
Dexamethasone-Induced Fatty Acid Oxidation and Autophagy/Mitophagy Are Essential for T-ALL Glucocorticoid Resistance

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell lines and Culture Conditions
2.3. Resazurin-Based Metabolic (Cytotoxicity) Assay
2.4. Assessment of Cell Viability by Cell Count (Trypan Blue Exclusion Test)
2.5. Cell Death Analysis by Flow Cytometry
2.6. Proliferation Assay by Flow Cytometry
2.7. Dex Uptake Assay
2.8. Glucose Uptake Assay
2.9. Glutamate Production Assay
2.10. Glycerol Production Assay
2.11. Lactate Production Assay
2.12. Evaluation of Mitochondrial Status by Mitochondria Staining
2.13. Estimation of Mitochondrial Ca2+ by CEPIA3mt
2.14. Simultaneous Monitoring of Changes in Cytosolic and Mitochondrial Ca2+
2.15. Estimation of ROS Production
2.16. Estimation of Autophagic Flux
2.17. pHAGE-mt-mKeima Transformation
2.18. Mitophagy Analysis in Leukemic Cells Transfected with pHAGE-mt-mKeima Construct
2.19. Transmission Electron Microscopy
2.20. Bidimensional Co-Culture of MSCs and T-ALL Cells
2.21. Tridimensional Co-Culture of MSCs and T-ALL Cells
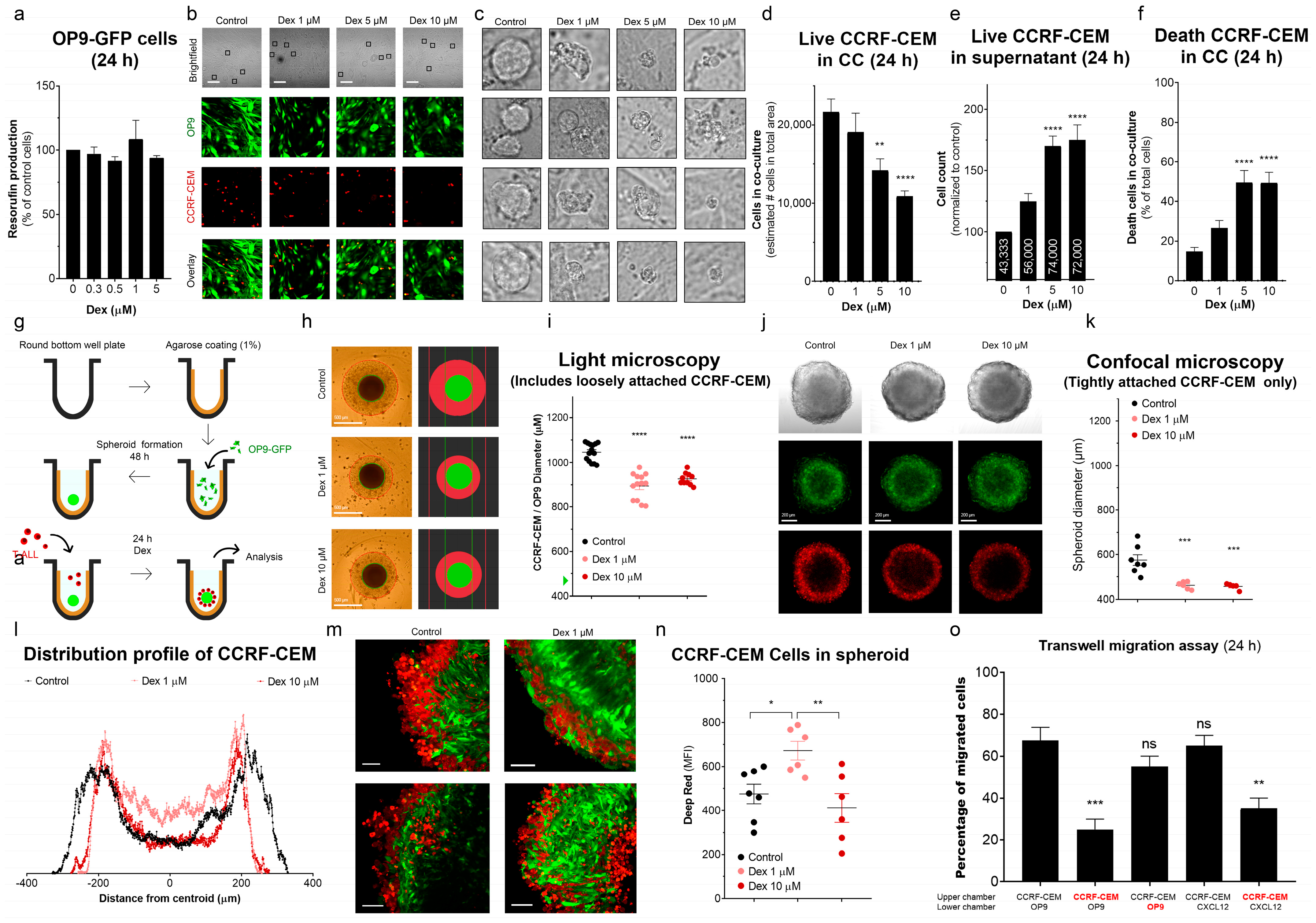
2.22. Transwell Migration Assay
3. Results
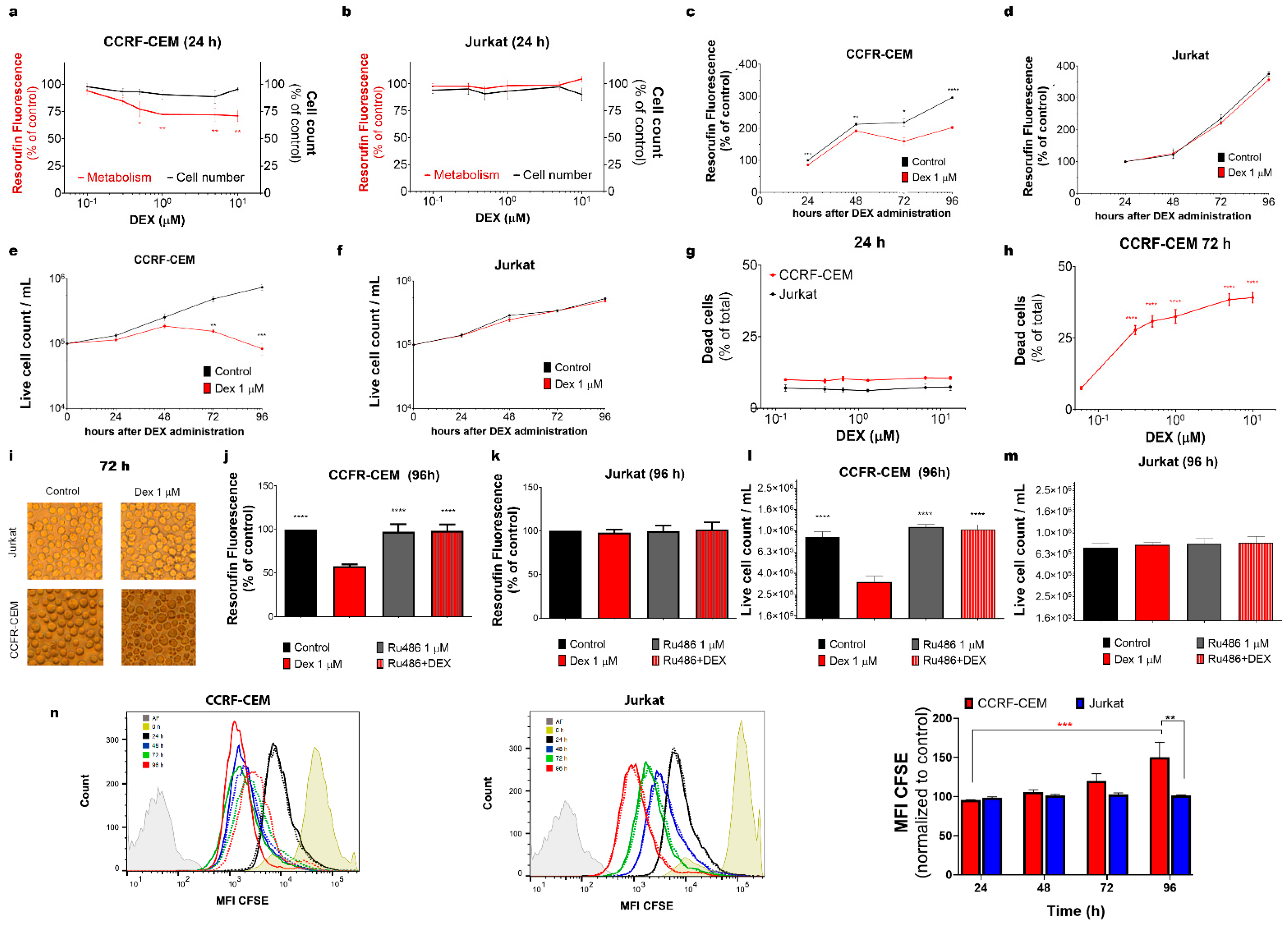
3.1. Two T-ALL Cell Lines Display Contrasting Sensitivity to Dex
3.1.1. Dex Affects the Metabolism and Viability of CCRF-CEM but Not of Jurkat Cells
3.1.2. GR Mediates the Effects of Dex on CCRF-CEM Cells
3.1.3. Dex Inhibits the Proliferation of CCRF-CEM Cells
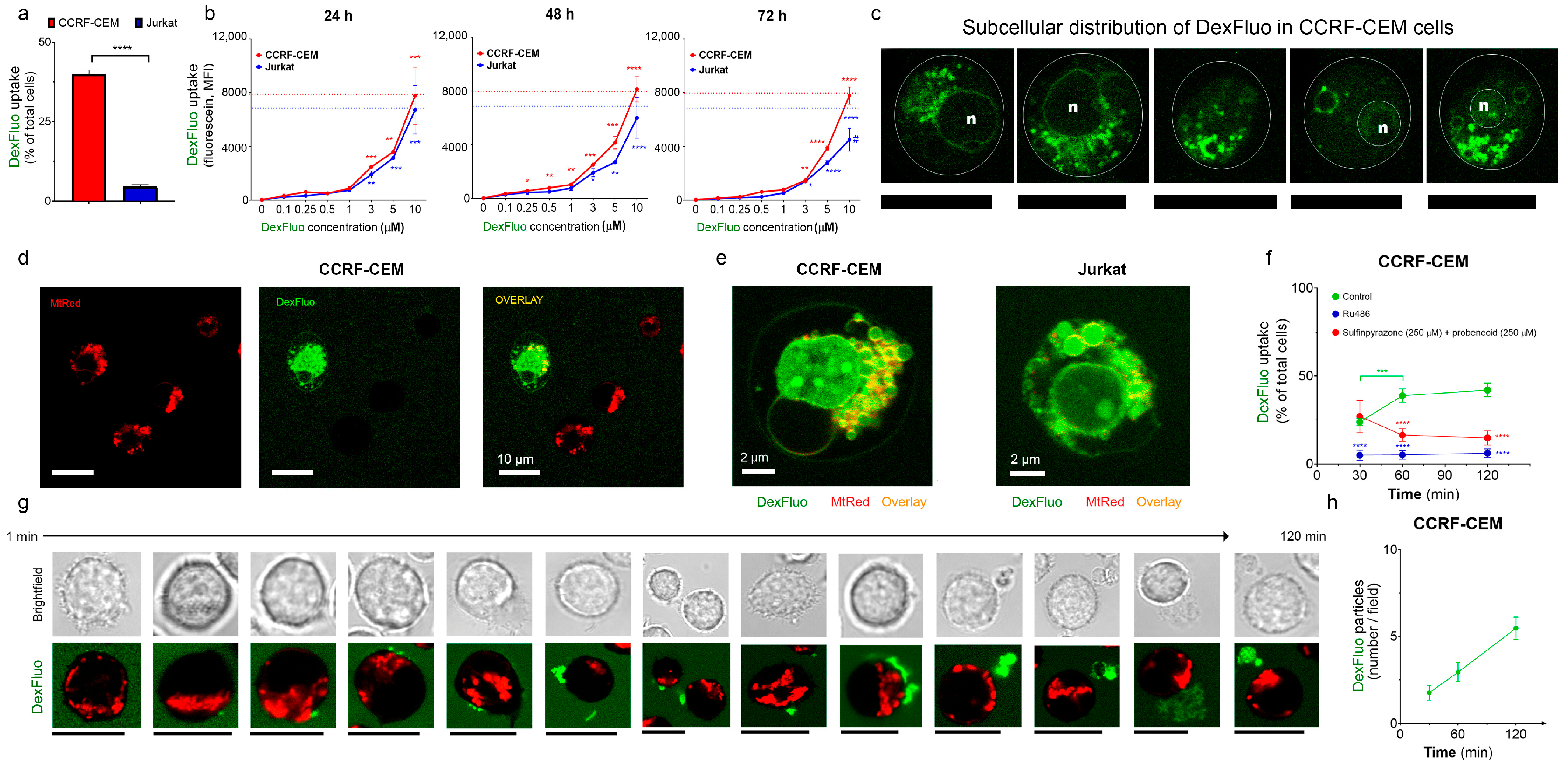
3.2. T-ALL Cells Can Efficiently Take Up and Retain Dex
3.2.1. Jurkat and CCRF-CEM Cells Retain Dex Differently
3.2.2. Dex Preferentially Targets Mitochondria
3.2.3. T-ALL Cells Can Extrude Dex by Alternative Mechanisms
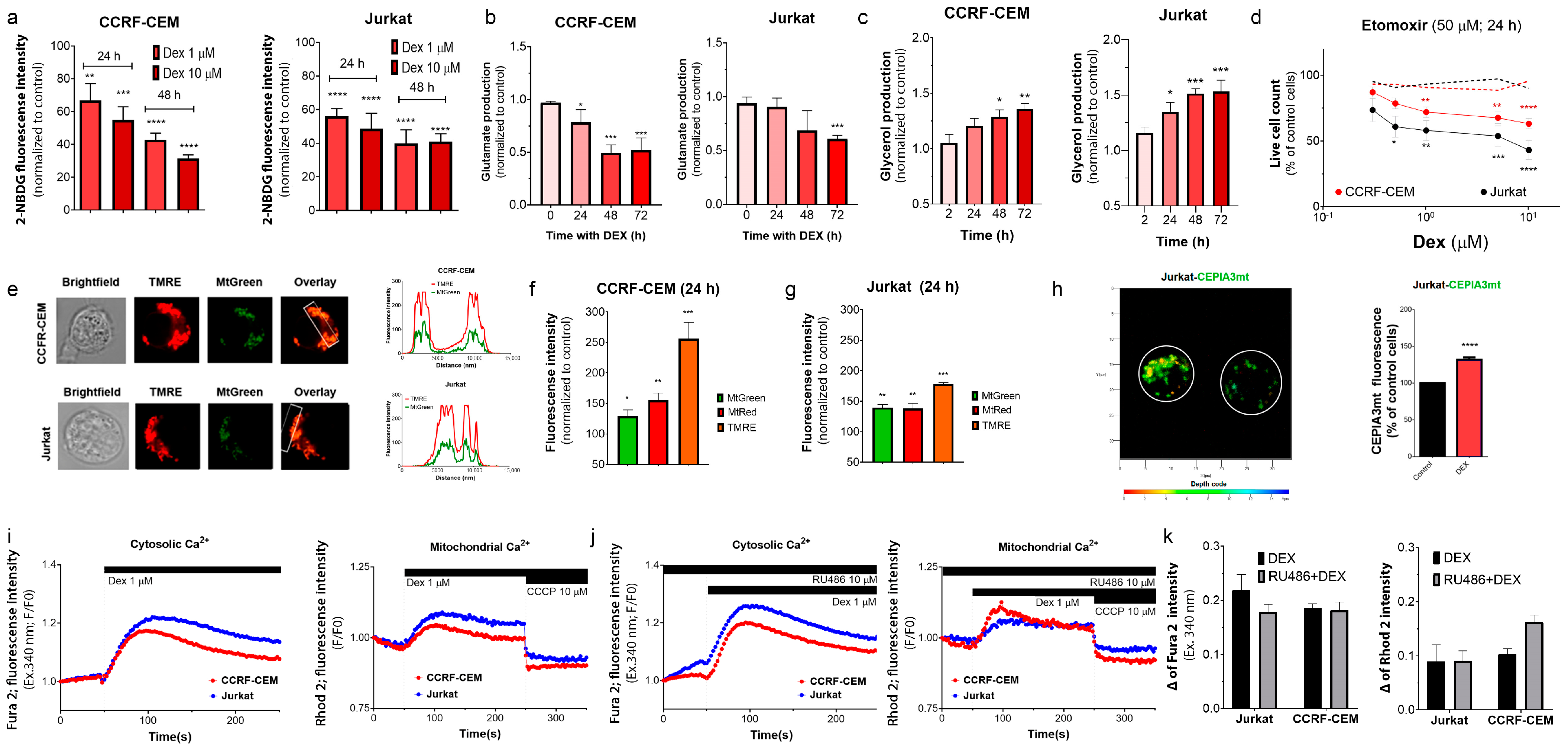
3.3. Dex Induces Metabolic Reprogramming in T-ALL Cells
3.3.1. Dex Reduces Glucose Uptake in T-ALL
3.3.2. Dex Reduces Glutaminolysis in T-ALL Cells
3.3.3. Dex Promotes Lipolysis in T-ALL
3.3.4. Selective Inhibition of Long-Chain FAO Overcomes GC Resistance in T-ALL
3.3.5. Dex Increases Retention of Mitochondrial Indicators in T-ALL
3.3.6. Dex Modulates Intracellular and Mitochondrial Ca2+ Levels in T-ALL
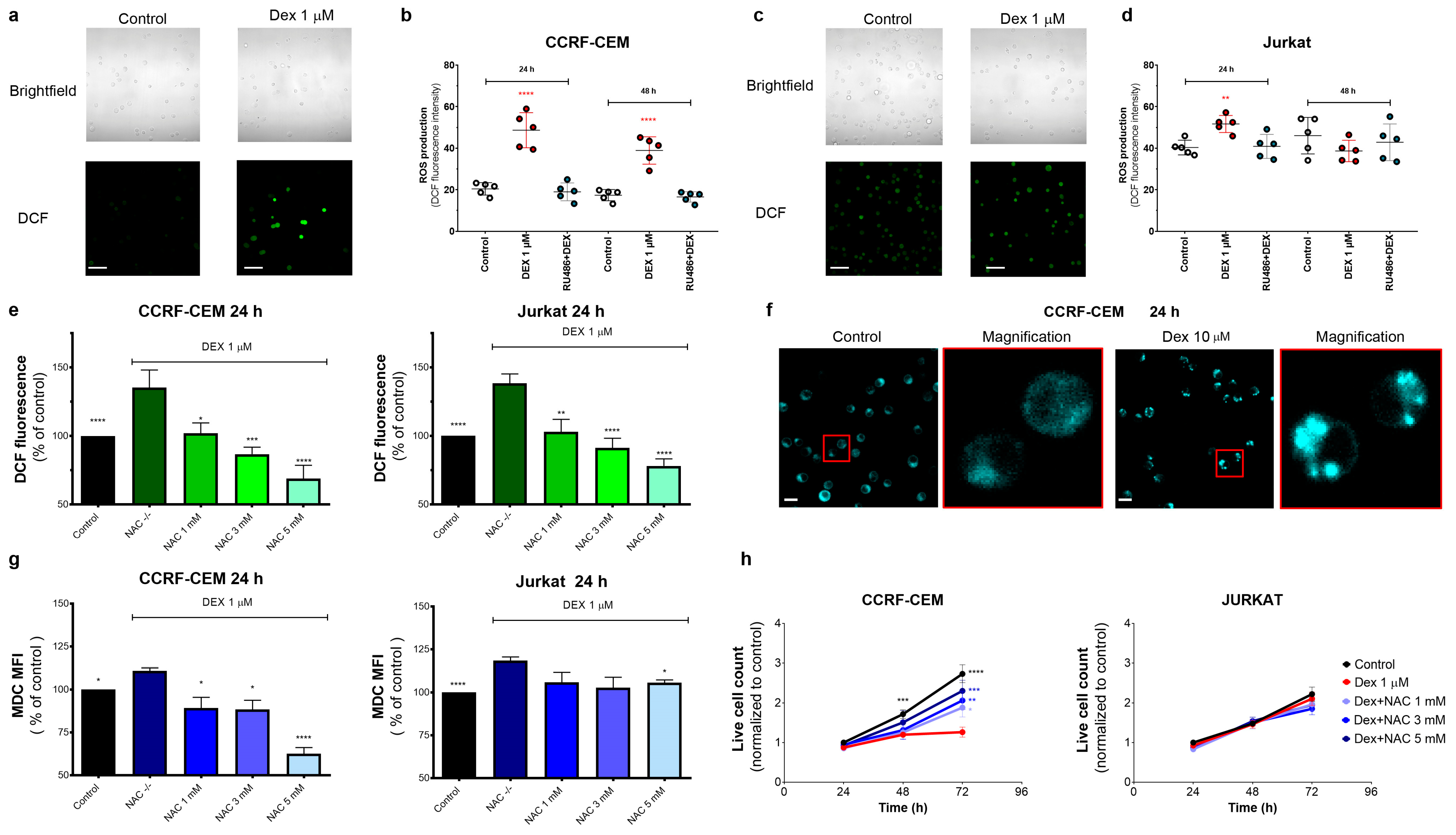
3.4. Oxidative Stress Caused by Dex Contributes to GC Sensitivity in T-ALL Cells
3.4.1. Dex Increases the Production of Reactive Oxygen Species (ROS) by T-ALL Cells
3.4.2. Oxidative Stress Caused by Dex Promotes Autophagy in T-ALL Cells
3.4.3. ROS-Mediated Autophagy Influences GC Sensitivity
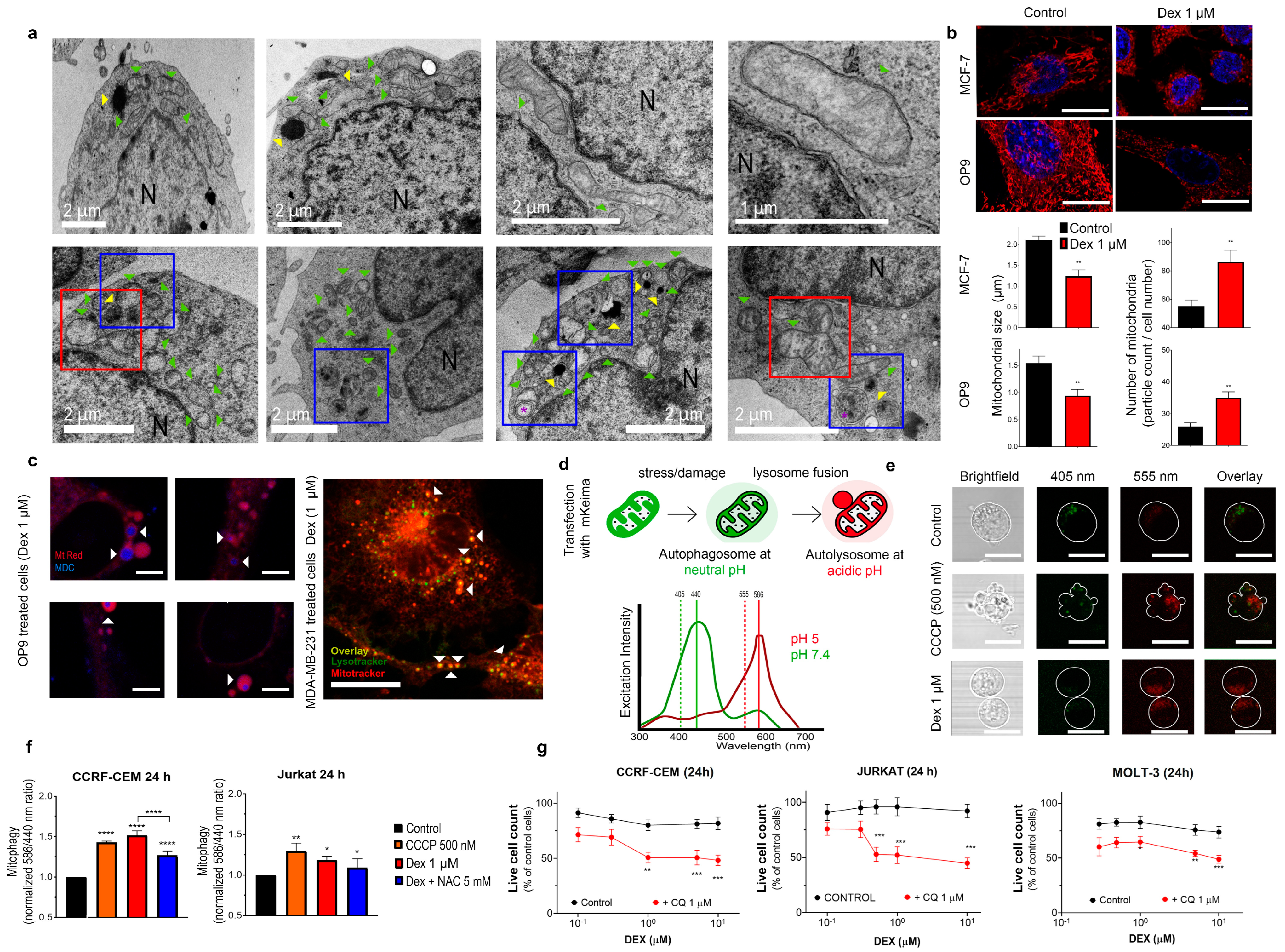
3.5. Dex Promotes Mitochondrial Fragmentation and Mitophagy
3.5.1. TEM Monitoring of Dex-Induced Morphological Changes in Sensitive CCRF-CEM Cells
3.5.2. Dex-Dependent Reduction in Mitochondrial Size and Increase in Mitochondrial Number Is Not Restricted to T-ALL Cells
