
A Systemic and Integrated Analysis of p63-Driven Regulatory Networks in Mouse Oral Squamous Cell Carcinoma

 ,
,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
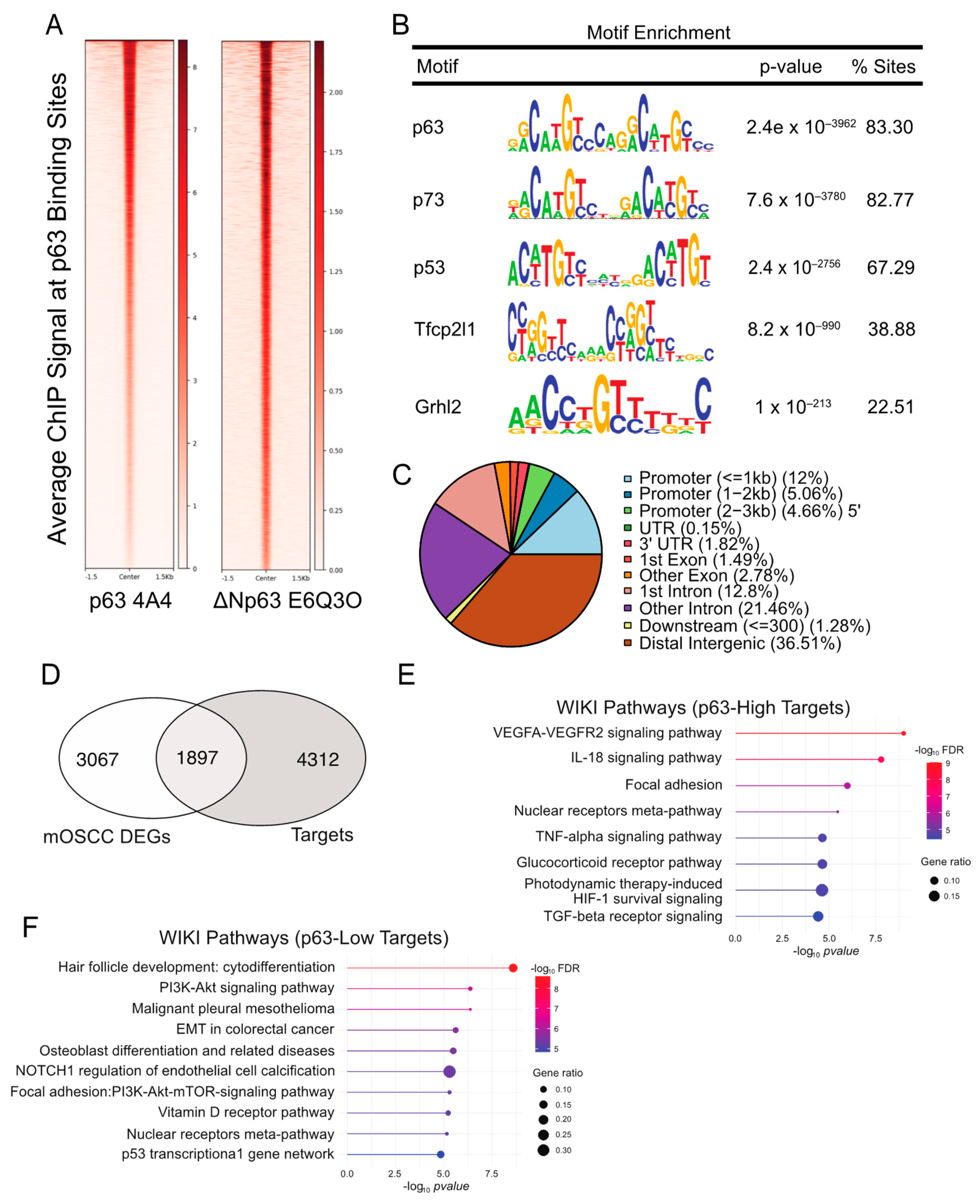{kind=link}
{kind=link}
{kind=link}
{kind=link}
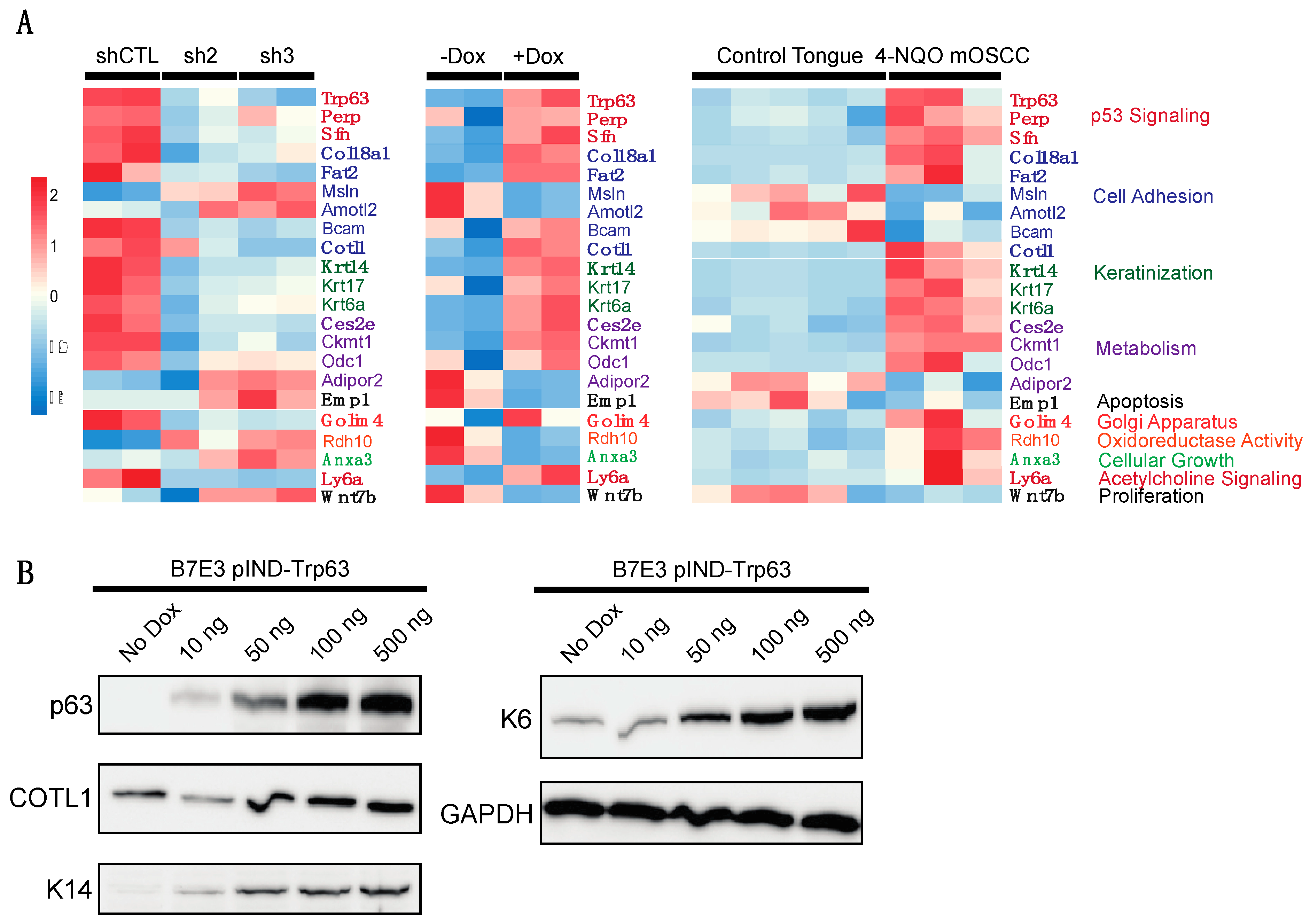{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture Studies
2.2. Generation of Knockdown and Overexpression of Cell Lines
2.3. Western Blot Analysis
2.4. ChIP-seq of p63/ΔNp63
2.5. ChIP-seq Analysis
2.6. RNA Isolation and Library Preparation for RNA-seq
2.7. RNA-seq Analysis
2.8. qRT-PCR Analysis
2.9. HNSCC and mOSCC Dataset Analysis
2.10. Invasion Assay
2.11. Spheroid Assay
2.12. Motif Enrichment Analysis of Enhancers
2.13. Gene Ontology/Pathway Enrichment Analysis
2.14. Immunofluorescence and Immunohistochemistry
2.15. Statistics
3. Results
3.1. The Global Transcriptome of 4NQO-Generated mOSCC Cell Lines
3.2. Generating a p63 Cistrome in the 4NQO-Transformed B7E11 mOSCC Cell Line
3.3. Generating a p63-Driven mOSCC Transcriptomic Signature
3.4. Evaluating the p63-Driven Gene Signature in an Independent 4NQO-Induced mOSCC Model
3.5. scRNA-seq Analysis of 4NQO-Induced Mouse ESCC
3.6. p63-Driven mOSCC Gene Signature Enriched in the Human TCGA Dataset
3.7. p63 Affects mOSCC Cell Line Migration and Proliferation
3.8. COTL1 Is a Novel Target of p63 and Is Driven by p63 Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leemans, C.R.; Braakhuis, B.J.M.; Brakenhoff, R.H. The Molecular Biology of Head and Neck Cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and Neck Squamous Cell Carcinoma. Nat. Rev. Dis. Prim. 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.; Agrawal, N.; Gooi, Z. Lip and Oral Cavity Squamous Cell Carcinoma. Hematol. Oncol. Clin. North Am. 2021, 35, 895–911. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G.; Ally, A.; Balasundaram, M.; Birol, I.; et al. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kürten, C.H.L.; Kulkarni, A.; Cillo, A.R.; Santos, P.M.; Roble, A.K.; Onkar, S.; Reeder, C.; Lang, S.; Chen, X.; Duvvuri, U.; et al. Investigating Immune and Non-Immune Cell Interactions in Head and Neck Tumors by Single-Cell RNA Sequencing. Nat. Commun. 2021, 12, 7338. [Google Scholar] [CrossRef]
- Vered, M.; Yarom, N.; Dayan, D. 4NQO Oral Carcinogenesis: Animal Models, Molecular Markers and Future Expectations. Oral Oncol. 2005, 41, 337–339. [Google Scholar] [CrossRef]
- Kanojia, D.; Vaidya, M.M. 4-Nitroquinoline-1-Oxide Induced Experimental Oral Carcinogenesis. Oral Oncol. 2006, 42, 655–667. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, V.H.; Allevato, M.M.; Gilardi, M.; He, Y.; Luis Callejas-Valera, J.; Vitale-Cross, L.; Martin, D.; Amornphimoltham, P.; Mcdermott, J.; et al. Syngeneic Animal Models of Tobacco-Associated Oral Cancer Reveal the Activity of in Situ Anti-CTLA-4. Nat. Commun. 2019, 10, 5546. [Google Scholar] [CrossRef] [Green Version]
