google Scholar] [CrossRef]
- Aliee, M.; Röper, J.C.; Landsberg, K.P.; Pentzold, C.; Widmann, T.J.; Jülicher, F.; Dahmann, C. Physical mechanisms shaping the Drosophila dorsoventral compartment boundary. Curr. Biol. 2012, 22, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Irvine, K.D. Cellular organization and cytoskeletal regulation of the Hippo signaling network. Trends Cell Biol. 2016, 26, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, T.; Yap, A.S. E-cadherin junctions as active mechanical integrators in tissue dynamics. Nat. Cell Biol. 2015, 17, 533. [Google Scholar] [CrossRef]
- Hong, S.; Troyanovsky, R.B.; Troyanovsky, S.M. Cadherin exits the junction by switching its adhesive bond. J. Cell Biol. 2011, 192, 1073–1083. [Google Scholar] [CrossRef]
- Angulo-Urarte, A.; van der Wal, T.; Huveneers, S. Cell-cell junctions as sensors and transducers of mechanical forces. Biochim. Biophys. Acta-Bba-Biomembr. 2020, 1862, 183316. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.M.; Stow, J.L. The ins and outs of E-cadherin trafficking. Trends in Cell Biology 2004, 14, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Wirtz-Peitz, F.; Zallen, J.A. Junctional trafficking and epithelial morphogenesis. Curr. Opin. Genet. Dev. 2009, 19, 350–356. [Google Scholar] [CrossRef]
- Röper, J.C.; Mitrossilis, D.; Stirnemann, G.; Waharte, F.; Brito, I.; Fernandez-Sanchez, M.E.; Baaden, M.; Salamero, J.; Farge, E. The major β-catenin/E-cadherin junctional binding site is a primary molecular mechano-transductor of differentiation in vivo. Elife 2018, 7, e33381. [Google Scholar] [CrossRef] [PubMed]
- Efimova, N.; Svitkina, T.M. Branched actin networks push against each other at adherens junctions to maintain cell–cell adhesion. J. Cell Biol. 2018, 217, 1827–1845. [Google Scholar] [CrossRef]
- Röper, K. Integration of cell–cell adhesion and contractile actomyosin activity during morphogenesis. Curr. Top. Dev. Biol. 2015, 112, 103–127. [Google Scholar]
- Bosveld, F.; Wang, Z.; Bellaïche, Y. Tricellular junctions: A hot corner of epithelial biology. Curr. Opin. Cell Biol. 2018, 54, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Bosveld, F.; Markova, O.; Guirao, B.; Martin, C.; Wang, Z.; Pierre, A.; Balakireva, M.; Gaugue, I.; Ainslie, A.; Christophorou, N.; et al. Epithelial tricellular junctions act as interphase cell shape sensors to orient mitosis. Nature 2016, 530, 495. [Google Scholar] [CrossRef]
- Zhou, Z.; Alégot, H.; Irvine, K.D. Oriented Cell Divisions Are Not Required for Drosophila Wing Shape. Curr. Biol. 2019, 29, 856–864. [Google Scholar] [CrossRef]
- Hirata, H.; Tatsumi, H.; Sokabe, M. Zyxin emerges as a key player in the mechanotransduction at cell adhesive structures. Commun. Integr. Biol. 2008, 1, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; Van Der Krogt, G.; Twiss, F.; Bongaarts, A.; Habani, Y.; Slotman, J.A.; Houtsmuller, A.; Huveneers, S.; De Rooij, J. VASP, zyxin and TES are tension-dependent members of Focal Adherens Junctions independent of the α-catenin-vinculin module. Sci. Rep. 2015, 5, 17225. [Google Scholar] [CrossRef]
- Sala, S.; Van Troys, M.; Medves, S.; Catillon, M.; Timmerman, E.; Staes, A.; Schaffner-Reckinger, E.; Gevaert, K.; Ampe, C. Expanding the Interactome of TES by Exploiting TES Modules with Different Subcellular Localizations. J. Proteome Res. 2017, 16, 2054–2071. [Google Scholar] [CrossRef]