3.5.3. Autophagic Components Are in Close Contact with Fragmented Mitochondria in Dex-Treated Cells: In Vivo Studies
3.5.4. Dex Promotes Mitophagy in T-ALL Cells: mtKeima-Based In Vivo Monitoring
3.5.5. Autophagy Inhibition and Mitochondria-Targeted Drugs Sensitize T-ALL Cells to Dex
3.6. Dex Reduces the Ability of T-ALL to Migrate and Colonize MSC Niches
Dex Inhibits T-ALL Interaction with MSCs and Promotes Cell Death
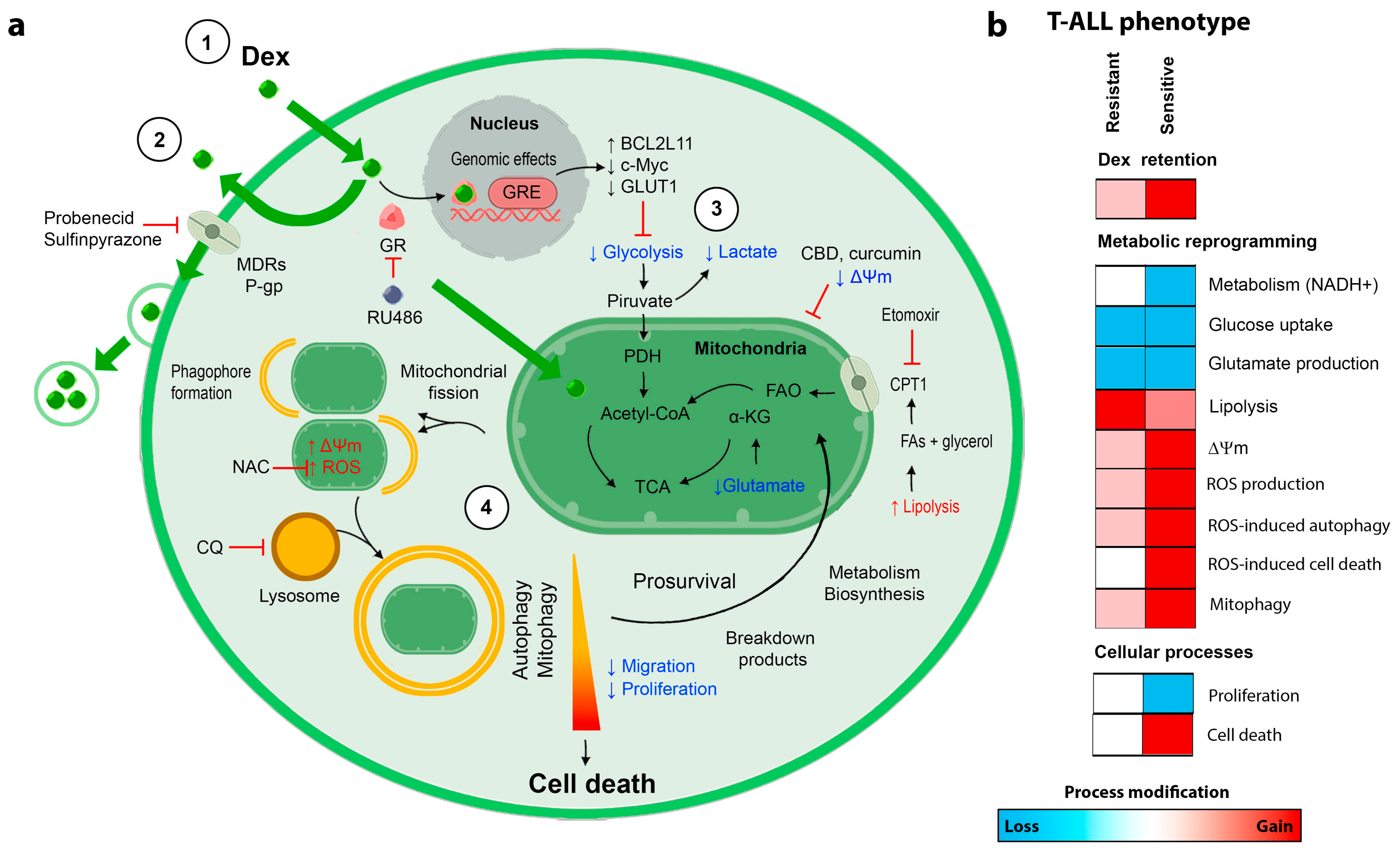
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raetz, E.A.; Teachey, D.T. T-cell acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2016, 16, 580–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrooman, L.M.; Silverman, L.B. Treatment of childhood acute lymphoblastic leukemia: Prognostic factors and clinical advances. Curr. Hematol. Malig. Rep. 2016, 11, 285–394. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Manabe, A. Treatment and biology of pediatric acute lymphoblastic leukemia. Pediatr. Int. 2018, 60, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Rafei, H.; Kantarjian, H.M.; Jabbour, E.J. Recent advances in the treatment of acute lymphoblastic leukemia. Leuk. Lymphoma 2019, 60, 2606–2621. [Google Scholar] [CrossRef] [PubMed]
- Reiter, A.; Schrappe, M.; Parwaresch, R.; Henze, G.; Müller-Weihrich, S.; Sauter, S.; Riehm, H. Non-Hodgkin’s lymphomas of childhood and adolescence: Results of a treatment stratified for biologic subtypes and stage--a report of the Berlin-Frankfurt-Münster Group. J. Clin. Oncol. 1995, 13, 359–372. [Google Scholar] [CrossRef]
- Beesley, A.; Palmer, M.L.; Ford, J.; Weller, R.E.; Cummings, A.J.; Freitas, J.R.; Firth, M.J.; Perera, K.U.; de Klerk, N.H.; Kees, U.R. Authenticity and drug resistance in a panel of acute lymphoblastic leukaemia cell lines. Br. J. Cancer 2006, 95, 1537–1544. [Google Scholar] [CrossRef] [Green Version]
- Olivas-Aguirre, M.; Torres-López, L.; Pottosin, I.; Dobrovinskaya, O. Overcoming glucocorticoid resistance in acute lymphoblastic leukemia: Repurposed drugs can improve the protocol. Front. Oncol. 2021, 11, 647. [Google Scholar] [CrossRef]
- Beesley, A.H.; Firth, M.J.; Ford, J.; Weller, R.E.; Freitas, J.R.; Perera, K.U.; Kees, U.R. Glucocorticoid resistance in T-lineage acute lymphoblastic leukaemia is associated with a proliferative metabolism. Br. J. Cancer 2009, 100, 1926–1936. [Google Scholar] [CrossRef] [Green Version]
- Beesley, A.H.; Weller, R.E.; Senanayake, S.; Welch, M.; Kees, U.R. Receptor mutation is not a common mechanism of naturally occurring glucocorticoid resistance in leukaemia cell lines. Leuk. Res. 2009, 33, 321–325. [Google Scholar] [CrossRef]
- Murani, E.; Trakooljul, N.; Hadlich, F.; Ponsuksili, S.; Wimmers, K. Transcriptome responses to dexamethasone depending on dose and glucocorticoid receptor sensitivity in the liver. Front. Genet. 2019, 10, 559. [Google Scholar] [CrossRef]
