
The Combined Inhibition of Autophagy and Diacylglycerol Acyltransferase-Mediated Lipid Droplet Biogenesis Induces Cancer Cell Death during Acute Amino Acid Starvation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Treatments
2.3. Gene Silencing Using Small Interfering RNA (siRNA)
2.4. Flow Cytometry Analysis of Cellular LD Content
2.5. Cyto-ID Autophagy Detection Flow Cytometry Assay
2.6. LC3 Immunostaining
2.7. Live Cell Imaging of LDs
2.8. Live Cell Imaging of Autophagosomes Stained with Cyto-ID
2.9. Pulse-Chase Imaging Experiments for Detection of LD Formation
2.10. LD Counting and Diameter Analysis
2.11. Western Blot Analysis
2.12. TMRM/YO-PRO-1 Cell Death Assay
2.13. Statistical Analyses
3. Results
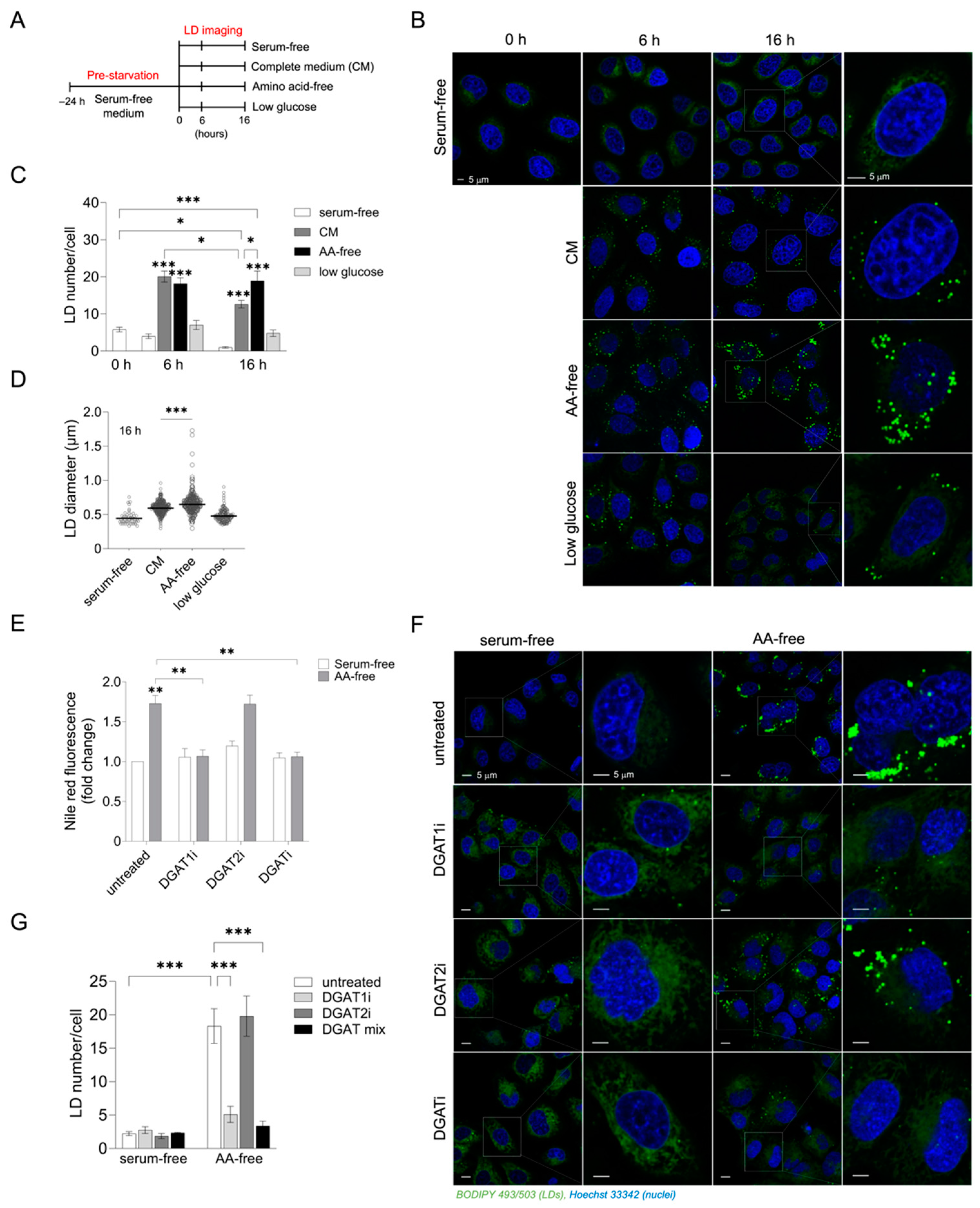
3.1. Acute Amino Acid Depletion Increases DGAT1-Dependent LD Accumulation in HeLa Cells
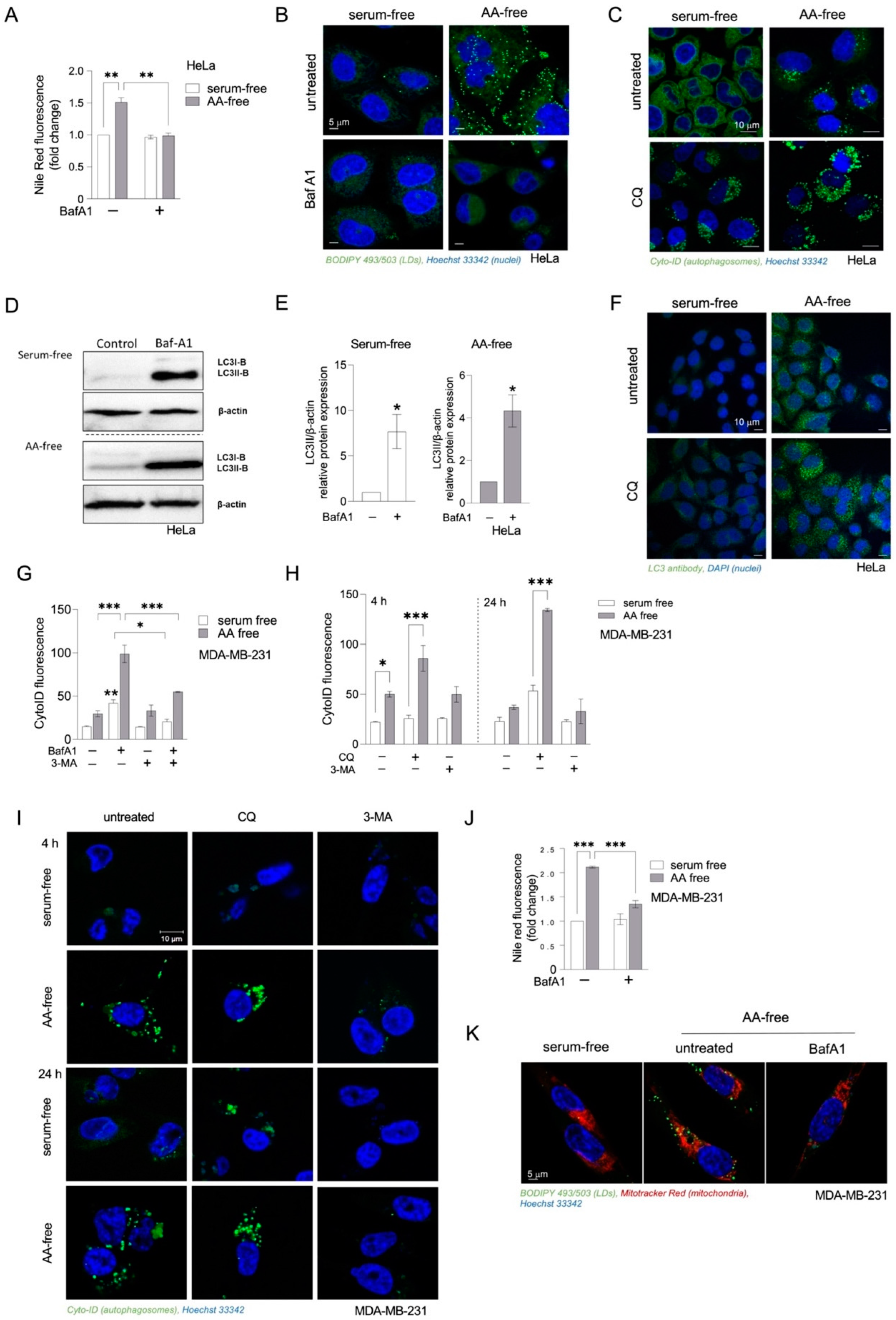
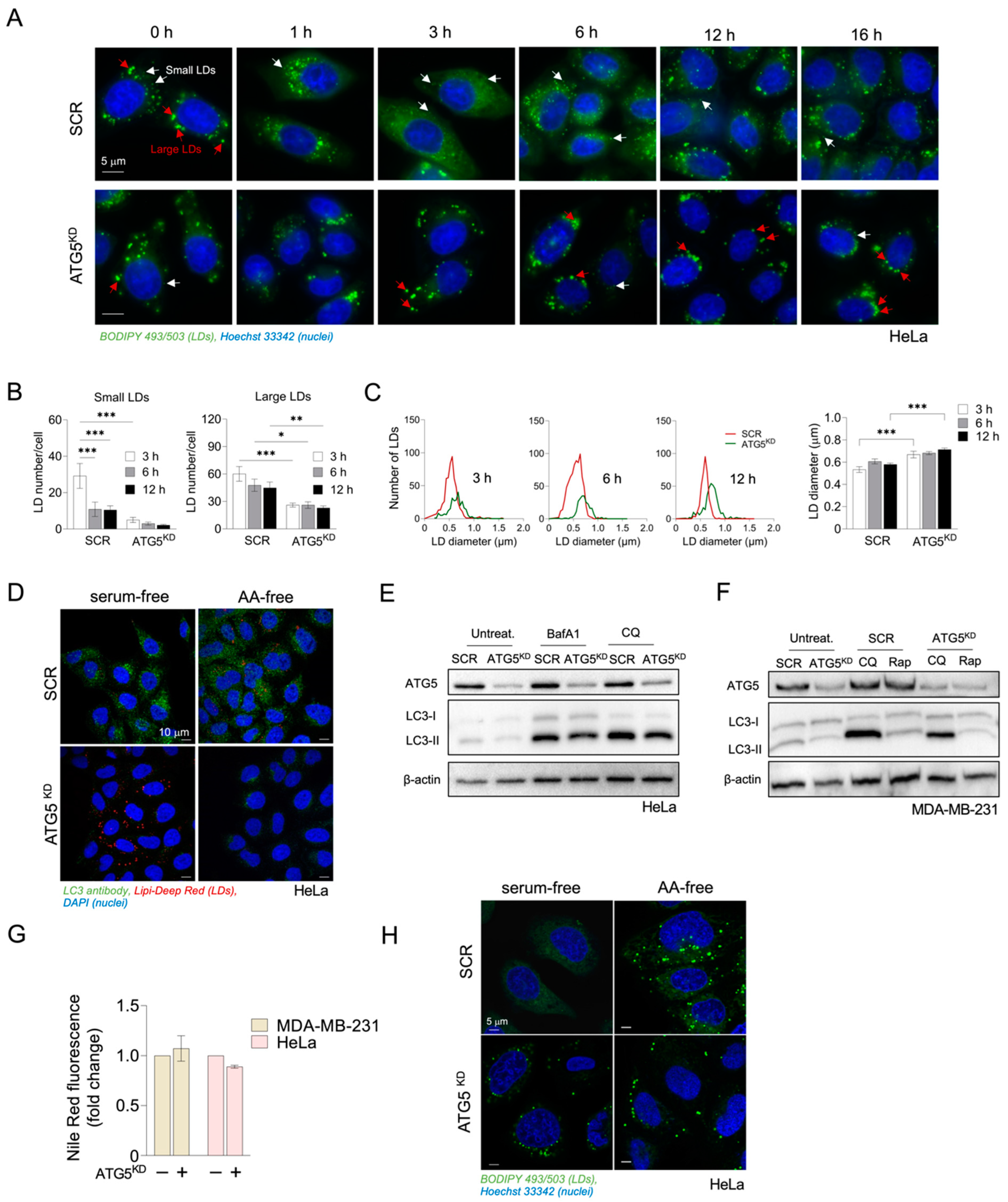
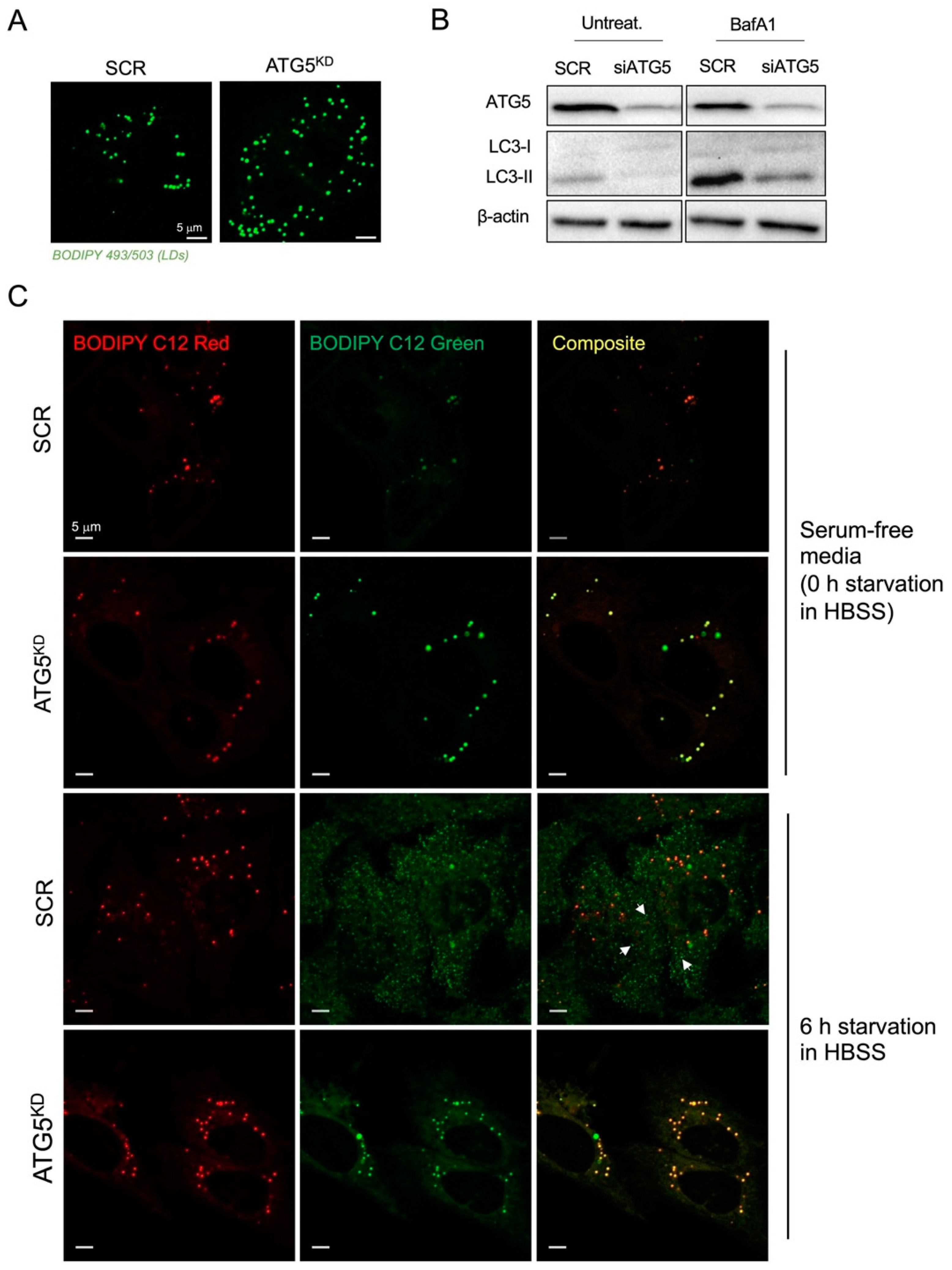
3.2. Inhibition of Autophagy Suppresses Amino-Acid-Starvation-Induced LD Biogenesis in Cancer Cells
3.3. Autophagy-Driven and DGAT-Mediated LD Biogenesis Protect HeLa Cells from Cell Death during Severe Amino Acid Starvation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cheng, H.; Wang, M.; Su, J.; Li, Y.; Long, J.; Chu, J.; Wan, X.; Cao, Y.; Li, Q. Lipid Metabolism and Cancer. Life 2022, 12, 784. [Google Scholar] [CrossRef]