- Kondo, S. A Test for Mutation Theory of Cancer: Carcinogenesis by Misrepair of Dna Damaged by 4-Nitroquinoline 1-Oxide. Br. J. Cancer 1977, 35, 595–601. [Google Scholar] [CrossRef]
- Tang, X.H.; Osei-Sarfo, K.; Urvalek, A.M.; Zhang, T.; Scognamiglio, T.; Gudas, L.J. Combination of Bexarotene and the Retinoid CD1530 Reduces Murine Oral-Cavity Carcinogenesis Induced by the Carcinogen 4-Nitroquinoline 1-Oxide. Proc. Natl. Acad. Sci. USA 2014, 111, 8907–8912. [Google Scholar] [CrossRef] [Green Version]
- Ishida, K.; Tomita, H.; Kanayama, T.; Noguchi, K.; Niwa, A.; Kawaguchi, M.; Miyai, M.; Matsuo, M.; Imaizumi, Y.; Kato, K.; et al. Specific Deletion of P16INK4a with Retention of P19ARF Enhances the Development of Invasive Oral Squamous Cell Carcinoma. Am. J. Pathol. 2020, 190, 1332–1342. [Google Scholar] [CrossRef]
- Chen, W.; Kang, K.L.; Alshaikh, A.; Varma, S.; Lin, Y.; Shin, K.; Kim, R.; Wang, C.; Park, N.; Walentin, K.; et al. Grainyhead-like 2 (GRHL2) Knockout Abolishes Oral Cancer Development through Reciprocal Regulation of the MAP Kinase and TGF-β Signaling Pathways. Oncogenesis 2018, 7, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Romano, R.A.; Si, H.; Mattox, A.; Bian, Y.; Yang, X.; Sinha, S.; Van Waes, C.; Chen, Z. Epidermal Overexpression of Transgenic Δnp63 Promotes Type 2 Immune and Myeloid Inflammatory Responses and Hyperplasia via NF-ΚB Activation. J. Pathol. 2014, 232, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Sethi, I.; Gluck, C.; Zhou, H.; Buck, M.J.; Sinha, S. Evolutionary Re-Wiring of P63 and the Epigenomic Regulatory Landscape in Keratinocytes and Its Potential Implications on Species-Specific Gene Expression and Phenotypes. Nucleic Acids Res. 2017, 45, 8208–8224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, R.; Smalley, K.; Magraw, C.; Serna, V.A.; Kurita, T.; Raghavan, S.; Sinha, S. DNp63 Knockout Mice Reveal Its Indispensable Role as a Master Regulator of Epithelial Development and Differentiation. Development 2012, 139, 772–782. [Google Scholar] [CrossRef] [Green Version]
- Keyes, W.M.; Pecoraro, M.; Aranda, V.; Vernersson-Lindahl, E.; Li, W.; Vogel, H.; Guo, X.; Garcia, E.L.; Michurina, T.V.; Enikolopov, G.; et al. ΔNp63α Is an Oncogene That Targets Chromatin Remodeler Lsh to Drive Skin Stem Cell Proliferation and Tumorigenesis. Cell Stem Cell 2011, 8, 164–176. [Google Scholar] [CrossRef] [Green Version]
- Botchkarev, V.; Flores, E. P53/P63/P73 in the Epidermis in Health and Disease. Cold Spring Harb. Perspect. Med. 2014, 4, 1260–1266. [Google Scholar] [CrossRef]