- Kale, G.R.; Yang, X.; Philippe, J.M.; Mani, M.; Lenne, P.F.; Lecuit, T. Distinct contributions of tensile and shear stress on E-cadherin levels during morphogenesis. Nat. Commun. 2018, 9, 5021. [Google Scholar] [CrossRef] [PubMed]
- Baum, B.; Georgiou, M. Dynamics of adherens junctions in epithelial establishment, maintenance, and remodeling. J. Cell Biol. 2011, 192, 907–917. [Google Scholar] [CrossRef]
- Fletcher, G.C.; Borreguero-Muñoz, N.; Holder, M.; Aguilar-Aragon, M.; Thompson, B.J. Mechanical strain regulates the Hippo pathway in Drosophila. Development 2018, 145, dev–159467. [Google Scholar] [CrossRef]
- Huveneers, S.; de Rooij, J. Mechanosensitive systems at the cadherin–F-actin interface. J. Cell Sci. 2013, 126, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Sarpal, R.; Yan, V.; Kazakova, L.; Sheppard, L.; Yu, J.C.; Fernandez-Gonzalez, R.; Tepass, U. Role of α-Catenin and its mechanosensing properties in regulating Hippo/YAP-dependent tissue growth. PLOS Genet. 2019, 15, e1008454. [Google Scholar] [CrossRef] [PubMed]
- Alégot, H.; Markosian, C.; Rauskolb, C.; Yang, J.; Kirichenko, E.; Wang, Y.C.; Irvine, K.D. Recruitment of Jub by α-catenin promotes Yki activity and Drosophila wing growth. J. Cell Sci. 2019, 132, jcs222018. [Google Scholar] [CrossRef]
- Rauskolb, C.; Sun, S.; Sun, G.; Pan, Y.; Irvine, K.D. Cytoskeletal Tension Inhibits Hippo Signaling through an Ajuba-Warts Complex. Cell 2014, 158, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Duszyc, K.; Viasnoff, V.; Yap, A.S. Mechanosensing and mechanotransduction at cell–cell junctions. CSH Perspect. Biol. 2018, 10, a028761. [Google Scholar]
- Fletcher, G.C.; Elbediwy, A.; Khanal, I.; Ribeiro, P.S.; Tapon, N.; Thompson, B.J. The Spectrin cytoskeleton regulates the Hippo signalling pathway. EMBO J. 2015, 34, 940–954. [Google Scholar] [CrossRef]
- Nola, S.; Daigaku, R.; Smolarczyk, K.; Carstens, M.; Martin-Martin, B.; Longmore, G.; Bailly, M.; Braga, V.M. Ajuba is required for Rac activation and maintenance of E-cadherin adhesion. J. Cell Biol. 2011, 195, 855–871. [Google Scholar] [CrossRef]
- Marie, H.; Pratt, S.J.; Betson, M.; Epple, H.; Kittler, J.T.; Meek, L.; Moss, S.J.; Troyanovsky, S.; Attwell, D.; Longmore, G.D.; et al. The LIM Protein Ajuba Is Recruited to Cadherin-dependent Cell Junctions through an Association with α-Catenin*. J. Biol. Chem. 2003, 278, 1220–1228. [Google Scholar] [CrossRef]
- Rauskolb, C.; Cervantes, E.; Madere, F.; Irvine, K.D. Organization and function of tension-dependent complexes at adherens junctions. J. Cell Sci. 2019, 132, jcs224063. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Vanderzalm, P.J.; Ludwig, M.; Su, T.; Tokamov, S.A.; Fehon, R.G. Yorkie Functions at the Cell Cortex to Promote Myosin Activation in a Non-transcriptional Manner. Dev. Cell 2018, 46, 271–284.e5. [Google Scholar] [CrossRef]
- Bax, N.A.; Wang, A.; Huang, D.L.; Pokutta, S.; Weis, W.I.; Dunn, A.R. Multi-level Force-dependent Allosteric Enhancement of αE-catenin Binding to F-actin by Vinculin. J. Mol. Biol. 2023, 435, 167969. [Google Scholar] [CrossRef]
- Bell, G.I. Models for the specific adhesion of cells to cells. Science 1978, 200, 618–627. [Google Scholar] [CrossRef]
- Jagannathan, B.; Marqusee, S. Protein folding and unfolding under force. Biopolymers 2013, 99, 860–869. [Google Scholar] [CrossRef]
- Strunz, T.; Oroszlan, K.; Schäfer, R.; Güntherodt, H.J. Dynamic force spectroscopy of single DNA molecules. Proc. Natl. Acad. Sci. USA 1999, 96, 11277–11282. [Google Scholar] [CrossRef]