- Fernández, L.P.; Gomez de Cedron, M.; Ramirez de Molina, A. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10, 577420. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zou, T.; Shen, X.; Nelson, P.J.; Li, J.; Wu, C.; Yang, J.; Zheng, Y.; Bruns, C.; Zhao, Y.; et al. Lipid metabolism in cancer progression and therapeutic strategies. MedComm 2021, 2, 27–59. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Finn, P.F.; Dice, J.F. Proteolytic and lipolytic responses to starvation. Nutrition 2006, 22, 830–844. [Google Scholar] [CrossRef]
- Bosma, M.; Dapito, D.H.; Drosatos-Tampakaki, Z.; Huiping-Son, N.; Huang, L.-S.; Kersten, S.; Drosatos, K.; Goldberg, I.J. Sequestration of fatty acids in triglycerides prevents endoplasmic reticulum stress in an in vitro model of cardiomyocyte lipotoxicity. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2014, 1841, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Kien, B.; Kolleritsch, S.; Kunowska, N.; Heier, C.; Chalhoub, G.; Tilp, A.; Wolinski, H.; Stelzl, U.; Haemmerle, G. Lipid droplet-mitochondria coupling via perilipin 5 augments respiratory capacity but is dispensable for FA oxidation. J. Lipid Res. 2022, 63, 100172. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Louie, S.M.; Daniele, J.R.; Tran, Q.; Dillin, A.; Zoncu, R.; Nomura, D.K.; Olzmann, J.A. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev. Cell 2017, 42, 9–21.e5. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, X.; Zhang, L.; Zhang, M.; Li, L.; Luo, D.; Zhong, Y. Perilipin5 protects against lipotoxicity and alleviates endoplasmic reticulum stress in pancreatic β-cells. Nutr. Metab. 2019, 16, 50. [Google Scholar] [CrossRef]
- Jarc, E.; Petan, T. Lipid Droplets and the Management of Cellular Stress. Yale J. Biol. Med. 2019, 92, 435–452. [Google Scholar] [PubMed]