- Sekeris, C.E. The mitochondrial genome: A possible primary site of action of steroid hormones. In Vivo 1990, 4, 317–320. [Google Scholar] [PubMed]
- Lee, S.R.; Kim, H.K.; Song, I.S.; Youm, J.; Dizon, L.A.; Jeong, S.H.; Ko, T.E.; Heo, H.J.; Ko, K.S.; Rhee, B.D.; et al. Glucocorticoids and their receptors: Insights into specific roles in mitochondria. Prog. Biophys. Mol. Biol. 2013, 112, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Kokkinopoulou, I.; Moutsatsou, P. Mitochondrial glucocorticoid receptors and their actions. Int. J. Mol. Sci. 2021, 22, 6054. [Google Scholar] [CrossRef] [PubMed]
- Sionov, R.V.; Cohen, O.; Kfir, S.; Zilberman, Y.; Yefenof, E. Role of mitochondrial glucocorticoid receptor in glucocorticoid-induced apoptosis. J. Exp. Med. 2006, 203, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Sionov, R.V.; Kfir, S.; Zafrir, E.; Cohen, O.; Zilberman, Y.; Yefenof, E. Glucocorticoid-induced apoptosis revisited: A novel role for glucocorticoid receptor translocation to the mitochondria. Cell Cycle 2006, 5, 1017–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prenek, L.; Boldizsár, F.; Kugyelka, R.; Ugor, E.; Berta, G.; Németh, P.; Berki, T. The regulation of the mitochondrial apoptotic pathway by glucocorticoid receptor in collaboration with Bcl-2 family proteins in developing T cells. Apoptosis 2017, 22, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Quah, B.J.C.; Warren, H.S.; Parish, C.R. Monitoring lymphocyte prolif-eration in vitro and in vivo with the intracellular fluorescent dyecarboxyfluorescein diacetate succinimidyl ester. Nat. Protoc. 2007, 2, 2049–2056. [Google Scholar] [CrossRef]
- Torres-López, L.; Maycotte, P.; Liñán-Rico, A.; Liñán-Rico, L.; Donis-Maturano, L.; Delgado-Enciso, I.; Meza-Robles, C.; Vásquez-Jiménez, C.; Hernández-Cruz, A.; Dobrovinskaya, O. Tamoxifen induces toxicity, causes autophagy, and partially reverses dexamethasone resistance in Jurkat T cells. J. Leukoc. Biol. 2019, 105, 983–998. [Google Scholar] [CrossRef]
- Olivas-Aguirre, M.; Torres-López, L.; Valle-Reyes, J.S.; Hernández-Cruz, A.; Pottosin, I.; Dobrovinskaya, O. Cannabidiol directly targets mitochondria and disturbs calcium homeostasis in acute lymphoblastic leukemia. Cell Death Dis. 2019, 10, 779. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1797280. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Wang, C.; Bunker, E.; Hao, L.; Maric, D.; Schiavo, G.; Randow, F.; Youle, R.J. Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol. Cell 2019, 74, 347–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, H.; Kogure, T.; Mizushima, N.; Yoshimori, T.; Miyawaki, A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem. Biol. 2011, 18, 1042–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balandrán, J.C.; Purizaca, J.; Enciso, J.; Dozal, D.; Sandoval, A.; Jiménez-Hernández, E.; Alemán-Lazarini, L.; Perez-Koldenkova, V.; Quintela-Nuñez Del Prado, H.; Rios de Los Rios, J.; et al. Pro-inflammatory-related loss of CXCL12 niche promotes acute lymphoblastic leukemic progression at the expense of normal lymphopoiesis. Front. Immunol. 2017, 7, 666. [Google Scholar] [CrossRef] [Green Version]