- Yao, M.; Qiu, W.; Liu, R.; Efremov, A.K.; Cong, P.; Seddiki, R.; Payre, M.; Lim, C.T.; Ladoux, B.; Mège, R.M.; et al. Force-dependent conformational switch of α-catenin controls vinculin binding. Nat. Commun. 2014, 5, 4525. [Google Scholar] [CrossRef]
- Sheppard, L.; Green, D.G.; Lerchbaumer, G.; Rothenberg, K.E.; Fernandez-Gonzalez, R.; Tepass, U. The α-Catenin mechanosensing M region is required for cell adhesion during tissue morphogenesis. J. Cell Biol. 2022, 222, e202108091. [Google Scholar] [CrossRef] [PubMed]
- Rauskolb, C.; Irvine, K.D. Localization of Hippo signaling components in drosophila by fluorescence and immunofluorescence. In The Hippo Pathway; Springer: Berlin/Heidelberg, Germany, 2019; pp. 61–73. [Google Scholar]
- Razzell, W.; Bustillo, M.E.; Zallen, J.A. The force-sensitive protein Ajuba regulates cell adhesion during epithelial morphogenesis. J. Cell Biol. 2018, 217, 3715–3730. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Matzavinos, A.; Othmer, H.G. A theoretical approach to actin filament dynamics. J. Stat. Phys. 2007, 128, 111–138. [Google Scholar] [CrossRef]
- Matzavinos, A.; Othmer, H.G. A stochastic analysis of actin polymerization in the presence of twinfilin and gelsolin. J. Theor. Biol. 2007, 249, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Blanchoin, L.; Boujemaa-Paterski, R.; Sykes, C.; Plastino, J. Actin dynamics, architecture, and mechanics in cell motility. Physiol. Rev. 2014, 94, 235–263. [Google Scholar] [CrossRef]
- Iyer, K.V.; Piscitello-Gómez, R.; Paijmans, J.; Jülicher, F.; Eaton, S. Epithelial viscoelasticity is regulated by mechanosensitive E-cadherin turnover. Curr. Biol. 2019, 29, 578–591. [Google Scholar] [CrossRef]
- Villa, C.; Chaplain, M.A.; Gerisch, A.; Lorenzi, T. Mechanical models of pattern and form in biological tissues: The role of stress–strain constitutive equations. Bull. Math. Biol. 2021, 83, 80. [Google Scholar] [CrossRef]
- Kollmannsberger, P.; Fabry, B. Linear and nonlinear rheology of living cells. Annu. Rev. Mater. Res. 2011, 41, 75–97. [Google Scholar] [CrossRef]
- Salbreux, G.; Barthel, L.K.; Raymond, P.A.; Lubensky, D.K. Coupling Mechanical Deformations and Planar Cell Polarity to Create Regular Patterns in the Zebrafish Retina. PLoS Comput. Biol. 2012, 8, e1002618. [Google Scholar] [CrossRef]
- Charras, G.; Yap, A.S. Tensile forces and mechanotransduction at cell–cell junctions. Curr. Biol. 2018, 28, R445–R457. [Google Scholar] [CrossRef]
- Fischer, S.C.; Blanchard, G.B.; Duque, J.; Adams, R.J.; Arias, A.M.; Guest, S.D.; Gorfinkiel, N. Contractile and Mechanical Properties of Epithelia with Perturbed Actomyosin Dynamics. PloS ONE 2014, 9, e95695. [Google Scholar] [CrossRef] [PubMed]
- Farhadifar, R.; Röper, J.C.; Aigouy, B.; Eaton, S.; Jülicher, F. The influence of cell mechanics, cell-cell interactions, and proliferation on epithelial packing. Curr. Biol. 2007, 17, 2095–2104. [Google Scholar] [CrossRef]
- Hannezo, E.; Prost, J.; Joanny, J.F. Theory of epithelial sheet morphology in three dimensions. Proc. Nat. Acad. Sci. USA 2014, 111, 27–32. [Google Scholar] [CrossRef]
- Fletcher, A.G.; Osborne, J.M.; Maini, P.K.; Gavaghan, D.J. Implementing vertex dynamics models of cell populations in biology within a consistent computational framework. Prog. Biophys. Mol. Biol. 2013, 113, 299–326. [Google Scholar] [CrossRef] [PubMed]