- Mashek, D.G. Hepatic lipid droplets: A balancing act between energy storage and metabolic dysfunction in NAFLD. Mol. Metab. 2020, 50, 101115. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Jarc, E.; Jusović, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef]
- Renne, M.F.; Hariri, H. Lipid Droplet-Organelle Contact Sites as Hubs for Fatty Acid Metabolism, Trafficking, and Metabolic Channeling. Front. Cell Dev. Biol. 2021, 9, 726261. [Google Scholar] [CrossRef]
- Gao, M.; Huang, X.; Song, B.-L.; Yang, H. The biogenesis of lipid droplets: Lipids take center stage. Prog. Lipid Res. 2019, 75, 100989. [Google Scholar] [CrossRef]
- Walther, T.C.; Chung, J.; Farese, R.V., Jr. Lipid Droplet Biogenesis. Annu. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef]
- Bozza, P.T.; Viola, J.P.B. Lipid droplets in inflammation and cancer. Prostaglandins Leukot. Essent. Fat. Acids (PLEFA) 2010, 82, 243–250. [Google Scholar] [CrossRef]
- Koizume, S.; Miyagi, Y. Lipid Droplets: A Key Cellular Organelle Associated with Cancer Cell Survival under Normoxia and Hypoxia. Int. J. Mol. Sci. 2016, 17, 1430. [Google Scholar] [CrossRef]
- Henne, W.M.; Reese, M.L.; Goodman, J.M. The assembly of lipid droplets and their roles in challenged cells. EMBO J. 2018, 37, e98947. [Google Scholar] [CrossRef]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L.; et al. Fatty Acid Uptake and Lipid Storage Induced by HIF-1α Contribute to Cell Growth and Survival after Hypoxia-Reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Chitraju, C.; Mejhert, N.; Haas, J.T.; Diaz-Ramirez, L.G.; Grueter, C.A.; Imbriglio, J.E.; Pinto, S.; Koliwad, S.K.; Walther, T.C.; Farese, R.V. Triglyceride Synthesis by DGAT1 Protects Adipocytes from Lipid-Induced ER Stress during Lipolysis. Cell Metab. 2017, 26, 407–418.e3. [Google Scholar] [CrossRef] [PubMed]