- Medh, R.D.; Webb, M.S.; Miller, A.L.; Johnson, B.H.; Fofanov, Y.; Li, T.; Wood, T.G.; Luxon, B.A.; Thompson, E.B. Gene expression profile of human lymphoid CEM cells sensitive and resistant to glucocorticoid-evoked apoptosis. Genomics 2003, 81, 543–555. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, P.S.; Gorman, R.; Papa, R.A.; Bardell, J.E.; Ford, J.; Kees, U.R.; Marshall, G.M.; Lock, R.B. Divergent mechanisms of glucocorticoid resistance in experimental models of pediatric acute lymphoblastic leukemia. Cancer Res. 2007, 67, 4482–4490. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Pan, S.; Xie, T.; Liu, H. MYC in T-cell acute lymphoblastic leukemia: Functional implications and targeted strategies. Blood Sci. 2021, 3, 65–70. [Google Scholar] [CrossRef]
- Dyczynski, M.; Vesterlund, M.; Björklund, A.C.; Zachariadis, V.; Janssen, J.; Gallart-Ayala, H.; Daskalaki, E.; Wheelock, C.E.; Lehtiö, J.; Grandér, D.; et al. Metabolic reprogramming of acute lymphoblastic leukemia cells in response to glucocorticoid treatment. Cell Death Dis. 2018, 9, 846. [Google Scholar] [CrossRef] [Green Version]
- Buentke, E.; Nordström, A.; Lin, H.; Björklund, A.C.; Laane, E.; Harada, M.; Lu, L.; Tegnebratt, T.; Stone-Elander, S.; Heyman, M.; et al. Glucocorticoid-induced cell death is mediated through reduced glucose metabolism in lymphoid leukemia cells. Blood Cancer J. 2011, 1, e312011. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Pemmari, A.; Leppänen, T.; Hämäläinen, M.; Moilanen, T.; Vuolteenaho, K.; Moilanen, E. Widespread regulation of gene expression by glucocorticoids in chondrocytes from patients with osteoarthritis as determined by RNA-Seq. Arthritis Res. Ther. 2020, 22, 271. [Google Scholar] [CrossRef] [PubMed]
- Raud, B.; Roy, D.G.; Divakaruni, A.S.; Tarasenko, T.N.; Franke, R.; Ma, E.H.; Samborska, B.; Hsieh, W.Y.; Wong, A.H.; Stüve, P.; et al. Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab. 2018, 28, 504–515.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, J.; Kanemaru, K.; Ishii, K.; Ohkura, M.; Okubo, Y.; Iino, M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014, 5, 4153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, R. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Abdoul-Azize, S.; Dubus, I.; Vannier, J.P. Improvement of dexamethasone sensitivity by chelation of intracellular Ca2+ in pediatric acute lymphoblastic leukemia cells through the prosurvival kinase ERK1/2 deactivation. Oncotarget 2017, 8, 27339–27352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Hao, D.; Hu, H. High doses of dexamethasone induce endoplasmic reticulum stress-mediated apoptosis by promoting calcium ion influx-dependent CHOP expression in osteoblasts. Mol. Biol. Rep. 2021, 48, 7841–7851. [Google Scholar] [CrossRef]