- Rauzi, M.; Verant, P.; Lecuit, T.; Lenne, P.F. Nature and anisotropy of cortical forces orienting Drosophila tissue morphogenesis. Nat. Cell Biol. 2008, 10, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Käfer, J.; Hayashi, T.; Marée, A.F.; Carthew, R.W.; Graner, F. Cell adhesion and cortex contractility determine cell patterning in the Drosophila retina. Proc. Nat. Acad. Sci. USA 2007, 104, 18549–18554. [Google Scholar] [CrossRef]
- Ma, X.; Lynch, H.E.; Scully, P.C.; Hutson, M.S. Probing embryonic tissue mechanics with laser hole drilling. Phys. Biol. 2009, 6, 036004. [Google Scholar] [CrossRef]
- Mao, Y.; Tournier, A.L.; Hoppe, A.; Kester, L.; Thompson, B.J.; Tapon, N. Differential proliferation rates generate patterns of mechanical tension that orient tissue growth. EMBO J. 2013, 32, 2790–2803. [Google Scholar] [CrossRef] [PubMed]
- Yonemura, S. Cadherin–actin interactions at adherens junctions. Curr. Opin. Cell Biol. 2011, 23, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Borghi, N.; Sorokina, M.; Shcherbakova, O.G.; Weis, W.I.; Pruitt, B.L.; Nelson, W.J.; Dunn, A.R. E-cadherin is under constitutive actomyosin-generated tension that is increased at cell–cell contacts upon externally applied stretch. Proc. Natl. Acad. Sci. USA 2012, 109, 12568–12573. [Google Scholar] [CrossRef]
- Li, X.; Zhai, W.; Guo, Z.; Chen, H. Force-dependent unfolding and folding dynamics of protein alpha-catenin modulation domains. J. Innov. Opt. Health Sci. 2018, 12, 1841001. [Google Scholar] [CrossRef]
- Clarke, D.N.; Martin, A.C. Actin-based force generation and cell adhesion in tissue morphogenesis. Curr. Biol. 2021, 31, R667–R680. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Dunn, A.R.; Weis, W.I.; Hill, C.P.; Pfeffer, S.R.; Yan, J. Mechanism of the cadherin–catenin F-actin catch bond interaction. eLife 2022, 11, e80130. [Google Scholar] [CrossRef]
- Silvestre, J.; Kenis, P.J.A.; Leckband, D.E. Cadherin and Integrin Regulation of Epithelial Cell Migration. Langmuir 2009, 25, 10092–10099. [Google Scholar] [CrossRef] [PubMed]
- Truong Quang, B.A.; Mani, M.; Markova, O.; Lecuit, T.; Lenne, P.F. Principles of E-Cadherin Supramolecular Organization In Vivo. Curr. Biol. 2013, 23, 2197–2207. [Google Scholar] [CrossRef]
- Yang, C.C.; Graves, H.K.; Moya, I.M.; Tao, C.; Hamaratoglu, F.; Gladden, A.B.; Halder, G. Differential regulation of the Hippo pathway by adherens junctions and apical–basal cell polarity modules. Proc. Natl. Acad. Sci. USA 2015, 112, 1785–1790. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Koontz, L.M.; Liu-Chittenden, Y.; Yin, F.; Zheng, Y.; Yu, J.; Huang, B.; Chen, Q.; Wu, S.; Pan, D. The Hippo Effector Yorkie Controls Normal Tissue Growth by Antagonizing Scalloped-Mediated Default Repression. Dev. Cell 2013, 25, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Irvine, K.D.; Harvey, K.F. Control of Organ Growth by Patterning and Hippo Signaling in Drosophila. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Eder, D.; Aegerter, C.; Basler, K. Forces controlling organ growth and size. Mech. Dev. 2017, 144, 53–61. [Google Scholar] [CrossRef]
- Sknepnek, R.; Djafer-Cherif, I.; Chuai, M.; Weijer, C.; Henkes, S.; Kruse, K.; Walczak, A.M. Generating active T1 transitions through mechanochemical feedback. eLife 2023, 12, e79862. [Google Scholar] [CrossRef]
- Pan, Y.; Heemskerk, I.; Ibar, C.; Shraiman, B.I.; Irvine, K.D. Differential growth triggers mechanical feedback that elevates Hippo signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E6974–E6983. [Google Scholar] [CrossRef]