- Jarc, E.; Kump, A.; Malavašič, P.; Eichmann, T.O.; Zimmermann, R.; Petan, T. Lipid droplets induced by secreted phospholipase A2 and unsaturated fatty acids protect breast cancer cells from nutrient and lipotoxic stress. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2018, 1863, 247–265. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Cabodevilla, A.G.; Sánchez-Caballero, L.; Nintou, E.; Boiadjieva, V.G.; Picatoste, F.; Gubern, A.; Claro, E. Cell Survival during Complete Nutrient Deprivation Depends on Lipid Droplet-fueled β-Oxidation of Fatty Acids*. J. Biol. Chem. 2013, 288, 27777–27788. [Google Scholar] [CrossRef] [PubMed]
- Minami, S.; Yamamoto, T.; Takabatake, Y.; Takahashi, A.; Namba, T.; Matsuda, J.; Kimura, T.; Kaimori, J.; Matsui, I.; Hamano, T.; et al. Lipophagy maintains energy homeostasis in the kidney proximal tubule during prolonged starvation. Autophagy 2017, 13, 1629–1647. [Google Scholar] [CrossRef] [PubMed]
- Pucer, A.; Brglez, V.; Payré, C.; Pungerčar, J.; Lambeau, G.; Petan, T. Group X secreted phospholipase A2 induces lipid droplet formation and prolongs breast cancer cell survival. Mol. Cancer 2013, 12, 111. [Google Scholar] [CrossRef]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty Acid Trafficking in Starved Cells: Regulation by Lipid Droplet Lipolysis, Autophagy, and Mitochondrial Fusion Dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef]