- Romano, R.; Ortt, K.; Birkaya, B.; Smalley, K.; Sinha, S. An Active Role of the D N Isoform of P63 in Regulating Basal Keratin Genes K5 and K14 and Directing Epidermal Cell Fate. PLoS ONE 2009, 4, e5623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Muzio, L.; Santarelli, A.; Caltabiano, R.; Rubini, C.; Pieramici, T.; Trevisiol, L.; Carinci, F.; Leonardi, R.; De Lillo, A.; Lanzafame, S.; et al. P63 Overexpression Associates With Poor Prognosis in Head and Neck Squamous Cell Carcinoma. Hum. Pathol. 2005, 36, 187–194. [Google Scholar] [CrossRef]
- Somerville, T.D.D.; Xu, Y.; Miyabayashi, K.; Tiriac, H.; Cleary, C.R.; Maia-Silva, D.; Milazzo, J.P.; Tuveson, D.A.; Vakoc, C.R. TP63-Mediated Enhancer Reprogramming Drives the Squamous Subtype of Pancreatic Ductal Adenocarcinoma. Cell Rep. 2018, 25, 1741–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, M.-H.; Choe, J.H.; Gadhvi, J.; Kim, Y.J.; Arguez, M.A.; Palmer, M.; Gerold, H.; Nowak, C.; Do, H.; Mazambani, S.; et al. P63 and SOX2 Dictate Glucose Reliance and Metabolic Vulnerabilities in Squamous Cell Carcinomas. Cell Rep. 2019, 28, 1860–1878.e9. [Google Scholar] [CrossRef] [Green Version]
- Sethi, I.; Romano, R.A.; Gluck, C.; Smalley, K.; Vojtesek, B.; Buck, M.J.; Sinha, S. A Global Analysis of the Complex Landscape of Isoforms and Regulatory Networks of P63 in Human Cells and Tissues. BMC Genomics 2015, 16, 584. [Google Scholar] [CrossRef] [Green Version]
- Lakshmanachetty, S.; Balaiya, V.; High, W.A.; Koster, M.I. Loss of TP63 Promotes the Metastasis of Head and Neck Squamous Cell Carcinoma by Activating MAPK and STAT3 Signaling. Mol. Cancer Res. 2019, 17, 1279–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, N.H.B.; Napoli, M.; Davis, A.J.; Abbas, H.A.; Rajapakshe, K.; Coarfa, C.; Flores, E.R. Spatiotemporal Regulation of ΔNp63 by TGFβ-Regulated MiRNAs Is Essential for Cancer Metastasis. Cancer Res. 2020, 80, 2833–2847. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.R.; Chen, Z.; Oechli, M.N.; Hendler, F.J.; Van Waes, C. Decreased Expression of CD80 Is a Marker for Increased Tumorigenicity in a New Murine Model of Oral Squamous-Cell Carcinoma. Int. J. Cancer 1999, 82, 377–384. [Google Scholar] [CrossRef]
- Oyelakin, A.; Nayak, K.B.; Glathar, A.R.; Gluck, C.; Wrynn, T.; Tugores, A.; Romano, R.; Sinha, S. EHF Is a Novel Regulator of Cellular Redox Metabolism and Predicts Patient Prognosis in HNSCC. NAR cancer 2022, 4, zcac017. [Google Scholar] [CrossRef]
- Gluck, C.; Glathar, A.; Tsompana, M.; Nowak, N.; Garrett-Sinha, L.A.; Buck, M.J.; Sinha, S. Molecular Dissection of the Oncogenic Role of ETS1 in the Mesenchymal Subtypes of Head and Neck Squamous Cell Carcinoma. PLoS Genet. 2019, 15, e1008250. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, J.M.; Oyelakin, A.; Min, S.; Smalley, K.; Bard, J.; Luo, W.; Nyquist, J.; Guttman-Yassky, E.; Yoshida, T.; De Benedetto, A.; et al. ΔNp63 Regulates IL-33 and IL-31 Signaling in Atopic Dermatitis. Cell Death Differ. 2016, 23, 1073–1085. [Google Scholar] [CrossRef]