- Herranz, H.; Weng, R.; Cohen, S.M. Crosstalk between epithelial and mesenchymal tissues in tumorigenesis and imaginal disc development. Curr. Biol. 2014, 24, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Milán, M. Tumor Models: Tumor–Stroma Interactions Drive Neoplastic Transformation in Drosophila. Curr. Biol. 2014, 24, R658–R659. [Google Scholar] [CrossRef] [PubMed]
- Buchmann, A.; Alber, M.; Zartman, J.J. Sizing it up: The mechanical feedback hypothesis of organ growth regulation. Semin. Cell Dev. Biol. 2014, 35, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.; Struhl, G. Scaling the Drosophila wing: TOR-dependent target gene access by the Hippo pathway transducer Yorkie. PLoS Biol. 2015, 13, e1002274. [Google Scholar] [CrossRef]
- Gou, J.; Stotsky, J.A.; Othmer, H.G. Growth control in the Drosophila wing disk. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1478. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, J.; Casares, F.; Iber, D. Growth and size control during development. Open Biol. 2017, 7, 170190. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.L. The Hippo pathway: Biology and pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef]
- van Soldt, B.J.; Cardoso, W.V. Hippo-Yap/Taz signaling: Complex network interactions and impact in epithelial cell behavior. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9, e371. [Google Scholar] [CrossRef]
- Fu, M.; Hu, Y.; Lan, T.; Guan, K.L.; Luo, T.; Luo, M. The Hippo signalling pathway and its implications in human health and diseases. Signal Transduct. Target. Ther. 2022, 7, 376. [Google Scholar] [CrossRef] [PubMed]
- Morishita, K.; Watanabe, K.; Ichijo, H. Cell volume regulation in cancer cell migration driven by osmotic water flow. Cancer Sci. 2019, 110, 2337–2347. [Google Scholar] [CrossRef]
- Catacuzzeno, L.; Sforna, L.; Esposito, V.; Limatola, C.; Franciolini, F. Ion channels in glioma malignancy. Transp. Malfunct. Cancer Spectr. 2020; 181, 223–267. [Google Scholar]
- Stock, C.; Schwab, A. Ion channels and transporters in metastasis. Biochim. Biophys. Acta-Bba-Biomembr. 2015, 1848, 2638–2646. [Google Scholar] [CrossRef]
- So, J.S.; Kim, H.; Han, K.S. Mechanisms of Invasion in Glioblastoma: Extracellular Matrix, Ca2+ Signaling, and Glutamate. Front. Cell. Neurosci. 2021, 15, 663092. [Google Scholar] [CrossRef]
- Stroka, K.M.; Jiang, H.; Chen, S.H.; Tong, Z.; Wirtz, D.; Sun, S.X.; Konstantopoulos, K. Water permeation drives tumor cell migration in confined microenvironments. Cell 2014, 157, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Saadoun, S. Key roles of aquaporins in tumor biology. Biochim. Biophys. Acta-BBA-Biomembr. 2015, 1848, 2576–2583. [Google Scholar] [CrossRef]
- Maki, K.; Han, S.W.; Hirano, Y.; Yonemura, S.; Hakoshima, T.; Adachi, T. Mechano-adaptive sensory mechanism of α-catenin under tension. Sci. Rep. 2016, 6, 24878. [Google Scholar] [CrossRef] [PubMed]
- Jukam, D.; Xie, B.; Rister, J.; Terrell, D.; Charlton-Perkins, M.; Pistillo, D.; Gebelein, B.; Desplan, C.; Cook, T. Opposite feedbacks in the Hippo pathway for growth control and neural fate. Science 2013, 342, 1238016. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Park, H.W.; Qin, B.; Chen, Q.; Meng, Z.; Plouffe, S.W.; Taniguchi, K.; Yu, F.X.; Karin, M.; Pan, D.; et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 2015, 29, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Baines, A.J. Evolution of spectrin function in cytoskeletal and membrane networks. Biochem. Soc. Trans. 2009, 37, 796–803. [Google Scholar] [CrossRef]