- Itagaki, K.; Menconi, M.; Antoniu, B.; Zhang, Q.; Gonnella, P.; Soybel, D.; Hauser, C.; Hasselgren, P.O. Dexamethasone stimulates store-operated calcium entry and protein degradation in cultured L6 myotubes through a phospholipase A2-dependent mechanism. Am. J. Physiol. Cell Physiol. 2010, 298, C1127–C1139. [Google Scholar] [CrossRef] [Green Version]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Gulttaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Niemann, A.; Takatsuki, A.; Elsässer, H.P. The lysosomotropic agent monodansylcadaverine also acts as a solvent polarity probe. J. Histochem. Cytochem. 2000, 48, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Laane, E.; Pokrovskaja Tamm, K.; Buentke, E.; Ito, K.; Khahariza, P.; Oscarsson, J.; Corcoran, M.; Björklund, A.C.; Hultenby, K.; Heyman, M.; et al. Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death Differ. 2009, 16, 1018–1029. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zhao, Z.; Na, Y.; Meng, C.; Wang, J.; Bai, R. Dexamethasone-induced production of reactive oxygen species promotes apoptosis via endoplasmic reticulum stress and autophagy in MC3T3-E1 cells. Int. J. Mol. Med. 2018, 41, 2028–2036. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, Y.; Liang, Q. Low-dose dexamethasone affects osteoblast viability by inducing autophagy via intracellular ROS. Mol. Med. Rep. 2018, 17, 4307–4316. [Google Scholar] [CrossRef] [Green Version]
- Chavez-Dominguez, R.; Perez-Medina, M.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Aguilar-Cazares, D. The double-edge sword of autophagy in cancer: From tumor suppression to pro-tumor activity. Front. Oncol. 2020, 10, 2064. [Google Scholar] [CrossRef]
- Olivas-Aguirre, M.; Torres-López, L.; Pottosin, I.; Dobrovinskaya, O. Phenolic compounds cannabidiol, curcumin and quercetin cause mitochondrial dysfunction and suppress acute lymphoblastic leukemia cells. Int. J. Mol. Sci. 2020, 22, 204. [Google Scholar] [CrossRef]
- Chiarini, F.; Lonetti, A.; Evangelisti, C.; Buontempo, F.; Orsini, E.; Evangelisti, C.; Cappellini, A.; Neri, L.M.; McCubrey, J.A.; Martelli, A.M. Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: From biology to therapeutic targeting. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 449–463. [Google Scholar] [CrossRef]
- Riml, S.; Schmidt, S.; Ausserlerchner, M.J.; Geley, S.; Kofler, R. Glucocorticoid receptor heterozygosity combined with lack of receptor auto-induction causes glucocorticoid resistance in Jurkat acute lymphoblastic leukemia cells. Cell Death Differ. 2004, 11, S65–S72. [Google Scholar] [CrossRef] [Green Version]