- Velázquez, A.P.; Tatsuta, T.; Ghillebert, R.; Drescher, I.; Graef, M. Lipid droplet–mediated ER homeostasis regulates autophagy and cell survival during starvation. J. Cell Biol. 2016, 212, 621–631. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic Control of Autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Madeo, F.; Kratky, D. Cytosolic lipolysis and lipophagy: Two sides of the same coin. Nat. Rev. Mol. Cell Biol. 2017, 18, 671–684. [Google Scholar] [CrossRef]
- Dupont, N.; Chauhan, S.; Arko-Mensah, J.; Castillo, E.F.; Masedunskas, A.; Weigert, R.; Robenek, H.; Proikas-Cezanne, T.; Deretic, V. Neutral Lipid Stores and Lipase PNPLA5 Contribute to Autophagosome Biogenesis. Curr. Biol. 2014, 24, 609–620. [Google Scholar] [CrossRef]
- Li, D.; Song, J.-Z.; Li, H.; Shan, M.-H.; Liang, Y.; Zhu, J.; Xie, Z. Storage lipid synthesis is necessary for autophagy induced by nitrogen starvation. FEBS Lett. 2015, 589, 269–276. [Google Scholar] [CrossRef]
- Shpilka, T.; Welter, E.; Borovsky, N.; Amar, N.; Mari, M.; Reggiori, F.; Elazar, Z. Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. EMBO J. 2015, 34, 2117–2131. [Google Scholar] [CrossRef]
- Drizyte-Miller, K.; Schott, M.B.; McNiven, M.A. Lipid Droplet Contacts With Autophagosomes, Lysosomes, and Other Degradative Vesicles. Contact 2020, 3, 2515256420910892. [Google Scholar] [CrossRef]
- Guštin, E.; Jarc, E.; Kump, A.; Petan, T. Lipid Droplet Formation in HeLa Cervical Cancer Cells Depends on Cell Density and the Concentration of Exogenous Unsaturated Fatty Acids. Acta Chim. Slov. 2017, 64, 549–554. [Google Scholar] [CrossRef]