- Glathar, A.R.; Oyelakin, A.; Gluck, C.; Bard, J.; Sinha, S. P63 Directs Subtype-Specific Gene Expression in HPV+ Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2022, 12, 879054. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-Based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [Green Version]
- Mclean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Wenger, A.M.; Bejerano, G. GREAT Improves Functional Interpretation of Cis-Regulatory Regions. Nat Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. DeepTools2: A next Generation Web Server for Deep-Sequencing Data Analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor Package for ChIP Peak Annotation, Comparison and Visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of MRNA Abundance Using RNA-Seq Data: RPKM Measure Is Inconsistent among Samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E. Yevgeniy Antipin; Boris Reva; Arthur P. Goldberg; Chris Sander; Nikolaus Schultz. The CBio Cancer Genomics. Cancer Discov. 2017, 32, 736–740. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2014, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repecka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Overcoming Barriers to Early Disease Intervention. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Cui, Q.; Fan, W.; Ma, Y.; Chen, Y.; Liu, T.; Zhang, X.; Xi, Y.; Wang, C.; Peng, L.; et al. Single-Cell Transcriptomic Analysis in a Mouse Model Deciphers Cell Transition States in the Multistep Development of Esophageal Cancer. Nat. Commun. 2020, 11, 3715. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Bailey, T.L.; MacHanick, P. Inferring Direct DNA Binding from ChIP-Seq. Nucleic Acids Res. 2012, 40, e128. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating Viruses and Cellular Organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics Enrichment Tools: Paths toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1α-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Mogi, S.; Dang, D.; Waes, C.V.A.N.; Ellis, D.; Atakilit, A.; Ramos, D.M. The Expression of Integrin AvB6 Promotes the Epithelial Cell Morphology and Suppresses Invasive Behavior in Transformed Oral Keratinocyte. Anticancer Res. 2005, 756, 751–755. [Google Scholar]
- Fisher, M.L.; Balinth, S.; Mills, A.A. ΔNp63α in Cancer: Importance and Therapeutic Opportunities. Trends Cell Biol. 2022. [Google Scholar] [CrossRef]
- Kumar, S.; Gabrilovich, D.; Chakrabarti, R.; Kumar, S.; Wilkes, D.W.; Samuel, N.; Blanco, M.A.; Nayak, A.; Alicea-torres, K.; Gluck, C.; et al. ΔNp63-Driven Recruitment of Myeloid-Derived Suppressor Cells Promotes Metastasis in Triple- Negative Breast Cancer. J. Clin. Invest. 2018, 128, 5095–5109. [Google Scholar] [CrossRef]