- Wu, I.; Shin, S.C.; Cao, Y.; Bender, I.K.; Jafari, N.; Feng, G.; Lin, S.; Cidlowski, R.P.; Schleimer, R.P.; Lu, N.Z. Selective glucocorticoid receptor translational isoforms reveal glucocorticoid-induced apoptotic transcriptomes. Cell Death Dis. 2013, 4, e453. [Google Scholar] [CrossRef]
- Beger, C.; Gerdes, K.; Lauten, M.; Tissing, W.J.; Fernandez-Munoz, I.; Schrappe, M.; Welte, K. Expression and structural analysis of glucocorticoid receptor isoform gamma in human leukaemia cells using an isoform-specific real-time polymerase chain reaction approach. Br. J. Haematol. 2003, 122, 245–252. [Google Scholar] [CrossRef]
- Haarman, E.G.; Kaspers, G.J.L.; Pieters, R.; Rottier, M.M.A.; Veerman, A.J.P. Glucocorticoid receptor alpha, beta and gamma expression vs in vitro glucocorticoid resistance in childhood leukemia. Leukemia 2004, 18, 530–537. [Google Scholar] [CrossRef] [Green Version]
- Morgan, D.J.; Poolman, T.M.; Williamson, A.J.; Wang, Z.; Clark, N.R.; Ma’ayan, A.; Whetto, A.D.; Brass, A.; Matthews, L.C.; Ray, D.W. Glucocorticoid receptor isoforms direct distinct mitochondrial programs to regulate ATP production. Sci. Rep. 2016, 6, 26419. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A hallmark of cancer metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef]
- Garcia, C.; de Oliveira, M.C.; Verlengia, R.; Curi, R.; Pithon-Curi, T.C. Effect of dexamethasone on neutrophil metabolism. Cell Biochem. Funct. 2003, 21, 105–111. [Google Scholar] [CrossRef]
- Tucci, J.; Chen, T.; Margulis, K.; Orgel, E.; Paszkiewicz, R.L.; Cohen, M.D.; Oberley, M.J.; Wahhab, R.; Jones, A.E.; Divakaruni, A.S.; et al. Adipocytes provide fatty acids to acute lymphoblastic leukemia cells. Front. Oncol. 2021, 11, 665763. [Google Scholar] [CrossRef]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat. Cancer. 2020, 1, 1176–1187. [Google Scholar] [CrossRef]
- Panina, S.B.; Pei, J.; Kirienko, N.V. Mitochondrial metabolism as a target for acute myeloid leukemia treatment. Cancer Metab. 2021, 9, 17. [Google Scholar] [CrossRef]
- Baran, N.; Lodi, A.; Dhungana, Y.; Herbrich, S.; Collins, M.; Sweeney, S.; Pandey, R.; Skwarska, A.; Patel, S.; Tremblay, M.; et al. Inhibition of mitochondrial complex I reverses NOTCH1-driven metabolic reprogramming in T-cell acute lymphoblastic leukemia. Nat. Commun. 2022, 13, 2801. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Hüttemann, M. Molecular mechanisms of ischemia–reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Gibson, S.B. Is mitochondrial generation of reactive oxygen species a trigger for autophagy? Autophagy 2008, 4, 246–248. [Google Scholar] [CrossRef] [Green Version]