- Deng, H.; Wang, W.; Yu, J.; Zheng, Y.; Qing, Y.; Pan, D. Spectrin regulates Hippo signaling by modulating cortical actomyosin activity. Elife 2015, 4, e06567. [Google Scholar] [CrossRef]
- England, J.L.; Cardy, J. Morphogen gradient from a noisy source. Phys. Rev. Lett. 2005, 94, 078101. [Google Scholar] [CrossRef]
- Shimmi, O.; O’Connor, M.B. Physical properties of Tld, Sog, Tsg and Dpp protein interactions are predicted to help create a sharp boundary in Bmp signals during dorsoventral patterning of the Drosophila embryo. Development 2003, 130, 4673–4682. [Google Scholar] [CrossRef] [PubMed]
- Kicheva, A.; Pantazis, P.; Bollenbach, T.; Kalaidzidis, Y.; Bittig, T.; Jülicher, F.; Gonzalez-Gaitan, M. Kinetics of morphogen gradient formation. Science 2007, 315, 521–525. [Google Scholar] [CrossRef]
- Pang, S.M.; Le, S.; Kwiatkowski, A.V.; Yan, J. Mechanical stability of αT-catenin and its activation by force for vinculin binding. Mol. Biol. Cell 2019, 30, 1930–1937. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Pokutta, S.; Cadwell, G.W.; Bobkov, A.A.; Bankston, L.A.; Liddington, R.C.; Weis, W.I. αE-catenin is an autoinhibited molecule that coactivates vinculin. Proc. Natl. Acad. Sci. USA 2012, 109, 8576–8581. [Google Scholar] [CrossRef]
- Mayer, M.; Depken, M.; Bois, J.S.; Jülicher, F.; Grill, S.W. Anisotropies in cortical tension reveal the physical basis of polarizing cortical flows. Nature 2010, 467, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Kassianidou, E.; Brand, C.A.; Schwarz, U.S.; Kumar, S. Geometry and network connectivity govern the mechanics of stress fibers. Proc. Natl. Acad. Sci. USA 2017, 114, 2622–2627. [Google Scholar] [CrossRef]
- De La Cruz, E.M.; Gardel, M.L. Actin Mechanics and Fragmentation*. J. Biol. Chem. 2015, 290, 17137–17144. [Google Scholar] [CrossRef]
- Hwang, Y.; Barakat, A.I. Dynamics of Mechanical Signal Transmission through Prestressed Stress Fibers. PLoS ONE 2012, 7, e35343. [Google Scholar] [CrossRef]
- Zimmermann, J.; Brunner, C.; Enculescu, M.; Goegler, M.; Ehrlicher, A.; Käs, J.; Falcke, M. Actin Filament Elasticity and Retrograde Flow Shape the Force-Velocity Relation of Motile Cells. Biophys. J. 2012, 102, 287–295. [Google Scholar] [CrossRef]
- Footer, M.J.; Kerssemakers, J.W.J.; Theriot, J.A.; Dogterom, M. Direct measurement of force generation by actin filament polymerization using an optical trap. Proc. Natl. Acad. Sci. USA 2007, 104, 2181–2186. [Google Scholar] [CrossRef]
- Jasnin, M.; Hervy, J.; Balor, S.; Bouissou, A.; Proag, A.; Voituriez, R.; Schneider, J.; Mangeat, T.; Maridonneau-Parini, I.; Baumeister, W.; et al. Elasticity of podosome actin networks produces nanonewton protrusive forces. Nat. Commun. 2022, 13, 3842. [Google Scholar] [CrossRef] [PubMed]

















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gou, J.; Zhang, T.; Othmer, H.G. The Interaction of Mechanics and the Hippo Pathway in Drosophila melanogaster. Cancers 2023, 15, 4840. https://doi.org/10.3390/cancers15194840
Gou J, Zhang T, Othmer HG. The Interaction of Mechanics and the Hippo Pathway in Drosophila melanogaster. Cancers. 2023; 15(19):4840. https://doi.org/10.3390/cancers15194840
Chicago/Turabian StyleGou, Jia, Tianhao Zhang, and Hans G. Othmer. 2023. "The Interaction of Mechanics and the Hippo Pathway in Drosophila melanogaster" Cancers 15, no. 19: 4840. https://doi.org/10.3390/cancers15194840
APA StyleGou, J., Zhang, T., & Othmer, H. G. (2023). The Interaction of Mechanics and the Hippo Pathway in Drosophila melanogaster. Cancers, 15(19), 4840. https://doi.org/10.3390/cancers15194840