- Jovičić, E.J.; Janež, A.P.; Eichmann, T.O.; Koren, Š.; Brglez, V.; Jordan, P.M.; Gerstmeier, J.; Lainšček, D.; Golob-Urbanc, A.; Jerala, R.; et al. Lipid Droplets Control Mitogenic Lipid Mediator Production in Human Cancer Cells. Mol. Metab. 2023, 76, 1–21. [Google Scholar] [CrossRef]
- Cao, J.; Zhou, Y.; Peng, H.; Huang, X.; Stahler, S.; Suri, V.; Qadri, A.; Gareski, T.; Jones, J.; Hahm, S.; et al. Targeting Acyl-CoA:Diacylglycerol Acyltransferase 1 (DGAT1) with Small Molecule Inhibitors for the Treatment of Metabolic Diseases. J. Biol. Chem. 2011, 286, 41838–41851. [Google Scholar] [CrossRef]
- Futatsugi, K.; Kung, D.W.; Orr, S.T.M.; Cabral, S.; Hepworth, D.; Aspnes, G.; Bader, S.; Bian, J.; Boehm, M.; Carpino, P.A.; et al. Discovery and Optimization of Imidazopyridine-Based Inhibitors of Diacylglycerol Acyltransferase 2 (DGAT2). J. Med. Chem. 2015, 58, 7173–7185. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef]
- Wang, H.; Wei, E.; Quiroga, A.D.; Sun, X.; Touret, N.; Lehner, R. Altered Lipid Droplet Dynamics in Hepatocytes Lacking Triacylglycerol Hydrolase Expression. Mol. Biol. Cell 2010, 21, 1991–2000. [Google Scholar] [CrossRef]
- Hussain, S.S.; Tran, T.-M.; Ware, T.B.; Luse, M.A.; Prevost, C.T.; Ferguson, A.N.; Kashatus, J.A.; Hsu, K.-L.; Kashatus, D.F. RalA and PLD1 promote lipid droplet growth in response to nutrient withdrawal. Cell Rep. 2021, 36, 109451. [Google Scholar] [CrossRef] [PubMed]
- Gubern, A.; Barceló-Torns, M.; Casas, J.; Barneda, D.; Masgrau, R.; Picatoste, F.; Balsinde, J.; Balboa, M.A.; Claro, E. Lipid Droplet Biogenesis Induced by Stress Involves Triacylglycerol Synthesis That Depends on Group VIA Phospholipase A2. J. Biol. Chem. 2009, 284, 5697–5708. [Google Scholar] [CrossRef] [PubMed]