- Filomeni, G.; Desideri, E.; Cardaci, S.; Rotilio, G.; Ciriolo, M.R. Under the ROS: Thiol network is the principal suspect for autophagy commitment. Autophagy 2010, 6, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Levonen, A.L.; Hill, B.G.; Kansanen, E.; Zhang, J.; Darley-Usmar, V.M. Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic. Biol. Med. 2014, 71, 196–207. [Google Scholar] [CrossRef]
- Jiang, L.; Xie, J.; Li, S.; Zhang, Y.; Hou, Z.; Guo, T.; Shu, X.; Wang, C.; Fan, W.; Si, Y.; et al. Inhibition of autophagy overcomes glucocorticoid resistance in lymphoid malignant cells. Cancer Biol. Ther. 2015, 16, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Bonapace, L.; Bornhauser, B.C.; Schmitz, M.; Cario, G.; Ziegler, U.; Niggli, F.K.; Schäfer, B.W.; Schrappe, M.; Stanulla, M.; Bourquin, J.P. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J. Clin. Investig. 2010, 120, 1310–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelisti, C.; Evangeliscti, C.; Chiarini, F.; Lonetti, A.; Buontempo, F.; Neri, L.M.; McCubrey, J.A.; Martelli, A.M. Autophagy in acute leukemias: A double-edged sword with important therapeutic implications. Biochim. Biophys. Acta 2015, 1853, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Troncoso, R.; Paredes, F.; Parra, V.; Gatica, D.; Vásquez-Trincado, C.; Quiroga, C.; Bravo-Sagua, R.; López-Crisosto, C.; Rodriguez, A.E.; Oyarzún, A.P.; et al. Dexamethasone-induced autophagy mediates muscle atrophy through mitochondrial clearance. Cell Cycle 2014, 13, 2281–2295. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Li, J.; Zhang, L.; Cheng, Y.; Yan, J.; Sun, Y.; Wang, J.; Jiang, H. Role of Parkin-mediated mitophagy in glucocorticoid-induced cardiomyocyte maturation. Life Sci. 2020, 255, 117817. [Google Scholar] [CrossRef]
- Rashidi, A.; DiPersio, J.F. Targeting the leukemia-stroma interaction in acute myeloid leukemia: Rationale and latest evidence. Ther. Adv. Hematol. 2016, 7, 40–51. [Google Scholar] [CrossRef]
- Shafat, M.S.; Gnaneswaran, B.; Bowles, K.M.; Rushworth, S. The bone marrow microenvironments- Home of the leukemic blasts. Blood Rev. 2017, 31, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Fallati, A.; Di Marzo, N.; D´Amico, G.; Dander, E. Mesenchymal stromal cells (MSCs): An ally of B-cell acute lymphoblastic leukemia (B-ALL) cells in disease maintenance and progression within the bone marrow hematopoietic niche. Cancers 2022, 14, 3303. [Google Scholar] [CrossRef]
- Hughes, A.M.; Kuek, V.; Kotecha, R.S.; Cheung, L.C. The bone marrow microenvironment in B-cell development and malignancy. Cancers 2022, 14, 2089. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, R.; Wang, B.; Chen, P.; Zheng, X.; Hou, D.; Wang, X.; Zhang, B.; Chen, L.; Li, D.; Li, X.; et al. Metformin sensitizes AML cells to chemotherapy through blocking mitochondrial transfer from stromal cells to AML cells. Cancer Lett. 2022, 532, 215582. [Google Scholar] [CrossRef] [PubMed]