- Kubo, T.; Ichimiya, S.; Tonooka, A.; Nagashima, T.; Kikuchi, T.; Sato, N. P63 Induces CD4+ T-Cell Chemoattractant TARC/CCL17 in Human Epithelial Cells. J. Interf. Cytokine Res. 2008, 28, 725–732. [Google Scholar] [CrossRef]
- Mitchell, M.J.; King, M.R. ΔNp63 Versatilely Regulates a Broad NF-ΚB Gene Program and Promotes Squamous Epithelial Proliferation, Migration and Inflammation. Cancer Res. 2014, 71, 3688–3700. [Google Scholar] [CrossRef] [Green Version]
- Soares, E.; Zhou, H. Master Regulatory Role of P63 in Epidermal Development and Disease. Cell. Mol. Life Sci. 2018, 75, 1179–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, R.; Birkaya, B.; Sinha, S. A Functional Enhancer of Keratin14 Is a Direct Transcriptional Target of D Np63. J. Invest. Dermatol. 2007, 127, 1175–1186. [Google Scholar] [CrossRef] [Green Version]
- Dang, T.T.; Westcott, J.M.; Maine, E.A.; Kanchwala, M.; Xing, C.; Pearson, G.W. ΔNp63α Induces the Expression of FAT2 and Slug to Promote Tumor Invasion. Oncotarget 2016, 7, 28592–28611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Tan, Y.; Wang, L.; Cai, J.; Li, X.; Zeng, Z.; Xiong, W.; Li, G.; Li, X.; Tan, P.; et al. TP63 Links Chromatin Remodeling and Enhancer Reprogramming to Epidermal Differentiation and Squamous Cell Carcinoma Development. Cell. Mol. Life Sci. 2020, 77, 4325–4346. [Google Scholar] [CrossRef]
- Hamdan, F.H.; Johnsen, S.A. DeltaNp63-Dependent Super Enhancers Define Molecular Identity in Pancreatic Cancer by an Interconnected Transcription Factor Network. Proc. Natl. Acad. Sci. USA 2018, 115, 201812915. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.H.; Knudsen, B.; Bemis, D.; Tickoo, S.; Gudas, L.J. Oral Cavity and Esophageal Carcinogenesis Modeled in Carcinogen-Treated Mice. Clin. Cancer Res. 2004, 10, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Devos, M.; Declercq, W. ΔNp63α Acts as a Lineage-Survival Oncogene in Squamous Cell Carcinoma. J. Med. Surg. Pathol. 2017, 2, 7–9. [Google Scholar] [CrossRef]
- Nekulova, M.; Holcakova, J.; Coates, P.; Vojtesek, B. The Role of P63 in Cancer, Stem Cells and Cancer Stem Cells. Cell. Mol. Biol. Lett. 2011, 16, 296–327. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Fiore, C.; Chutake, Y.; Verhaar, E.R.; Strasser, P.C.; Chen, M.W.; Farouq, D.; Das, S.; Li, X.; Eaton, E.N.; et al. ΔNp63/P73 Drive Metastatic Colonization by Controlling a Regenerative Epithelial Stem Cell Program in Quasi-Mesenchymal Cancer Stem Cells. Dev. Cell 2022, 57, 2714–2730.e8. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Tang, L.J.; Brown, K.A.; Pietenpol, J.A. Loss of P63 Leads to Increased Cell Migration and Up-Regulation of Genes Involved in Invasion and Metastasis. Cancer Res. 2006, 66, 7589–7597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provost, P.; Doucet, J.; Stock, A.; Gerisch, G.; Samuelsson, B.; Rådmark, O. Coactosin-like Protein, a Human F-Actin-Binding Protein: Critical Role of Lysine-75. Biochem. J. 2001, 359, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yin, Y.; Chen, J.; Fan, Y.; Ma, J.; Huang, Y.; Chen, C.; Dai, P.; Chen, S.; Zhao, S. Coactosin-like Protein 1 Inhibits Neuronal Migration during Mouse Corticogenesis. J. Vet. Sci. 2018, 19, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Hye-Cheol, J.; Gwang-Il, K.; Sang-Ho, C.; Kwang-Hyung, L.; Jung-Jae, K.; Jeong-Hee, K.; Kwang-Hoe, C. Proteomic Analysis of Human Small Cell Lung Cancer Tissues: Up-Regulation of Coactosin-like Protein-1. J. Proteome Res. 2011, 10, 269–276. [Google Scholar] [CrossRef]
- Shao, S.; Fan, Y.; Zhong, C.; Zhu, X.; Zhu, J. Coactosin-like Protein (Cotl1) Promotes Glioblastoma (Gbm) Growth in Vitro and in Vivo. Cancer Manag. Res. 2020, 12, 10909–10917. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Jia, Y.; Guo, F.; Zhengjun, S.; Shao, Z. Intratumoural Heterogeneity and Clone Evolution of Oral Squamous Cell Carcinoma. Mol. Carcinog. 2021, 60, 758–768. [Google Scholar] [CrossRef]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A Comparative Risk Assessment of Burden of Disease and Injury Attributable to 67 Risk Factors and Risk Factor Clusters in 21 Regions, 1990-2010: A Systematic Analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef] [Green Version]
- Agudo, A.; Bonet, C.; Travier, N.; González, C.A.; Vineis, P.; Bueno-de-Mesquita, H.B.; Trichopoulos, D.; Boffetta, P.; Clavel-Chapelon, F.; Boutron-Ruault, M.C.; et al. Impact of Cigarette Smoking on Cancer Risk in the European Prospective Investigation into Cancer and Nutrition Study. J. Clin. Oncol. 2012, 30, 4550–4557. [Google Scholar] [CrossRef]
- Li, Q.; Dong, H.; Yang, G.; Song, Y.; Mou, Y.; Ni, Y. Mouse Tumor-Bearing Models as Preclinical Study Platforms for Oral Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 212. [Google Scholar] [CrossRef] [PubMed]
- Judd, N.P.; Winkler, A.E.; Murillo-Sauca, O.; Brotman, J.J.; Law, J.H.; Lewis, J.S., Jr.; Dunn, G.P.; Bui, J.D.; Sunwoo, J.B.; Uppaluri, R. ERK1/2 Regulation of CD44 Modulates Oral Cancer Aggressiveness. Cancer Res. 2012, 72, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Zeng, Z.; Egloff, A.M.; Zhang, F.; Guo, F.; Campbell, K.M.; Du, P.; Fu, J.; Zolkind, P.; Ma, X.; et al. Checkpoint Blockade-Induced CD8+ T Cell Differentiation in Head and Neck Cancer Responders. J. Immunother. Cancer 2022, 10, e004034. [Google Scholar] [CrossRef] [PubMed]