- Herms, A.; Bosch, M.; Reddy, B.J.N.; Schieber, N.L.; Fajardo, A.; Rupérez, C.; Fernández-Vidal, A.; Ferguson, C.; Rentero, C.; Tebar, F.; et al. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat. Commun. 2015, 6, 7176. [Google Scholar] [CrossRef]
- Chitraju, C.; Walther, T.C.; Farese, R.V. The triglyceride synthesis enzymes DGAT1 and DGAT2 have distinct and overlapping functions in adipocytes. J. Lipid Res. 2019, 60, 1112–1120. [Google Scholar] [CrossRef]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Maycotte, P.; Gearheart, C.M.; Barnard, R.; Aryal, S.; Levy, J.M.M.; Fosmire, S.P.; Hansen, R.J.; Morgan, M.J.; Porter, C.C.; Gustafson, D.L.; et al. STAT3-Mediated Autophagy Dependence Identifies Subtypes of Breast Cancer Where Autophagy Inhibition Can Be Efficacious. Cancer Res. 2014, 74, 2579–2590. [Google Scholar] [CrossRef]
- Towers, C.G.; Fitzwalter, B.E.; Regan, D.; Goodspeed, A.; Morgan, M.J.; Liu, C.-W.; Gustafson, D.L.; Thorburn, A. Cancer Cells Upregulate NRF2 Signaling to Adapt to Autophagy Inhibition. Dev. Cell 2019, 50, 690–703.e6. [Google Scholar] [CrossRef] [PubMed]
- Towers, C.G.; Wodetzki, D.; Thorburn, A. Autophagy and cancer: Modulation of cell death pathways and cancer cell adaptations. J. Cell Biol. 2019, 219, e201909033. [Google Scholar] [CrossRef]
- Yoon, S.; Woo, S.U.; Kang, J.H.; Kim, K.; Kwon, M.-H.; Park, S.; Shin, H.-J.; Gwak, H.-S.; Chwae, Y.-J. STAT3 transcriptional factor activated by reactive oxygen species induces IL6 in starvation-induced autophagy of cancer cells. Autophagy 2010, 6, 1125–1138. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.A.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef]