- Burt, R.; Dey, A.; Aref, S.; Aguilar, M.; Akarca, A.; Baliley, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, B.; Polak, R.; Stalpers, F.; Pieters, R.; den Boer, M.L. Tunneling nanotubes facilitate autophagosome transfer in the leukemic niche. Leukemia 2017, 31, 1651–1654. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, X.; Qiu, Y.; Shi, Y.; Cai, J.; Wang, B.; Wei, X.; Ke, Q.; Sui, X.; Wang, Y.; et al. Cell adhesion—Mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J. Hematol. Oncol. 2018, 11, 11. [Google Scholar] [CrossRef]
- Bozok Cetintas, V.; Aktug, H.; Otlulu, F.; Keskinoglu, A.; Erer Del Castello, B.; Taskiran, D. The effects of mesenchymal stem cells on lymphoblastic leukemia cell proliferation. J. BUON 2014, 19, 1006–1017. [Google Scholar]
- Fathi, E.; Vietor, I. Mesenchymal stem cells promote caspase expression in Molt-4 leukemia cells via GSK-3α/B and ERK1/2 signaling pathways as a therapeutic strategy. Curr. Gene Ther. 2021, 21, 81–88. [Google Scholar] [CrossRef]
- Heidari, H.R.; Fathi, E.; Montazersaheb, S.; Mamandi, A.; Farahzadi, R.; Zalavi, S.; Charoudeh, H.N. Mesenchymal stem cells cause telomere length reduction of Molt-4 cells via caspase-3, BAD and P53 apoptotic pathway. Int. J. Mol. Cell Med. 2021, 10, 113–122. [Google Scholar] [CrossRef]
- Rahbaran, M.; Shojaei, S.; Mardasi, M.; Razeghian, E. MSCs modifies the proliferation of leukemia MOLT-4 cells and induces their apoptosis through up-regulating Bax, caspase-3, and-8, and down-regulating Bcl-2 expression. Ann. Cancer. Res. Ther. 2021, 29, 79–84. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivas-Aguirre, M.; Pérez-Chávez, J.; Torres-López, L.; Hernández-Cruz, A.; Pottosin, I.; Dobrovinskaya, O. Dexamethasone-Induced Fatty Acid Oxidation and Autophagy/Mitophagy Are Essential for T-ALL Glucocorticoid Resistance. Cancers 2023, 15, 445. https://doi.org/10.3390/cancers15020445
Olivas-Aguirre M, Pérez-Chávez J, Torres-López L, Hernández-Cruz A, Pottosin I, Dobrovinskaya O. Dexamethasone-Induced Fatty Acid Oxidation and Autophagy/Mitophagy Are Essential for T-ALL Glucocorticoid Resistance. Cancers. 2023; 15(2):445. https://doi.org/10.3390/cancers15020445
Chicago/Turabian StyleOlivas-Aguirre, Miguel, Jesús Pérez-Chávez, Liliana Torres-López, Arturo Hernández-Cruz, Igor Pottosin, and Oxana Dobrovinskaya. 2023. "Dexamethasone-Induced Fatty Acid Oxidation and Autophagy/Mitophagy Are Essential for T-ALL Glucocorticoid Resistance" Cancers 15, no. 2: 445. https://doi.org/10.3390/cancers15020445
APA StyleOlivas-Aguirre, M., Pérez-Chávez, J., Torres-López, L., Hernández-Cruz, A., Pottosin, I., & Dobrovinskaya, O. (2023). Dexamethasone-Induced Fatty Acid Oxidation and Autophagy/Mitophagy Are Essential for T-ALL Glucocorticoid Resistance. Cancers, 15(2), 445. https://doi.org/10.3390/cancers15020445