- Korpela, S.P.; Hinz, T.K.; Oweida, A.; Kim, J.; Calhoun, J.; Ferris, R.; Nemenoff, R.A.; Karam, S.D.; Clambey, E.T.; Heasley, L.E. Role of Epidermal Growth Factor Receptor Inhibitor-Induced Interferon Pathway Signaling in the Head and Neck Squamous Cell Carcinoma Therapeutic Response. J. Transl. Med. 2021, 19, 43. [Google Scholar] [CrossRef] [PubMed]
- Udani, M.; Zen, Q.; Cottman, M.; Leonard, N.; Jefferson, S.; Daymont, C.; Truskey, G.; Telen, M.J. Basal Cell Adhesion Molecule/Lutheran Protein: The Receptor Critical for Sickle Cell Adhesion to Laminin. J. Clin. Invest. 1998, 101, 2550–2558. [Google Scholar] [CrossRef] [Green Version]
- Bartolini, A.; Cardaci, S.; Lamba, S.; Oddo, D.; Marchiò, C.; Cassoni, P.; Amoreo, C.A.; Corti, G.; Testori, A.; Bussolino, F.; et al. BCAM and LAMA5 Mediate the Recognition between Tumor Cells and the Endothelium in the Metastatic Spreading of KRAS-Mutant Colorectal Cancer. Clin. Cancer Res. 2016, 22, 4923–4933. [Google Scholar] [CrossRef] [Green Version]
- Awais, R.; Spiller, D.G.; White, M.R.H.; Paraoan, L. P63 Is Required beside P53 for PERP-Mediated Apoptosis in Uveal Melanoma. Br. J. Cancer 2016, 115, 983–992. [Google Scholar] [CrossRef] [Green Version]
- Beaudry, V.G.; Jiang, D.; Dusek, R.L.; Park, E.J.; Knezevich, S.; Ridd, K.; Vogel, H.; Bastian, B.C.; Attardi, L.D. Loss of the P53/P63 Regulated Desmosomal Protein Perp Promotes Tumorigenesis. PLoS Genet. 2010, 6, e1001168. [Google Scholar] [CrossRef] [Green Version]
- Ralhan, R.; DeSouza, L.V.; Matta, A.; Tripathi, S.C.; Ghanny, S.; Datta Gupta, S.; Bahadur, S.; Siu, K.W.M. Discovery and Verification of Head-and-Neck Cancer Biomarkers by Differential Protein Expression Analysis Using ITRAQ Labeling, Multidimensional Liquid Chromatography, and Tandem Mass Spectrometry. Mol. Cell. Proteomics 2008, 7, 1162–1173. [Google Scholar] [CrossRef] [Green Version]
- Danilov, A.V.; Neupane, D.; Nagaraja, A.S.; Feofanova, E.V.; Humphries, L.A.; DiRenzo, J.; Korc, M. DeltaNp63alpha-Mediated Induction of Epidermal Growth Factor Receptor Promotes Pancreatic Cancer Cell Growth and Chemoresistance. PLoS ONE 2011, 6, e26815. [Google Scholar] [CrossRef]
- Guo, S.; Yang, P.; Jiang, X.; Li, X.; Wang, Y.; Zhang, X.; Sun, B.; Zhang, Y.; Jia, Y. Genetic and Epigenetic Silencing of MircoRNA-506-3p Enhances COTL1 Oncogene Expression to Foster Non-Small Lung Cancer Progression. Oncotarget 2017, 8, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Pei, B.; Li, T.; Qian, Q.; Fan, W.; He, X.; Zhu, Y.; Xu, L. Downregulation of MicroRNA-30c-5p Was Responsible for Cell Migration and Tumor Metastasis via COTL1-Mediated Microfilament Arrangement in Breast Cancer. Gland Surg. 2020, 9, 747–758. [Google Scholar] [CrossRef]