- Sharma, A.K.; Wang, T.; Othman, A.; Khandelwal, R.; Balaz, M.; Modica, S.; Zamboni, N.; Wolfrum, C. Basal re-esterification finetunes mitochondrial fatty acid utilization. Mol. Metab. 2023, 71, 101701. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jusović, M.; Starič, P.; Jarc Jovičić, E.; Petan, T. The Combined Inhibition of Autophagy and Diacylglycerol Acyltransferase-Mediated Lipid Droplet Biogenesis Induces Cancer Cell Death during Acute Amino Acid Starvation. Cancers 2023, 15, 4857. https://doi.org/10.3390/cancers15194857
Jusović M, Starič P, Jarc Jovičić E, Petan T. The Combined Inhibition of Autophagy and Diacylglycerol Acyltransferase-Mediated Lipid Droplet Biogenesis Induces Cancer Cell Death during Acute Amino Acid Starvation. Cancers. 2023; 15(19):4857. https://doi.org/10.3390/cancers15194857
Chicago/Turabian StyleJusović, Maida, Pia Starič, Eva Jarc Jovičić, and Toni Petan. 2023. "The Combined Inhibition of Autophagy and Diacylglycerol Acyltransferase-Mediated Lipid Droplet Biogenesis Induces Cancer Cell Death during Acute Amino Acid Starvation" Cancers 15, no. 19: 4857. https://doi.org/10.3390/cancers15194857
APA StyleJusović, M., Starič, P., Jarc Jovičić, E., & Petan, T. (2023). The Combined Inhibition of Autophagy and Diacylglycerol Acyltransferase-Mediated Lipid Droplet Biogenesis Induces Cancer Cell Death during Acute Amino Acid Starvation. Cancers, 15(19), 4857. https://doi.org/10.3390/cancers15194857