- Lv, M.; Shen, Y.; Yang, J.; Li, S.; Wang, B.; Chen, Z.; Li, P.; Liu, P.; Yang, J. Angiomotin Family Members: Oncogenes or Tumor Suppressors? Int. J. Biol. Sci. 2017, 13, 772–781. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, L.; Riedel, H.; Wang, K.; Yang, Y.; Dinu, C.Z.; Rojanasakul, Y. Mesothelin Promotes Epithelial-to-Mesenchymal Transition and Tumorigenicity of Human Lung Cancer and Mesothelioma Cells. Mol. Cancer 2017, 16, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.C.; Chen, C.A.; Chen, P.J.; Chiang, Y.C.; Chen, Y.L.; Mao, T.L.; Lin, H.W.; Lin Chiang, W.H.; Cheng, W.F. Mesothelin Enhances Invasion of Ovarian Cancer by Inducing MMP-7 through MAPK/ERK and JNK Pathways. Biochem. J. 2012, 442, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, L.; Li, D.; Wang, H.B.; Chen, Q.F. Mesothelin Promotes Invasion and Metastasis in Breast Cancer Cells. J. Int. Med. Res. 2012, 40, 2109–2116. [Google Scholar] [CrossRef]
- Kim, J.; Shapiro, M.J.; Bamidele, A.O.; Gurel, P.; Thapa, P.; Higgs, H.N.; Hedin, K.E.; Shapiro, V.S.; Billadeau, D.D. Coactosin-like 1 Antagonizes Cofilin to Promote Lamellipodial Protrusion at the Immune Synapse. PLoS ONE 2014, 9, e85090. [Google Scholar] [CrossRef]
- Vidotto, A.; Polachini, G.M.; De Paula-Silva, M.; Oliani, S.M.; Henrique, T.; López, R.V.M.; Cury, P.M.; Nunes, F.D.; Góis-Filho, J.F.; De Carvalho, M.B.; et al. Differentially Expressed Proteins in Positive versus Negative HNSCC Lymph Nodes. BMC Med. Genomics 2018, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nekulova, M.; Holcakova, J.; Gu, X.; Hrabal, V.; Galtsidis, S.; Orzol, P.; Liu, Y.; Logotheti, S.; Zoumpourlis, V.; Nylander, K.; et al. ΔNp63α Expression Induces Loss of Cell Adhesion in Triple-Negative Breast Cancer Cells. BMC Cancer 2016, 16, 782. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glathar, A.R.; Oyelakin, A.; Nayak, K.B.; Sosa, J.; Romano, R.-A.; Sinha, S. A Systemic and Integrated Analysis of p63-Driven Regulatory Networks in Mouse Oral Squamous Cell Carcinoma. Cancers 2023, 15, 446. https://doi.org/10.3390/cancers15020446
Glathar AR, Oyelakin A, Nayak KB, Sosa J, Romano R-A, Sinha S. A Systemic and Integrated Analysis of p63-Driven Regulatory Networks in Mouse Oral Squamous Cell Carcinoma. Cancers. 2023; 15(2):446. https://doi.org/10.3390/cancers15020446
Chicago/Turabian StyleGlathar, Alexandra Ruth, Akinsola Oyelakin, Kasturi Bala Nayak, Jennifer Sosa, Rose-Anne Romano, and Satrajit Sinha. 2023. "A Systemic and Integrated Analysis of p63-Driven Regulatory Networks in Mouse Oral Squamous Cell Carcinoma" Cancers 15, no. 2: 446. https://doi.org/10.3390/cancers15020446
APA StyleGlathar, A. R., Oyelakin, A., Nayak, K. B., Sosa, J., Romano, R. -A., & Sinha, S. (2023). A Systemic and Integrated Analysis of p63-Driven Regulatory Networks in Mouse Oral Squamous Cell Carcinoma. Cancers, 15(2), 446. https://doi.org/10.3390/cancers15020446